
Trichome-Specific Analysis and Weighted Gene Co-Expression Correlation Network Analysis (WGCNA) Reveal Potential Regulation Mechanism of Artemisinin Biosynthesis in Artemisia annua

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Assembly and Annotation of the Transcriptome Data
2.2. Trichome-Specific Gene Analysis in A. annua
2.3. Trichome-Specific Gene Involved in Terpenoid Metabolism
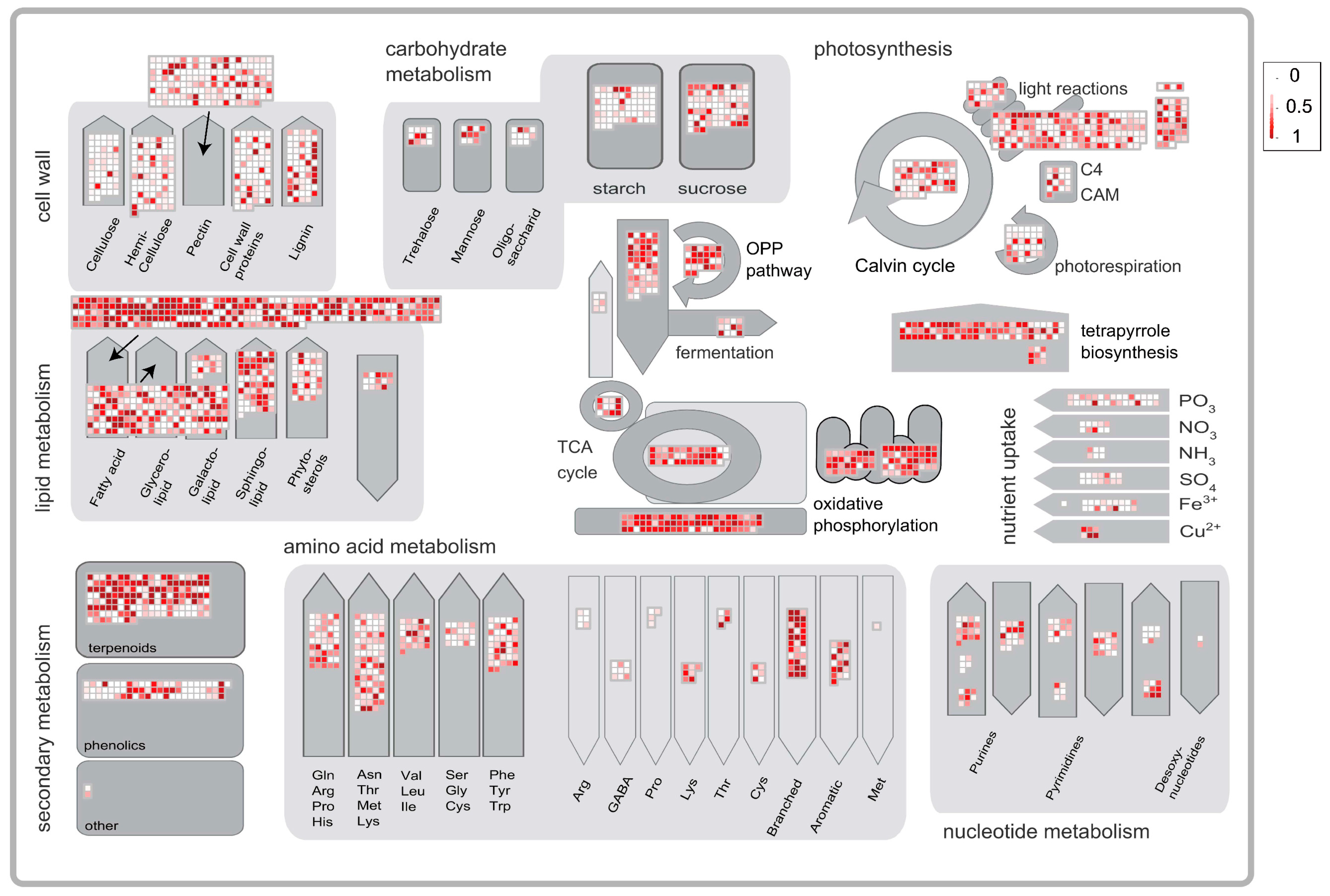
2.4. Enrichment Analysis of the Trichome-Specific Expression Genes
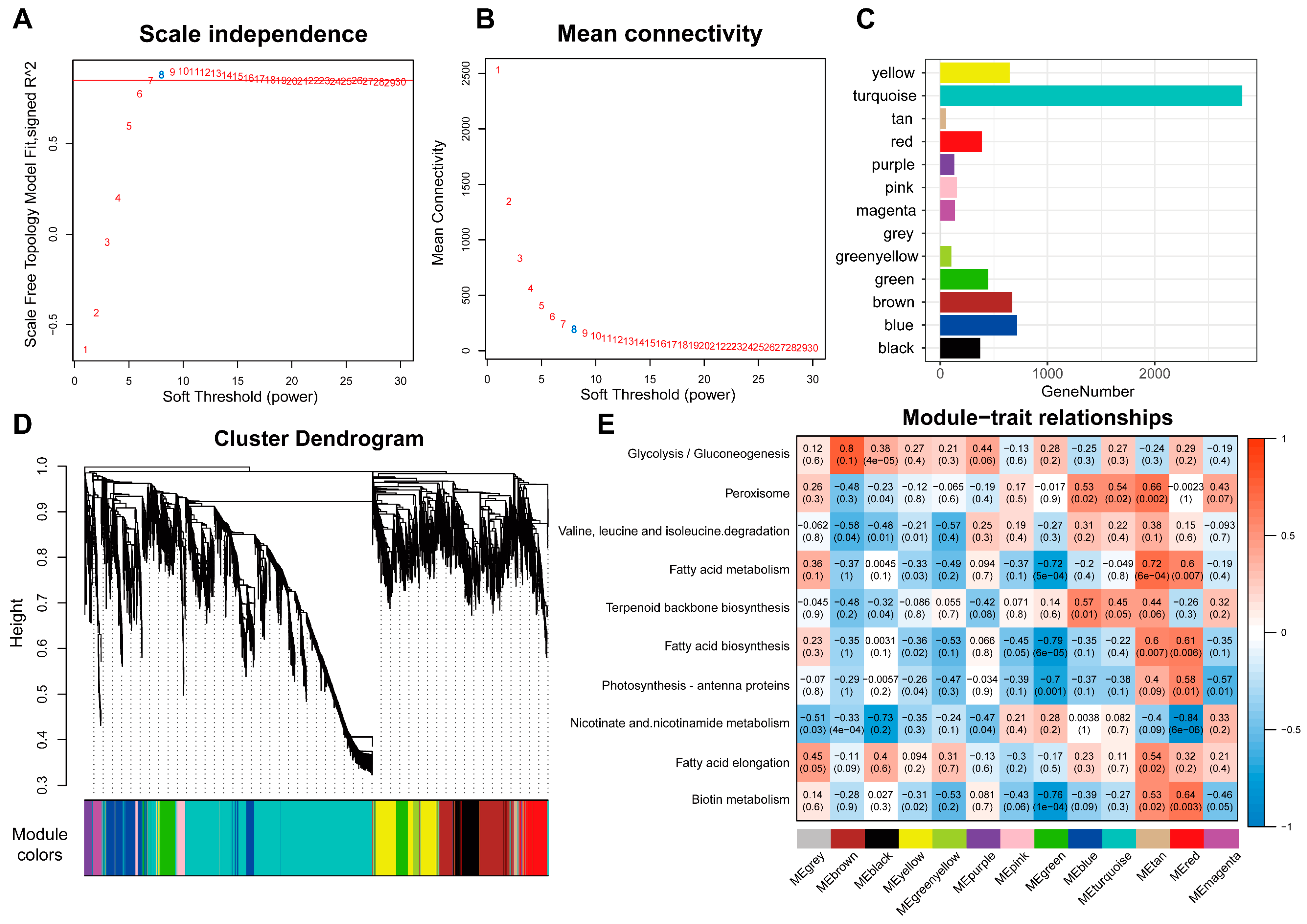
2.5. Weighted Gene Co-Expression Network Analysis
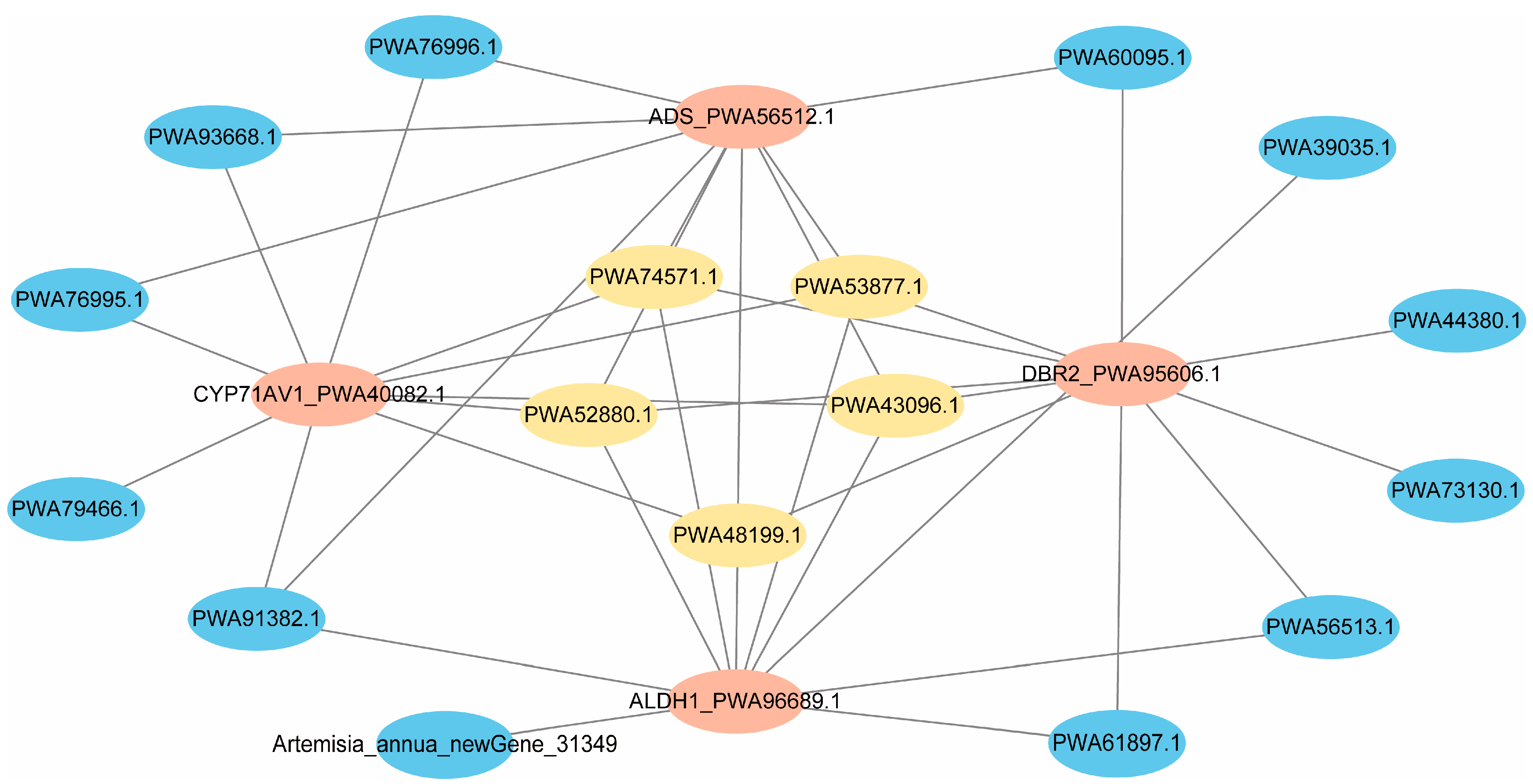
2.6. Identification of Trichome-Specific Hub Genes Affecting Artemisinin Synthesis
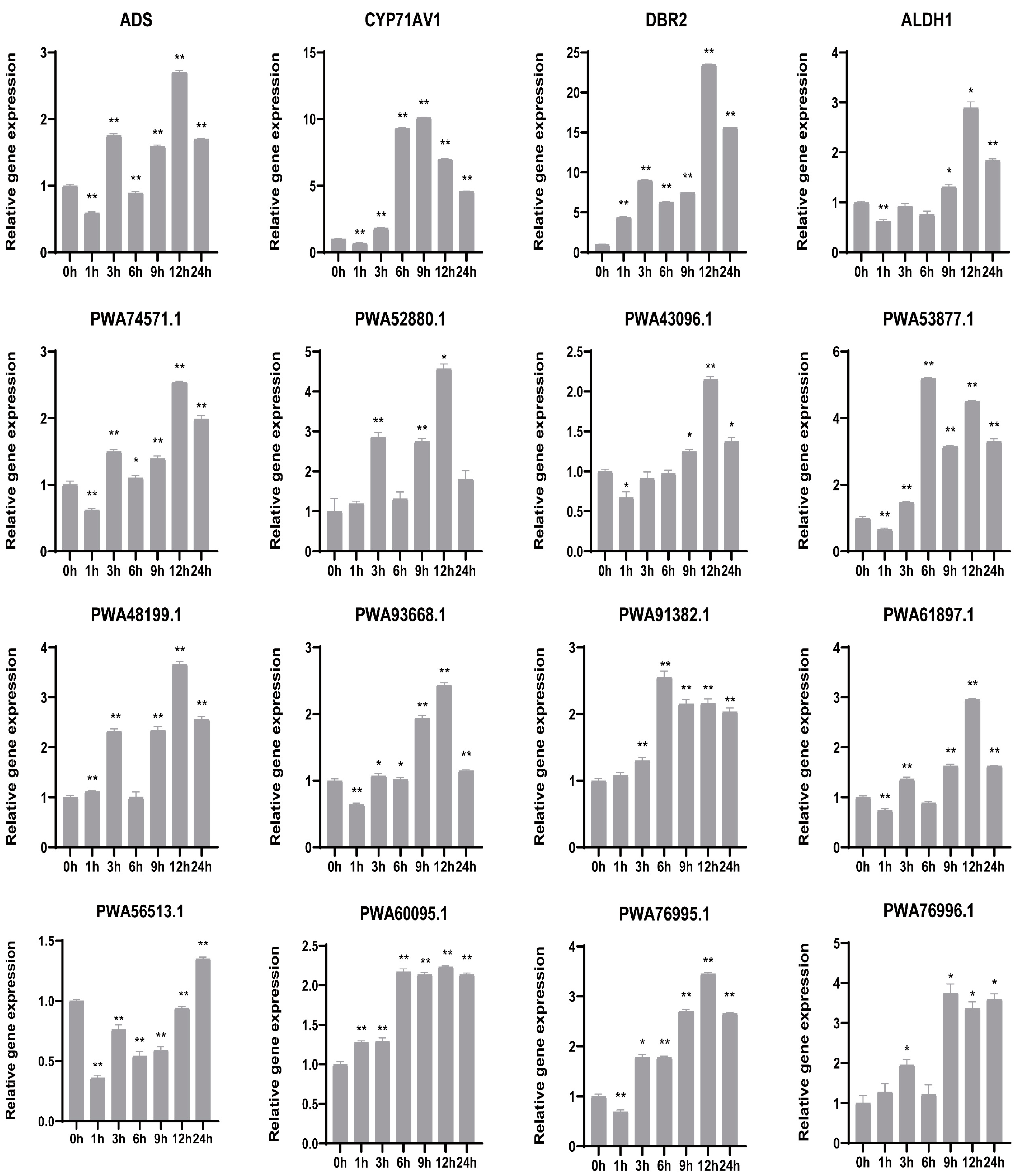
2.7. Quantitative Real Time (qRT)-PCR Validation of Candidate Genes
3. Discussion
4. Materials and Methods
4.1. Resources Used for Data Analysis
4.2. Plant Material Collection and RNAseq
4.3. Quantitative Real Time (qRT)-PCR
4.4. Quality Control and Alignment of Sequencing Datasets
4.5. Gene Annotation and Enrichment Analysis
4.6. Calculation of Trichome Specificity Index
4.7. Expression Pattern Analysis
4.8. Weighted Gene Co-Expression Network Analysis
4.9. Identification of Hub Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, S.H.; Chang, Y.J.; Kim, S.U. Tissue specificity and developmental pattern of amorpha-4,11-diene synthase (ADS) proved by ADS promoter-driven GUS expression in the heterologous plant, Arabidopsis thaliana. Planta Med. 2008, 74, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, J.; Kanagarajan, S.; Lundgren, A.; Brodelius, P.E. Trichome-specific expression of the amorpha-4,11-diene 12-hydroxylase (cyp71av1) gene, encoding a key enzyme of artemisinin biosynthesis in Artemisia annua, as reported by a promoter-GUS fusion. Plant Mol. Biol. 2013, 81, 119–138. [Google Scholar] [CrossRef]
- Zhang, Y.; Teoh, K.H.; Reed, D.W.; Maes, L.; Goossens, A.; Olson, D.J.; Ross, A.R.; Covello, P.S. The molecular cloning of artemisinic aldehyde Delta11(13) reductase and its role in glandular trichome-dependent biosynthesis of artemisinin in Artemisia annua. J. Biol. Chem. 2008, 283, 21501–21508. [Google Scholar] [CrossRef] [PubMed]
- Duke, M.V.; Paul, R.N.; Elsohly, H.N.; Sturtz, G.; Duke, S.O. Localization of Artemisinin and Artemisitene in Foliar Tissues of Glanded and Glandless Biotypes of Artemisia annua L. Int. J. Plant Sci. 1994, 155, 365–372. [Google Scholar] [CrossRef]
- Ishida, T.; Kurata, T.; Okada, K.; Wada, T. A Genetic Regulatory Network in the Development of Trichomes and Root Hairs. Annu. Rev. Plant Biol. 2008, 59, 365–386. [Google Scholar] [CrossRef]
- Maurya, S.; Chandra, M.; Yadav, R.K.; Narnoliya, L.K.; Sangwan, R.S.; Bansal, S.; Sandhu, P.; Singh, U.; Kumar, D.; Sangwan, N.S. Interspecies comparative features of trichomes in Ocimum reveal insights for biosynthesis of specialized essential oil metabolites. Protoplasma 2019, 256, 893–907. [Google Scholar] [CrossRef]
- Ma, Y.N.; Xu, D.B.; Yan, X.; Wu, Z.K.; Kayani, S.I.; Shen, Q.; Fu, X.Q.; Xie, L.H.; Hao, X.L.; Hassani, D.; et al. Jasmonate- and abscisic acid-activated AaGSW1-AaTCP15/AaORA transcriptional cascade promotes artemisinin biosynthesis in Artemisia annua. Plant Biotechnol. J. 2021, 19, 1412–1428. [Google Scholar] [CrossRef]
- Chalvin, C.; Drevensek, S.; Dron, M.; Bendahmane, A.; Boualem, A. Genetic Control of Glandular Trichome Development. Trends Plant Sci. 2020, 25, 477–487. [Google Scholar] [CrossRef]
- Shi, P.; Fu, X.; Shen, Q.; Liu, M.; Pan, Q.; Tang, Y.; Jiang, W.; Lv, Z.; Yan, T.; Ma, Y.; et al. The roles of AaMIXTA1 in regulating the initiation of glandular trichomes and cuticle biosynthesis in Artemisia annua. New Phytol. 2018, 217, 261–276. [Google Scholar] [CrossRef]
- Yan, T.; Li, L.; Xie, L.; Chen, M.; Shen, Q.; Pan, Q.; Fu, X.; Shi, P.; Tang, Y.; Huang, H.; et al. A novel HD-ZIP IV/MIXTA complex promotes glandular trichome initiation and cuticle development in Artemisia annua. New Phytol. 2018, 218, 567–578. [Google Scholar] [CrossRef]
- Yan, T.; Chen, M.; Shen, Q.; Li, L.; Fu, X.; Pan, Q.; Tang, Y.; Shi, P.; Lv, Z.; Jiang, W.; et al. HOMEODOMAIN PROTEIN 1 is required for jasmonate-mediated glandular trichome initiation in Artemisia annua. New Phytol. 2017, 213, 1145–1155. [Google Scholar] [CrossRef]
- Chen, T.T.; Liu, H.; Li, Y.P.; Yao, X.H.; Qin, W.; Yan, X.; Wang, X.Y.; Peng, B.W.; Zhang, Y.J.; Shao, J.; et al. AaSEPALLATA1 integrates JA and light-regulated glandular secretory trichome initiation in Artemisia annua. Plant Physiol. 2023, kiad113. [Google Scholar] [CrossRef]
- Wang, C.; Chen, T.; Li, Y.; Liu, H.; Qin, W.; Wu, Z.; Peng, B.; Wang, X.; Yan, X.; Fu, X.; et al. AaWIN1, an AP2/ERF protein, positively regulates glandular secretory trichome initiation in Artemisia annua. Plant Sci. Int. J. Exp. Plant Biol. 2023, 329, 111602. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fu, X.; Li, L.; Sun, X.; Tang, K.; Zhao, J. AaSPL9 affects glandular trichomes initiation by positively regulating expression of AaHD1 in Artemisia annua L. Plant Sci. 2022, 317, 111172. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Y.; Zhang, Q.; Qi, Y.; Guo, D. Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genom. 2009, 10, 465. [Google Scholar] [CrossRef]
- Graham, I.A.; Besser, K.; Blumer, S.; Branigan, C.A.; Czechowski, T.; Elias, L.; Guterman, I.; Harvey, D.; Isaac, P.G.; Khan, A.M.; et al. The Genetic Map of Artemisia annua L. Identifies Loci Affecting Yield of the Antimalarial Drug Artemisinin. Science 2010, 327, 328–331. [Google Scholar] [CrossRef]
- Soetaert, S.S.; Van Neste, C.M.; Vandewoestyne, M.L.; Head, S.R.; Goossens, A.; Van Nieuwerburgh, F.C.; Deforce, D.L. Differential transcriptome analysis of glandular and filamentous trichomes in Artemisia annua. BMC Plant Biol. 2013, 13, 220. [Google Scholar] [CrossRef]
- Shen, Q.; Zhang, L.; Liao, Z.; Wang, S.; Yan, T.; Shi, P.; Liu, M.; Fu, X.; Pan, Q.; Wang, Y.; et al. The Genome of Artemisia annua Provides Insight into the Evolution of Asteraceae Family and Artemisinin Biosynthesis. Mol. Plant 2018, 11, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Kryuchkova-Mostacci, N.; Robinson-Rechavi, M. A benchmark of gene expression tissue-specificity metrics. Brief. Bioinform. 2016, 18, 205–214. [Google Scholar] [CrossRef]
- Huminiecki, L.; Lloyd, A.T.; Wolfe, K.H. Congruence of tissue expression profiles from Gene Expression Atlas, SAGEmap and TissueInfo databases. BMC Genom. 2003, 4, 31. [Google Scholar] [CrossRef]
- Vandenbon, A.; Nakai, K. Modeling tissue-specific structural patterns in human and mouse promoters. Nucleic Acids Res. 2010, 38, 17–25. [Google Scholar] [CrossRef]
- Xiao, S.J.; Zhang, C.; Zou, Q.; Ji, Z.L. TiSGeD: A database for tissue-specific genes. Bioinformatics 2010, 26, 1273–1275. [Google Scholar] [CrossRef]
- Wu, T.; Wang, Y.; Guo, D. Investigation of glandular trichome proteins in Artemisia annua L. using comparative proteomics. PloS ONE 2012, 7, e41822. [Google Scholar] [CrossRef]
- Czechowski, T.; Larson, T.R.; Catania, T.M.; Harvey, D.; Brown, G.D.; Graham, I.A. Artemisia annua mutant impaired in artemisinin synthesis demonstrates importance of nonenzymatic conversion in terpenoid metabolism. Proc. Natl. Acad. Sci. USA 2016, 113, 15150–15155. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Ma, Y.N.; Xu, D.B.; Li, L.; Zhang, F.; Fu, X.Q.; Shen, Q.; Lyu, X.Y.; Wu, Z.K.; Pan, Q.F.; Shi, P.; et al. Jasmonate promotes artemisinin biosynthesis by activating the TCP14-ORA complex in Artemisia annua. Sci. Adv. 2018, 4, eaas9357. [Google Scholar] [CrossRef]
- Murfitt, L.M.; Kolosova, N.; Mann, C.J.; Dudareva, N. Purification and Characterization of S-Adenosyl-l-methionine: Benzoic Acid Carboxyl Methyltransferase, the Enzyme Responsible for Biosynthesis of the Volatile Ester Methyl Benzoate in Flowers of Antirrhinum majus. Arch. Biochem. Biophys. 2000, 382, 145–151. [Google Scholar] [CrossRef]
- Zhao, H.; Ma, H.; Yu, L.; Wang, X.; Zhao, J. Genome-wide survey and expression analysis of amino acid transporter gene family in rice (Oryza sativa L.). PLoS ONE 2012, 7, e49210. [Google Scholar] [CrossRef]
- Crater, A.K.; Roscoe, S.; Fahim, A.; Ananvoranich, S. Toxoplasma ubiquitin-like protease 1, a key enzyme in sumoylation and desumoylation pathways, is under the control of non-coding RNAs. Int. J. Parasitol. 2018, 48, 867–880. [Google Scholar] [CrossRef]
- Lai, C.P.; Lee, C.L.; Chen, P.H.; Wu, S.H.; Yang, C.C.; Shaw, J.F. Molecular analyses of the Arabidopsis TUBBY-like protein gene family. Plant Physiol. 2004, 134, 1586–1597. [Google Scholar] [CrossRef]
- Boggon, T.J.; Shan, W.S.; Santagata, S.; Myers, S.C.; Shapiro, L. Implication of tubby proteins as transcription factors by structure-based functional analysis. Science 1999, 286, 2119–2125. [Google Scholar] [CrossRef]
- Bateman, A.; Finn, R.D.; Sims, P.J.; Wiedmer, T.; Biegert, A.; Söding, J. Phospholipid scramblases and Tubby-like proteins belong to a new superfamily of membrane tethered transcription factors. Bioinformatics 2009, 25, 159–162. [Google Scholar] [CrossRef]
- Xiao, W.; Jang, J. F-box proteins in Arabidopsis. Trends Plant Sci. 2000, 5, 454–457. [Google Scholar] [CrossRef]
- Wang, B.; Kashkooli, A.B.; Sallets, A.; Ting, H.-M.; de Ruijter, N.C.A.; Olofsson, L.; Brodelius, P.; Pottier, M.; Boutry, M.; Bouwmeester, H.; et al. Transient production of artemisinin in Nicotiana benthamiana is boosted by a specific lipid transfer protein from A. annua. Metab. Eng. 2016, 38, 159–169. [Google Scholar] [CrossRef]
- Hao, X.; Zhong, Y.; Fu, X.; Lv, Z.; Shen, Q.; Yan, T.; Shi, P.; Ma, Y.; Chen, M.; Lv, X.; et al. Transcriptome Analysis of Genes Associated with the Artemisinin Biosynthesis by Jasmonic Acid Treatment under the Light in Artemisia annua. Front. Plant Sci. 2017, 8, 971. [Google Scholar] [CrossRef]
- Fu, X.; Peng, B.; Hassani, D.; Xie, L.; Liu, H.; Li, Y.; Chen, T.; Liu, P.; Tang, Y.; Li, L.; et al. AaWRKY9 contributes to light- and jasmonate-mediated to regulate the biosynthesis of artemisinin in Artemisia annua. New Phytol. 2021, 231, 1858–1874. [Google Scholar] [CrossRef]
- Liu, H.; Li, L.; Fu, X.; Li, Y.; Chen, T.; Qin, W.; Yan, X.; Wu, Z.; Xie, L.; Kayani, S.l.; et al. AaMYB108 is the core factor integrating light and jasmonic acid signaling to regulate artemisinin biosynthesis in Artemisia annua. New Phytol. 2022, 237. [Google Scholar] [CrossRef]
- Zheng, H.; Fu, X.; Shao, J.; Tang, Y.; Yu, M.; Li, L.; Huang, L.; Tang, K. Transcriptional regulatory network of high-value active ingredients in medicinal plants. Trends Plant Sci. 2023, 28, 429–446. [Google Scholar] [CrossRef]
- Sallets, A.; Beyaert, M.; Boutry, M.; Champagne, A. Comparative proteomics of short and tall glandular trichomes of Nicotiana tabacum reveals differential metabolic activities. J. Proteome Res. 2014, 13, 3386–3396. [Google Scholar] [CrossRef]
- Jin, J.; Panicker, D.; Wang, Q.; Kim, M.J.; Liu, J.; Yin, J.L.; Wong, L.; Jang, I.C.; Chua, N.H.; Sarojam, R. Next generation sequencing unravels the biosynthetic ability of spearmint (Mentha spicata) peltate glandular trichomes through comparative transcriptomics. BMC Plant Biol. 2014, 14, 292. [Google Scholar] [CrossRef]
- Balcke, G.U.; Bennewitz, S.; Bergau, N.; Athmer, B.; Henning, A.; Majovsky, P.; Jiménez-Gómez, J.M.; Hoehenwarter, W.; Tissier, A. Multi-Omics of Tomato Glandular Trichomes Reveals Distinct Features of Central Carbon Metabolism Supporting High Productivity of Specialized Metabolites. Plant Cell 2017, 29, 960–983. [Google Scholar] [CrossRef]
- Schilmiller, A.L.; Miner, D.P.; Larson, M.; McDowell, E.; Gang, D.R.; Wilkerson, C.; Last, R.L. Studies of a biochemical factory: Tomato trichome deep expressed sequence tag sequencing and proteomics. Plant Physiol. 2010, 153, 1212–1223. [Google Scholar] [CrossRef]
- Harada, E.; Kim, J.-A.; Meyer, A.J.; Hell, R.; Clemens, S.; Choi, Y.-E. Expression Profiling of Tobacco Leaf Trichomes Identifies Genes for Biotic and Abiotic Stresses. Plant Cell Physiol. 2010, 51, 1627–1637. [Google Scholar] [CrossRef]
- Salminen, T.A.; Blomqvist, K.; Edqvist, J. Lipid transfer proteins: Classification, nomenclature, structure, and function. Planta 2016, 244, 971–997. [Google Scholar] [CrossRef]
- Adhikari, P.B.; Han, J.Y.; Ahn, C.H.; Choi, Y.E. Lipid Transfer Proteins (AaLTP3 and AaLTP4) Are Involved in Sesquiterpene Lactone Secretion from Glandular Trichomes in Artemisia annua. Plant Cell Physiol. 2019, 60, 2826–2836. [Google Scholar] [CrossRef]
- Sugimoto, K.; Zager, J.J.; Aubin, B.S.; Lange, B.M.; Howe, G.A. Flavonoid deficiency disrupts redox homeostasis and terpenoid biosynthesis in glandular trichomes of tomato. Plant Physiol. 2022, 188, 1450–1468. [Google Scholar] [CrossRef]
- Bedon, F.; Bomal, C.; Caron, S.; Levasseur, C.; Boyle, B.; Mansfield, S.D.; Schmidt, A.; Gershenzon, J.; Grima-Pettenati, J.; Séguin, A.; et al. Subgroup 4 R2R3-MYBs in conifer trees: Gene family expansion and contribution to the isoprenoid- and flavonoid-oriented responses. J. Exp. Bot. 2010, 61, 3847–3864. [Google Scholar] [CrossRef]
- Matías-Hernández, L.; Jiang, W.; Yang, K.; Tang, K.; Brodelius, P.E.; Pelaz, S. AaMYB1 and its orthologue AtMYB61 affect terpene metabolism and trichome development in Artemisia annua and Arabidopsis thaliana. Plant J. 2017, 90, 520–534. [Google Scholar] [CrossRef]
- Perez de Souza, L.; Garbowicz, K.; Brotman, Y.; Tohge, T.; Fernie, A.R. The Acetate Pathway Supports Flavonoid and Lipid Biosynthesis in Arabidopsis1. Plant Physiol. 2019, 182, 857–869. [Google Scholar] [CrossRef]
- Deribe, Y.L.; Pawson, T.; Dikic, I. Post-translational modifications in signal integration. Nat. Struct. Mol. Biol. 2010, 17, 666–672. [Google Scholar] [CrossRef]
- Cappadocia, L.; Lima, C.D. Ubiquitin-like Protein Conjugation: Structures, Chemistry, and Mechanism. Chem. Rev. 2018, 118, 889–918. [Google Scholar] [CrossRef]
- Stankovic-Valentin, N.; Melchior, F. Control of SUMO and Ubiquitin by ROS: Signaling and disease implications. Mol. Asp. Med. 2018, 63, 3–17. [Google Scholar] [CrossRef]
- Arito, M.; Horiba, T.; Hachimura, S.; Inoue, J.; Sato, R. Growth factor-induced phosphorylation of sterol regulatory element-binding proteins inhibits sumoylation, thereby stimulating the expression of their target genes, low density lipoprotein uptake, and lipid synthesis. J. Biol. Chem. 2008, 283, 15224–15231. [Google Scholar] [CrossRef]
- Zhang, H.; Lang, Z.; Zhu, J.-K. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Biol. 2018, 19, 489–506. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Camargo, A.P.; Vasconcelos, A.; Fiamenghi, M.; Pereira, G.; Carazzolle, M. tspex: A tissue-specificity calculator for gene expression data. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Thimm, O.; Bläsing, O.; Gibon, Y.; Nagel, A.; Meyer, S.; Krüger, P.; Selbig, J.; Müller, L.A.; Rhee, S.Y.; Stitt, M. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004, 37, 914–939. [Google Scholar] [CrossRef]
- Schwacke, R.; Ponce-Soto, G.Y.; Krause, K.; Bolger, A.M.; Arsova, B.; Hallab, A.; Gruden, K.; Stitt, M.; Bolger, M.E.; Usadel, B. MapMan4: A Refined Protein Classification and Annotation Framework Applicable to Multi-Omics Data Analysis. Mol. Plant 2019, 12, 879–892. [Google Scholar] [CrossRef]
- Villanueva, R.A.; Chen, Z. ggplot2: Elegant Graphics for Data Analysis. In Measurement: Interdisciplinary Research and Perspectives, 2nd ed.; Lawrence Erlbaum Associates, Inc.: Mahwah, NJ, USA, 2019; Volume 17, pp. 160–167. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, D.; Zhong, G.; Zhang, S.; Jiang, K.; Wang, C.; Wu, J.; Wang, B. Trichome-Specific Analysis and Weighted Gene Co-Expression Correlation Network Analysis (WGCNA) Reveal Potential Regulation Mechanism of Artemisinin Biosynthesis in Artemisia annua. Int. J. Mol. Sci. 2023, 24, 8473. https://doi.org/10.3390/ijms24108473
Huang D, Zhong G, Zhang S, Jiang K, Wang C, Wu J, Wang B. Trichome-Specific Analysis and Weighted Gene Co-Expression Correlation Network Analysis (WGCNA) Reveal Potential Regulation Mechanism of Artemisinin Biosynthesis in Artemisia annua. International Journal of Molecular Sciences. 2023; 24(10):8473. https://doi.org/10.3390/ijms24108473
Chicago/Turabian StyleHuang, Dawei, Guixian Zhong, Shiyang Zhang, Kerui Jiang, Chen Wang, Jian Wu, and Bo Wang. 2023. "Trichome-Specific Analysis and Weighted Gene Co-Expression Correlation Network Analysis (WGCNA) Reveal Potential Regulation Mechanism of Artemisinin Biosynthesis in Artemisia annua" International Journal of Molecular Sciences 24, no. 10: 8473. https://doi.org/10.3390/ijms24108473