
Potential Clinically Relevant Effects of Sialylation on Human Serum AAG-Drug Interactions Assessed by Isothermal Titration Calorimetry: Insight into Pharmacoglycomics?
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
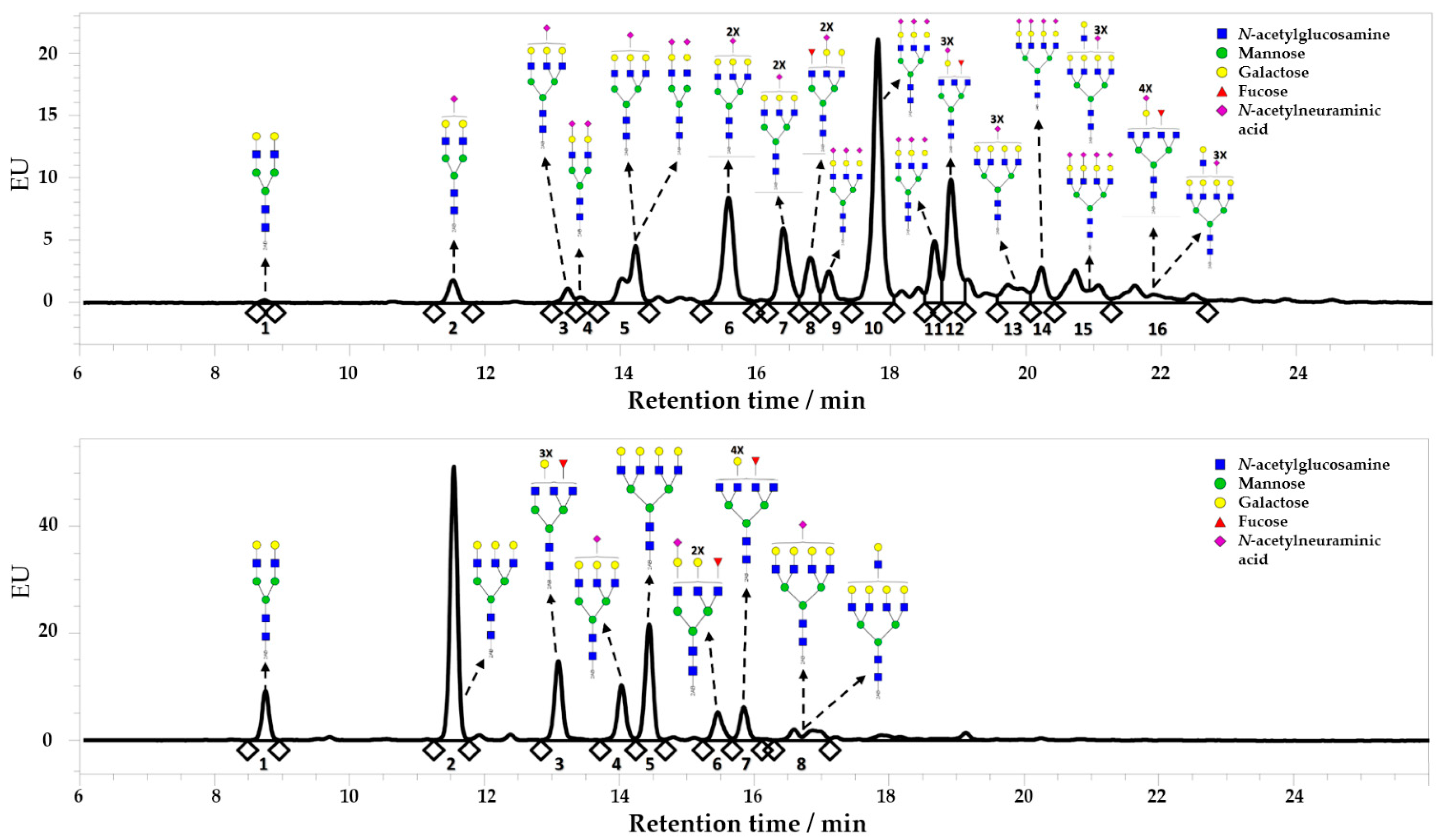
2.1. Enzymatic Desialylation of Human AAG
2.2. Analysis of Binding Affinity to Native and Desialylated AAG
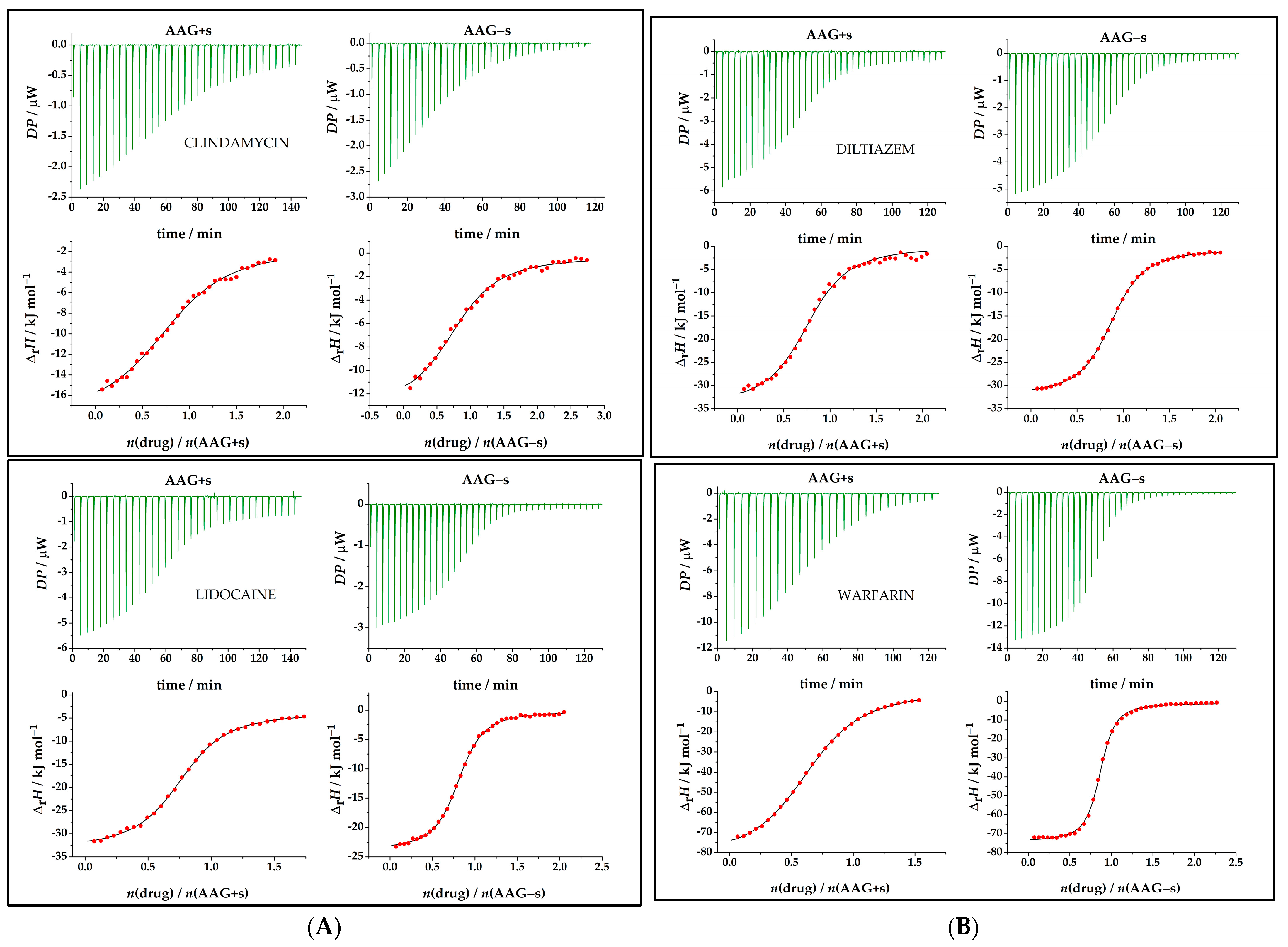
2.3. Thermodynamic Analysis of Drug Binding for the Native and Desialylated AAG
2.4. Driving Forces for Drug Binding to AAG
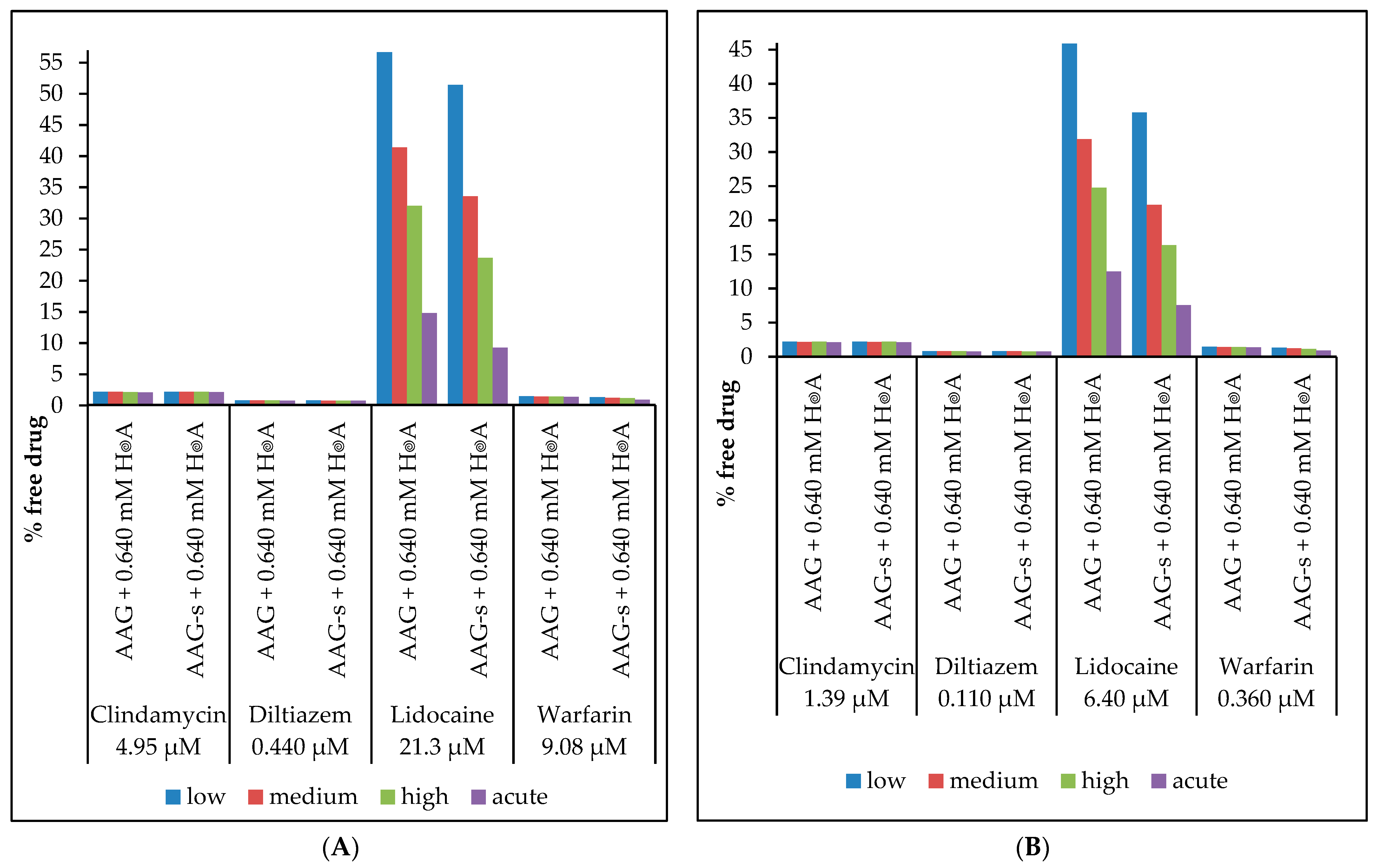
2.5. Determination of the Free Fraction of Drugs
2.6. Pharmacoglycomics
3. Materials and Methods
3.1. Materials and Solution Preparation
3.2. Desialylation of Human AAG
3.3. UV/Vis Spectrophotometry
3.4. Microcalorimetric Titrations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Otagiri, M. A Molecular Functional Study on the Interactions of Drugs with Plasma Proteins. Drug Metab. Pharmacokinet. 2005, 20, 309–323. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Molecular aspects of ligand binding to serum albumin. Pharmacol. Rev. 1981, 33, 17–53. [Google Scholar] [PubMed]
- Mehvar, R. Role of Protein Binding in Pharmacokinetics. Am. J. Pharm. Educ. 2005, 69, 1526. [Google Scholar] [CrossRef]
- Israili, Z.H.; Dayton, P.G. Human alpha-1-glycoprotein and its interaction with drugs. Drug Metab. Rev. 2001, 33, 161–235. [Google Scholar] [CrossRef] [PubMed]
- Kremer, J.M.; Wilting, J.; Janssen, L.H. Drug binding to human alpha-1-acid glycoprotein in health and disease. Pharmacol. Rev. 1988, 40, 1–47. [Google Scholar]
- Bi, C.; Zheng, X.; Hage, D.S. Analysis of free drug fractions in serum by ultrafast affinity extraction and two-dimensional affinity chromatography using α1-acid glycoprotein microcolumns. J. Chromatogr. A 2016, 1432, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Yasgar, A.; Furdas, S.D.; Maloney, D.J.; Jadhav, A.; Jung, M.; Simeonov, A. High-Throughput 1,536-Well Fluorescence Polarization Assays for α1-Acid Glycoprotein and Human Serum Albumin Binding. PLoS ONE 2012, 7, e45594. [Google Scholar] [CrossRef]
- Fitos, I.; Visy, J.; Zsila, F.; Mády, G.; Simonyi, M. Selective binding of imatinib to the genetic variants of human α1-acid glycoprotein. Biochim. Biophys. Acta Gen. Subj. 2016, 1760, 1704–1712. [Google Scholar] [CrossRef]
- Beeram, S.; Bi, C.; Zheng, X.; Hage, D.S. Chromatographic studies of drug interactions with alpha1-acid glycoprotein by ultrafast affinity extraction and peak profiling. J. Chromatogr. A 2017, 1497, 92–101. [Google Scholar] [CrossRef]
- Fournier, T.; Medjoubi-N, N.; Porquet, D. Alpha-1-acid glycoprotein. Biochim. Biophys. Acta 2000, 1482, 157–171. [Google Scholar] [CrossRef]
- Pj, M.; Rc, J.; Mn, G. The interaction of alpha-1-acid glycoprotein with endogenous autocoids, in particular, platelet activating factor (PAF). Prog. Clin. Biol. Res. 1989, 300, 307–319. [Google Scholar]
- Schonfeld, D.L.; Raimond, B.G.R.; Uwe, M.; Skera, A. The 1.8-A crystal structure of alpha1-acid glycoprotein (Orosomucoid) solved by UV RIP reveals the broad drug-binding activity of this human plasma lipocalin. J. Mol. Biol. 2008, 384, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Dente, L.; Pizza, M.G.; Metspalu, A.; Cortese, R. Structure and expression of the genes coding for human alpha 1-acid glycoprotein. EMBO J. 1987, 6, 2289–2296. [Google Scholar] [CrossRef] [PubMed]
- Dente, L.; Rüther, U.; Tripodi, M.; Wagner, E.F.; Cortese, R. Expression of human alpha 1-acid glycoprotein genes in cultured cells and in transgenic mice. Genes Dev. 1988, 2, 259–266. [Google Scholar] [CrossRef]
- Eap, C.B.; Baumann, P. Isoelectric focusing of alpha-1 acid glycoprotein (orosomucoid) in immobilized pH-gradients with 8M urea: Detection of its desialylated variants using an alkaline phosphatase-linked secondary antibody system. Electrophoresis 1988, 9, 650–654. [Google Scholar] [CrossRef]
- Hervé, F.; Caron, G.; Duché, J.-C.; Gaillard, P.; Rahman, N.A.; Tsantili-Kakoulidou, A.; Carrupt, P.-A.; D’athis, P.; Tillement, J.-P.; Testa, B. Ligand Specificity of the Genetic Variants of Human α1-Acid Glycoprotein: Generation of a Three-Dimensional Quantitative Structure-Activity Relationship Model for Drug Binding to the A Variant. Mol. Pharmacol. 1998, 54, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Hervé, F.; Gomas, E.; Duché, J.-C.; Tillement, J.-P. Fractionation of the genetic variants of human α1-acid glycoprotein in the native form by chromatography on an immobilized copper(II) affinity adsorbent: Heterogeneity of the separate variants by isoelectrofocusing and by concanavalin A affinity chromatography. J. Chromatogr. B Biomed. Sci. Appl. 1993, 615, 47–57. [Google Scholar]
- Schmid, K.; Nimberg, R.; Kimura, A.; Yamaguchi, H.; Binette, J. The carbohydrate units of human plasma α1-Acid glycoprotein. Biochim. Biophys. Acta Protein Struct. 1977, 492, 291–302. [Google Scholar] [CrossRef]
- Shiono, H.; Shibukawa, A.; Kuroda, Y.; Nakagawa, T. Effect of sialic acid residues of human α1-acid glycoprotein on stereoselectivity in basic drug-protein binding. Chirality 1997, 9, 291–296. [Google Scholar] [CrossRef]
- Friedman, M.L.; Wermeling, J.R.; Halsall, H.B. The influence of N-acetylneuraminic acid on the properties of human orosomucoid. Biochem. J. 1986, 236, 149–153. [Google Scholar] [CrossRef]
- Primozic, S.; McNamara, P. Effect of the Sialylation State of α1-Acid Glycoprotein on Propranolol Binding. J. Pharm. Sci. 1985, 74, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Bordas, M.C.; Serbource-Goguel, N.S.; Feger, J.M.; Maccario, J.M.; Agneray, J.M.; Durand, G.M. Evaluation of the degree of desialylation of serum α1-acid glycoprotein and α1-antitrypsin. Clin. Chim. Acta 1982, 125, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.S.; Williams, S. Serum inhibitors of desialylated glycoprotein binding to hepatocyte membranes. Biochim. Biophys. Acta Gen. Subj. 1978, 543, 41–52. [Google Scholar] [CrossRef] [PubMed]
- De Graaf, T.W.; Van der Stelt, M.E.; Anbergen, M.G.; van Dijk, W. Inflammation-induced expression of sialyl Lewis X-containing glycan structures on alpha 1-acid glycoprotein (orosomucoid) in human sera. J. Exp. Med. 1993, 177, 657–666. [Google Scholar] [CrossRef]
- Trbojević-Akmačić, I.; Lageveen-Kammeijer, G.S.M.; Heijs, B.; Petrović, T.; Deriš, H.; Wuhrer, M.; Lauc, G. High-Throughput Glycomic Methods. Chem. Rev. 2022, 122, 15865–15913. [Google Scholar] [CrossRef]
- Keser, T.; Tijardović, M.; Gornik, I.; Lukić, E.; Lauc, G.; Gornik, O.; Novokmet, M. High-Throughput and Site-Specific N-Glycosylation Analysis of Human Alpha-1-Acid Glycoprotein Offers a Great Potential for New Biomarker Discovery. Mol. Cell. Proteom. 2021, 20, 100044. [Google Scholar] [CrossRef]
- Higai, K.; Azuma, Y.; Aoki, Y.; Matsumoto, K. Altered glycosylation of α1-acid glycoprotein in patients with inflammation and diabetes mellitus. Clin. Chim. Acta 2003, 329, 117–125. [Google Scholar] [CrossRef]
- Gornik, O.; Wagner, J.; Pučić, M.; Knežević, A.; Redžić, I.; Lauc, G. Stability of N-glycan profiles in human plasma. Glycobiology 2009, 19, 1547–1553. [Google Scholar] [CrossRef]
- Taguchi, K.; Nishi, K.; Chuang, V.; Maruyama, T.; Otagiri, M. Molecular Aspects of Human Alpha-1 Acid Glycoprotein—Structure and Function. In Acute Phase Proteins; Janciauskiene, S., Ed.; InTech: New York, NY, USA, 2013; pp. 139–162. [Google Scholar]
- Bteich, M. An overview of albumin and alpha-1-acid glycoprotein main characteristics: Highlighting the roles of amino acids in binding kinetics and molecular interactions. Heliyon 2019, 5, e02879. [Google Scholar] [CrossRef]
- Fischer, K.; Kettunen, J.; Würtz, P.; Haller, T.; Havulinna, A.S.; Kangas, A.; Soininen, P.; Esko, T.; Tammesoo, M.-L.; Mägi, R.; et al. Biomarker Profiling by Nuclear Magnetic Resonance Spectroscopy for the Prediction of All-Cause Mortality: An Observational Study of 17,345 Persons. PLoS Med. 2014, 11, e1001606. [Google Scholar] [CrossRef]
- Walle, U.; Fagan, T.; Topmiller, M.; Conradi, E.; Walle, T. The influence of gender and sex steroid hormones on the plasma binding of propranolol enantiomers. Br. J. Clin. Pharmacol. 1994, 37, 21–25. [Google Scholar] [CrossRef] [PubMed]
- Tsen, L.C.; Tarshis, J.; Denson, D.D.; Osathanondh, R.; Datta, S.; Bader, A.M. Measurements of Maternal Protein Binding of Bupivacaine Throughout Pregnancy. Anesth. Analg. 1999, 89, 965. [Google Scholar] [CrossRef] [PubMed]
- Friganović, T.; Tomašić, A.; Šeba, T.; Biruš, I.; Kerep, R.; Borko, V.; Šakić, D.; Gabričević, M.; Weitner, T. Low-pressure chromatographic separation and UV/Vis spectrophotometric characterization of the native and desialylated human apo-transferrin. Heliyon 2021, 7, e08030. [Google Scholar] [CrossRef] [PubMed]
- Saldova, R.; Asadi Shehni, A.; Haakensen, V.D.; Steinfeld, I.; Hilliard, M.; Kifer, I.; Helland, A.; Yakhini, Z.; Børresen-Dale, A.-L.; Rudd, P.M.A. Analysis of Lidocaine Interactions with Serum Proteins Using High-Performance Affinity Chrotion quantitative UPLC. J. Proteome Res. 2014, 13, 2314–2327. [Google Scholar] [CrossRef]
- Olsson, T.S.G.; Williams, M.A.; Pitt, W.R.; Ladbury, J.E. The Thermodynamics of Protein–Ligand Interaction and Solvation: Insights for Ligand Design. J. Mol. Biol. 2008, 384, 1002–1017. [Google Scholar] [CrossRef] [PubMed]
- Qiang, Z.; Adams, C. Potentiometric determination of acid dissociation constants (pKa) for human and veterinary antibiotics. Water Res. 2004, 38, 2874–2890. [Google Scholar] [CrossRef]
- Settimo, L.; Bellman, K.; Knegtel, R.M.A. Comparison of the accuracy of experimental and predicted pKa values of basic and acidic compounds. Pharm. Res. 2014, 31, 1082–1095. [Google Scholar] [CrossRef]
- Huang, R.Y.-C.; Hudgens, J.W. Effects of Desialylation on Human α1-Acid Glycoprotein–Ligand Interactions. Biochemistry 2013, 52, 7127–7136. [Google Scholar] [CrossRef]
- Nowak, P.; Olechowska, P.; Mitoraj, M.; Woźniakiewicz, M.; Kościelniak, P. Determination of acid dissociation constants of warfarin and hydroxywarfarins by capillary electrophoresis. J. Pharm. Biomed. Anal. 2015, 112, 89–97. [Google Scholar] [CrossRef]
- Claveria-Gimeno, R.; Vega, S.; Abian, O.; Velazquez-Campoy, A. A look at ligand binding thermodynamics in drug discovery. Expert Opin. Drug Discov. 2017, 12, 363–377. [Google Scholar] [CrossRef]
- Bianconi, M.L. Avoiding Buffer Interference in ITC Experiments: A Case Study from the Analysis of Entropy-Driven Reactions of Glucose-6-Phosphate Dehydrogenase. Methods Enzymol. 2016, 567, 237–256. [Google Scholar] [PubMed]
- Jelesarov, I.; Bosshard, H.R. Isothermal titration calorimetry and differential scanning calorimetry as complementary tools to investigate the energetics of biomolecular recognition. J. Mol. Recognit. 1999, 12, 3–18. [Google Scholar] [CrossRef]
- Alderighi, L.; Gans, P.; Ienco, A.; Peters, D.; Sabatini, A.; Vacca, A. Hyperquad simulation and speciation (HySS): A utility program for the investigation of equilibria involving soluble and partially soluble species. Coord. Chem. Rev. 1999, 184, 311–318. [Google Scholar] [CrossRef]
- Putnam, F.W. Alpha, Beta, Gamma, Omega—The Structure of the Plasma Proteins. In The Plasma Proteins, 2nd ed.; Putnam, F.W., Ed.; Academic Press: Orlando, FL, USA, 1984; Volume 4, pp. 45–166. [Google Scholar]
- Lidocaine Level: Reference Range, Interpretation, Collection and Panels. Available online: https://emedicine.medscape.com/article/2090152-overview (accessed on 7 June 2022).
- Cardizem LA (Diltiazem): Uses, Dosage, Side Effects, Interactions, Warning. RxList. Available online: https://www.rxlist.com/cardizem-la-drug.htm (accessed on 7 June 2022).
- Cleocin Hydrochloride. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/050162s098s099lbl.pdf (accessed on 7 June 2022).
- Sun, S.; Wang, M.; Su, L.; Li, J.; Li, H.; Gu, D. Study on warfarin plasma concentration and its correlation with international normalized ratio. J. Pharm. Biomed. Anal. 2006, 42, 218–222. [Google Scholar] [CrossRef]
- Albumin. Available online: https://www.forthwithlife.co.uk/our-service/biomarkers/alb/ (accessed on 10 April 2023).
- Lauc, G.; Rudan, I.; Campbell, H.; Rudd, P.M. Complex genetic regulation of protein glycosylation. Mol. Biosyst. 2010, 6, 329–335. [Google Scholar] [CrossRef]
- Štambuk, T.; Klasić, M.; Zoldoš, V.; Lauc, G. N-glycans as functional effectors of genetic and epigenetic disease risk. Mol. Aspects Med. 2021, 79, 100891. [Google Scholar] [CrossRef]
- Klarić, L.; Tsepilov, Y.A.; Stanton, C.M.; Mangino, M.; Sikka, T.T.; Esko, T.; Pakhomov, E.; Salo, P.; Deelen, J.; McGurnaghan, S.J.; et al. Glycosylation of immunoglobulin G is regulated by a large network of genes pleiotropic with inflammatory diseases. Sci. Adv. 2020, 6, eaax0301. [Google Scholar] [CrossRef]
- Landini, A.; Trbojević-Akmačić, I.; Navarro, P.; Tsepilov, Y.A.; Sharapov, S.Z.; Vučković, F.; Polašek, O.; Hayward, C.; Petrović, T.; Vilaj, M.; et al. Genetic regulation of post-translational modification of two distinct proteins. Nat. Commun. 2022, 13, 1586. [Google Scholar] [CrossRef]
- Krištić, J.; Zaytseva, O.; Ram, R.; Nguyen, Q.; Novokmet, M.; Vučković, F.; Vilaj, M.; Trbojević-Akmačić, I.; Pezer, M.; Davern, K.M.; et al. Profiling and genetic control of the murine immunoglobulin G glycome. Nat. Chem. Biol. 2018, 14, 516–524. [Google Scholar] [CrossRef]
- Wu, D.; Struwe, W.B.; Harvey, D.J.; Ferguson, M.A.J.; Robinson, C.V. N-glycan microheterogeneity regulates interactions of plasma proteins. Proc. Natl. Acad. Sci. USA 2018, 115, 8763–8768. [Google Scholar] [CrossRef]
- Soman, S.; Yoo, M.J.; Jang, Y.J.; Hage, D.S. Analysis of Lidocaine Interactions with Serum Proteins Using High-Performance Affinity Chromatography. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 705–708. [Google Scholar] [CrossRef]
- Otagiri, M.; Maruyama, T.; Imai, T.; Suenaga, A.; Imamura, Y. A comparative study of the interaction of warfarin with human alpha 1-acid glycoprotein and human albumin. J Pharm Pharmacol. 1987, 39, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Wulkersdorfer, B.; Wicha, S.G.; Kurdina, E.; Carrion Carrera, S.F.; Matzneller, P.; Al Jalali, V.; Vossen, M.G.; Riesenhuber, S.; Lackner, E.; Dorn, C.; et al. Protein binding of clindamycin in vivo by means of intravascular microdialysis in healthy volunteers. J. Antimicrob. Chemother. 2021, 76, 2106–2113. [Google Scholar] [CrossRef] [PubMed]
- SialEXO Genovis. Available online: https://www.genovis.com/products/exoglycosidases/sialexo/immobilized-sialexo/ (accessed on 18 November 2022).
- Štambuk, T.; Cvetko, A.; Weitner, T. Protocol for ultra performance liquid chromatography-mass spectrometry N-glycan analysis of the native and desialylated human apo-transferrin. Heliyon 2021, 9, e08030. [Google Scholar] [CrossRef]
- Šeba, T.; Friganović, T.; Weitner, T. Protocol for spectrophotometric determination of native and desialylated apo-transferrin molar absorption coefficients. Heliyon 2021, 7, e08030. [Google Scholar] [CrossRef]
- T.DIST.2T Function. Available online: https://support.microsoft.com/en-us/office/t-dist-2t-function-198e9340-e360-4230-bd21-f52f22ff5c28 (accessed on 7 June 2022).
- Di, L. An Update on the Importance of Plasma Protein Binding in Drug Discovery and Development. Expert Opin. Drug Discov. 2021, 16, 1453–1465. [Google Scholar] [CrossRef]
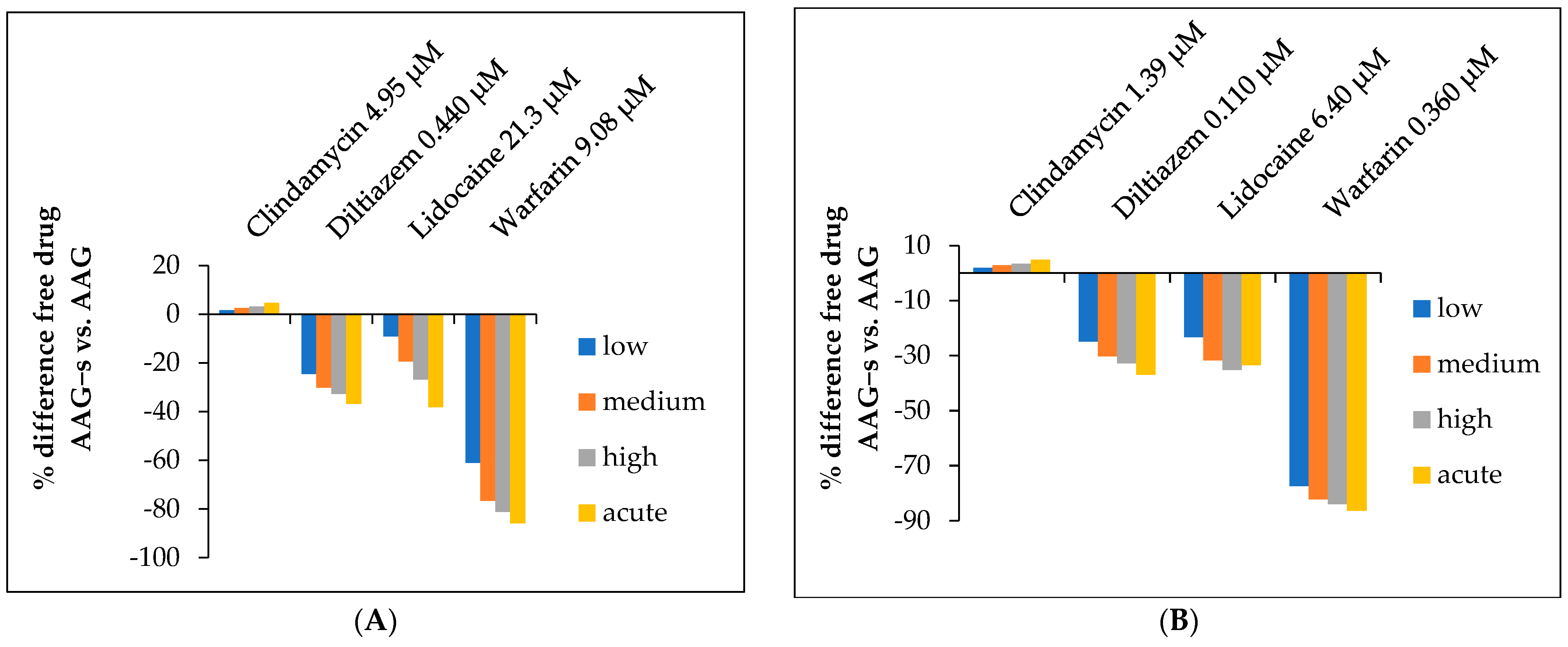
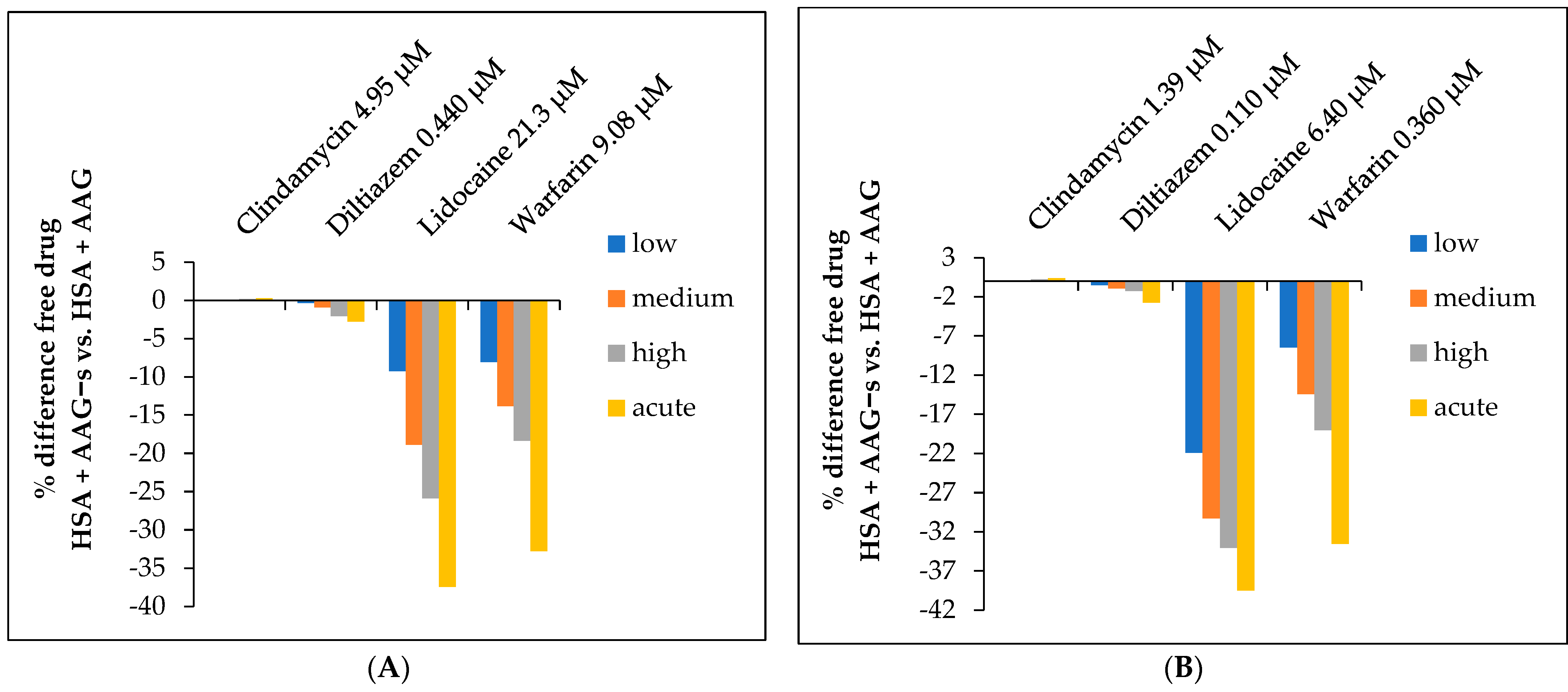

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | AAGs * (µM) | AAGs * (µM) |
|---|---|---|
| Clindamycin | 31.0 2.40 | 32.8 3.32 |
| Diltiazem | 13.2 0.490 | 7.75 1.45 |
| Lidocaine | 9.43 0.950 | 5.21 0.930 |
| Warfarin | 14.4 2.83 | 1.65 0.150 |
| Drug | AAGs | |||
|---|---|---|---|---|
| N * | H° * (kJ mol−1) | S° * (J K−1 mol−1) | G° * (kJ mol−1) | |
| Clindamycin ** | 0.880 0.169 | 18.1 2.48 | 27.9 8.48 | 26.8 0.141 |
| Diltiazem ** | 0.804 0.238 | 31.5 4.24 | 7.80 1.74 | .9 1.130 |
| Lidocaine ** | 0.810 0.0990 | 29.6 0.212 | 0.725 1.53 | 29.9 0.283 |
| Warfarin ** | 0.654 0.117 | 81.4 1.56 | 5.42 | 28.9 0.424 |
| AAGs | ||||
| Clindamycin | 0.846 0.0280 | 23.3 1.07 | 11.1 4.42 | 26.7 0.283 |
| Diltiazem | 0.868 0.0290 | 31.8 1.83 | 4.09 0.103 | 30.4 0.414 |
| Lidocaine | 0.818 0.226 | 26.5 3.61 | 15.9 3.17 | 31.4 0.424 |
| Warfarin | 0.795 0.0290 | .1 4.31 | 168 5.86 | 34.3 0.212 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kerep, R.; Šeba, T.; Borko, V.; Weitner, T.; Keser, T.; Lauc, G.; Gabričević, M. Potential Clinically Relevant Effects of Sialylation on Human Serum AAG-Drug Interactions Assessed by Isothermal Titration Calorimetry: Insight into Pharmacoglycomics? Int. J. Mol. Sci. 2023, 24, 8472. https://doi.org/10.3390/ijms24108472
Kerep R, Šeba T, Borko V, Weitner T, Keser T, Lauc G, Gabričević M. Potential Clinically Relevant Effects of Sialylation on Human Serum AAG-Drug Interactions Assessed by Isothermal Titration Calorimetry: Insight into Pharmacoglycomics? International Journal of Molecular Sciences. 2023; 24(10):8472. https://doi.org/10.3390/ijms24108472
Chicago/Turabian StyleKerep, Robert, Tino Šeba, Valentina Borko, Tin Weitner, Toma Keser, Gordan Lauc, and Mario Gabričević. 2023. "Potential Clinically Relevant Effects of Sialylation on Human Serum AAG-Drug Interactions Assessed by Isothermal Titration Calorimetry: Insight into Pharmacoglycomics?" International Journal of Molecular Sciences 24, no. 10: 8472. https://doi.org/10.3390/ijms24108472
APA StyleKerep, R., Šeba, T., Borko, V., Weitner, T., Keser, T., Lauc, G., & Gabričević, M. (2023). Potential Clinically Relevant Effects of Sialylation on Human Serum AAG-Drug Interactions Assessed by Isothermal Titration Calorimetry: Insight into Pharmacoglycomics? International Journal of Molecular Sciences, 24(10), 8472. https://doi.org/10.3390/ijms24108472