Abstract
Herein we report a comprehensive laboratory synthesis of a series of energetic azidonitrate derivatives (ANDP, SMX, AMDNNM, NIBTN, NPN, 2-nitro-1,3-dinitro-oxypropane) starting from the readily available nitroisobutylglycerol. This simple protocol allows obtaining the high-energy additives from the available precursor in yields higher than those reported using safe and simple operations not presented in previous works. A detailed characterization of the physical, chemical, and energetic properties including impact sensitivity and thermal behavior of these species was performed for the systematic evaluation and comparison of the corresponding class of energetic compounds.
1. Introduction
High-energy fillers and additives are indispensable components of energetic compositions necessary to render them desirable physico-chemical, mechanical, and energy properties [1], e.g., the addition of fillers reduces the melt viscosity, elastic modulus, and glass transition temperature (Tg) of the compositions [2]. In addition, these additives improve the energetics and oxygen balance of a mixture and affect the combustion temperature and burning rate [3]. High-energy binders can be especially useful in the production of cast-cured composites by softening and increasing the flexibility of polymer matrices, as well as improving tensile strength, elongation, toughness, and glass transition temperature [4]. Among them, the compounds with high thermal stability and low impact sensitivity are of particular interest [5,6]. It should also be noted that particularly low-molecular-weight fillers are especially promising for applications due to their simple affordable synthesis resulting in an excellent price/performance ratio [7,8].
The greatest efficiency can be achieved using the molecules containing high-energy moieties that are structurally similar to the main components of fuels, e.g., nitro (—C—NO2), nitramino (—N—NO2), difluoroamino (—NF2), azido (—N3), and nitrate ester (—ONO2) functional groups [9].
Among the low-molecular high-energy derivatives of the widely available nitroisobutylglycerol [3,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29] 1, several compounds can be identified as promising fillers and components of various widely used composites (Figure 1).
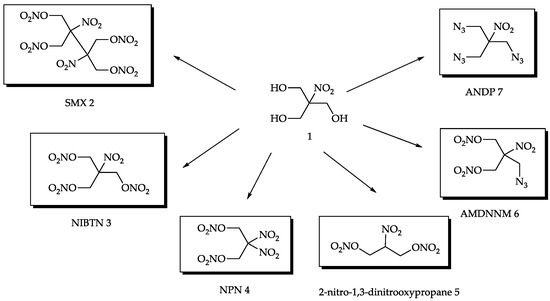
Figure 1.
High-energy compounds derived from nitroisobutylglycerol.
More specifically, these are 2,3-hydroxymethyl-2,3-dinitro-1,4-butanediol tetranitrate (SMX) 2 [15,16,17], nitroisobutametriol trinitrate (NIBTN) 3 [18,19,20,21], 2,2-dinitro-1,3-bis-nitrooxy-Propane (NPN) 4 [3,22,23], 2-nitro-1,3-dinitro-oxypropane 5 [24], azidomethyl-dinitroxydimethyl-nitromethane (AMDNNM) 6 [25,26], and tris(azidomethyl)nitromethane (ANDP) 7 [27,28,29]. All of them can easily be obtained in a few simple synthetic steps from nitroisobutylglycerol 1 using cheap reagents and appear to be promising low-molecular high-energy additives according to their properties.
Usually, in a comprehensive study of high-energy additives, the following parameters should be determined [30,31]: (1) Physico-chemical properties, i.e., density, nitrogen content, oxygen balance, and melting point; (2) safety evaluation: Decomposition temperature, impact, and friction sensitivities; (3) energetic performance: Enthalpy of formation, explosion temperature, detonation pressure, and velocity. We found that these data are missing or require clarification for a number of compounds under study. For example, the heat of explosion is not available for SMX [15,16,17], and in the case of ANDP, the melting temperature and sensitivities toward mechanical stimuli are missing [27,28,29]. Apart from this, there is no information on the melting point of AMDNNM [25,26]. NPN requires the clarification of a melting point, enthalpy of formation, and detonation parameters [3,22,23]. While NIBTN has been fully characterized [18,19,20,21], the data on sensitivity and detonation parameters for 2-nitro-1,3-dinitro-oxypropane are not available at all [24]. In addition, the solid-state formation enthalpies of the compounds studied can be refined using a recently proposed combined experimental and theoretical approach [32].
All compounds 2–7 contain common explosophoric groups in their structure and are used as energetic additives in various compositions [3,15,18,26]. Thus, in the present contribution, we attempted to describe and systematize their synthetic procedures and determine a comprehensive set of their safety and performance parameters.
2. Results and Discussion
2.1. Synthesis
An overview of the syntheses is given in Figure 2. All energetic derivatives were synthesized starting from nitroisobutylglycerol 1 obtained directly from nitromethane using a well-known procedure. SMX 2 [15] and 2-nitro-1,3-dinitrooxypropane 5 [24,33] were synthesized by known methods. In turn, the synthetic procedures for NIBTN 3, AMDNNM 6, ANDP 7, and NPN 4 were improved to obtain optimal yields and better working conditions.
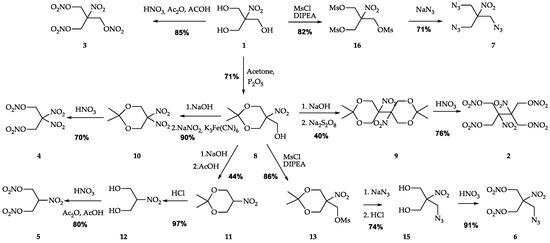
Figure 2.
Synthesis of high-energy compounds from nitroisobutylglycerol discussed in the present work.
The mixtures of concentrated nitric and sulfuric acids and oleum are commonly used in NIBTN 3 synthesis [18]. In this work, we used a mixture of nitric acid, acetic acid, and acetic anhydride as a nitrating agent. Ethyl ether was used for product extraction to avoid the formation of persistent emulsions when washing (which is often observed in methods mentioned before [18]). Flash chromatography on silica gel was used for further purification from byproducts and residual water. The traces of acetic acid in the product can be easily detected by 1H NMR and completely removed in vacuo. This method is more expensive, but it is reliable and convenient for obtaining a small number of pure samples in the laboratory.
For the first time, we describe a complete three-stage synthesis of NPN 4 from 1 with the 45% overall yield of the target compound. In the first step, 2,2-dimethyl-5-hydroxymethyl-5-nitro-1,3-dioxane 8 was obtained from 1 using phosphorus pentoxide [34], then 2,2-dimethyl-5,5-dinitro-1,3-dioxane 10 was prepared from 8 by adding NaNO2 and K3Fe(CN)6 under alkaline conditions [35]. In the third stage, 4 was isolated after treating 10 with 90% nitric acid.
The previously reported methods of 4 preparation included the treatment of 1 with concentrated nitric and sulfuric acids [22] or 100% nitric acid [36]. In our work, we used more accessible 90% nitric acid for treatment of 2,2-dimethyl-5,5-dinitro-1,3-dioxane 10. Furthermore, the conversion of nitromethane into 2,2-dinitropropanol was described in two steps: making potassium nitromethane from undesirable chloronitromethane in the first stage and mixing it with an aqueous solution of formaldehyde [23] in the second stage with an overall 16.5% yield, which is much lower than the one reported here. AMDNNM 6 is also made from nitroisobutylglycerol 1 requiring five steps with an overall yield of 41%. Regarding the previous work on this compound [26], we increased the yield of AMDNMM from 57% to 62% (starting from (2,2-dimethyl-5-nitro-1,3-dioxan-5-yl)methanol 8 in comparison). The increase was achieved by using an additional purification stage of 2-(azidomethyl)-2-nitropropane-1,3-diol 15. The azide derivative ANDP 7 is obtained from compound 1 in two steps using the nucleophilic substitution reaction to insert three azido groups into the trimesylate derivative of nitroisobutylglycerol. Unfortunately, there was no information on the yield of ANDP 7 in the literature [27,29]. However, it is worth mentioning that the mesylate derivative yield significantly increased from 65–68% [37,38] to 82% by using the DIPEA in DCM instead of pyridine.
All synthesized compounds were fully characterized by 1H, 13C, 15N NMR, and IR spectroscopies; the NMR spectra of the intermediates were also obtained.
It is also worth mentioning that the whole set of compounds 2–7 was synthesized and characterized in order to obtain a comprehensive comparison of their important physico-chemical properties.
2.2. Physico-Chemical Properties
2.2.1. Crystal Structure Analysis
The crystal structures of 2-nitro-1,3-dinitrooxypropane 5 [24] and SMX 2 [17] are well known in the literature. ANDP 7, AMDNNM 6, NIBTN 3, and NPN 4 are liquid compounds at room temperature. The densities of 2 and 5 were calculated from the crystal structure (Figure 3).
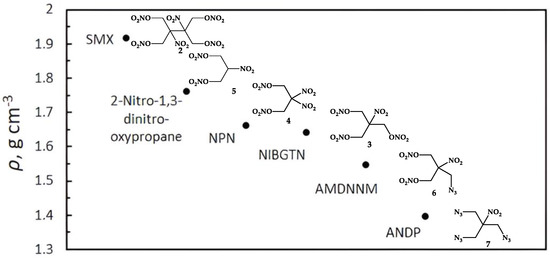
Figure 3.
The densities of nitroisobutylglycerol derivatives.
The densities obey the order 7 < 6 < 3 < 4 in line with the increase in the oxygen content (Figure 3, ADNP 14.15% O; AMDNNM 48.1% O; NIBGTN 61.51% O; NPN 62.48% O), which is typical of the nitro compounds [39]. Compounds 5 and 2 are solid at room temperature and do not follow this trend.
2.2.2. Thermal Analysis
The general trends of thermal behavior of the investigated compounds are similar to the previously reported nitroesters [40,41]. More specifically, compound 4, being a liquid at room temperature, exhibits the only thermal feature under linear heating, namely, the extrapolated onset exothermic decomposition lies at 167 °C (Figure 4). Other compounds containing the nitroester moieties, 6 and 3, also undergo thermal decomposition within a temperature range of 160–210 °C. Compound 7 shows the best thermal stability among the species studied. Its thermolysis commences at 179 °C, but the main peak of heat release is at 217 °C, which is nearly 20 degrees higher than those of other nitroesters.
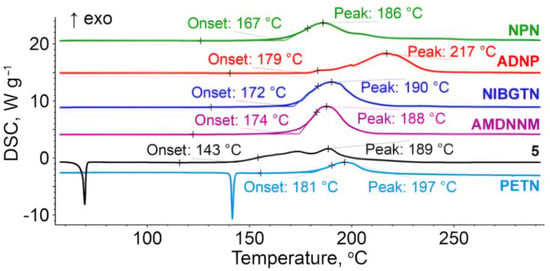
Figure 4.
DSC curves at a heating rate of 5 K min−1 for nitroisobutylglycerol derivatives.
2-Nitro-1,3-dinitro-oxypropane 5 melts at 68 °C with a complex decomposition profile, having at least two distinguishable stages. The thermolysis of 5 starts at the onset temperature, which is the lowest among the considered species, whilst the second (main) exothermic DSC peak is observed at 189 °C. The latter value is nearly the same for most of the analyzed compounds. As a reference compound, PETN is also shown in Figure 4, which decomposes after melting, and the temperature range of thermolysis is approximately the same as for investigated compounds bearing nitroester groups. It should be mentioned that all species with nitroester moieties behave similarly in terms of the decomposition onset. This fact is also in line with the thermal properties of other energetic nitroethers (e.g., PETN). The only compound without several ONO2-groups in its structure—ADNP 7—exhibits a retarded decomposition peak, but the decomposition onset is low due to the presence of azide groups.
In additional DSC tests with a liquid nitrogen cooling unit, we tried to crystallize the liquid nitro esters ANDP 7, AMDNNM 6, NIBTN 3, and NPN 4. All four compounds show vitrification behavior upon cooling with a 5 K min−1 rate to −150 °C (Figures S1–S4, Supporting information). In turn, once heated after cooling, ANDP 7 shows a sequence of thermally induced transformations: viz., a glass transition (Tg = −87 °C) followed by cold crystallization (−51 °C), and, finally, melting at Tm = 12 °C. All attempts to enhance the crystallization by the introduction of isothermal segments failed, and the vitrification and glass transition were the only thermal events observed for other samples. Thus, we see that the empirical rule Tg/Tm = 2/3 (T in Kelvins [42]) remains valid for ANDP. In a similar way, we estimated the melting points for the compounds where melting was not registered directly by DSC and utilized these values in the reparametrized Trouton rule [32] to obtain the sublimation/evaporation enthalpies for the compounds studied. These vaporization enthalpies were complemented by high-level quantum chemical-calculated gas-phase formation enthalpies, and the obtained standard state formation enthalpies were used for the calculations of detonation performance. As seen from Table 1, the detonation parameters of the compounds studied range from the low energetic ANDP 7 with sub-TNT performance to a highly energetic SMX 2, outperforming the benchmark powerful HMX explosive.

Table 1.
Physical and energetic properties of nitroisobutylglycerol derivatives 2–7 studied in the present work.
2.2.3. Detonation Parameters
In Table 1 the physico-chemical properties and detonation parameters of all 6 compounds are presented.
The parameters from Table 1 show that the compounds exhibit good oxygen balance and high densities. The high densities and good oxygen balance of SMX 2 and 2-nitro-1,3-dinitro-oxypropane 5 render them to be more powerful with a detonation velocity of 9.2 and 8.3 km s−1, whereas lower-density NIBTN 3 and AMDNNM 6 still exhibit a good detonation velocity of 8.1 and 7.8 kms−1, which is comparable to PETN (8.3 km s−1) with 1.77 g·cm−3 density [45]. Compound 2 shows the largest calculated detonation pressure of 37 GPa and heat of explosion of 6260 J/g. Apart from this, 2-nitro-1,3-dinitro-oxypropane 5 and AMDNNM 6 reach the detonation pressure of 25 GPa and heat of explosion above 5800 J/g. It should also be mentioned that the oxygen balance is better for 2 and 3, and 7 exhibits the worst oxygen balance of all compounds. In terms of sensitivity, all compounds are very sensitive towards impact (0.3–4 J) and friction (50–120 N) while PETN is classified as sensitive with 3.5 J IS and 50 N FS [46].
3. Materials and Methods
3.1. Materials
Caution: Work with explosive (or potentially explosive) materials generally requires protective apparel such as face shields, gloves, laboratory coats, and protective devices such as explosion shields, barriers, enclosed barricades, or even an isolated room. All compounds were characterized by 1H NMR and 13C NMR data. Chemicals and solvents were obtained from commercial sources and used without further purification. NMR spectra were obtained on a Bruker “Ascend™ 400” (400 MHz 1H, 101 MHz 13C, 40.55 MHz 15N). The chemical shifts are frequency-referenced relative to the residual undeuterated solvent peaks. Coupling constants J are given in Hertz as positive values regardless of their real individual signs. The multiplicity of the signals is indicated as ‘‘s”, ‘‘d”, “t”, or ‘‘m” for singlet, doublet, triplet, or multiplet, respectively. The abbreviation ‘‘br” is given for broadened signals. FT-IR spectra were obtained in a Bruker “Alpha-T” FTIR (KBr).
3.2. Synthetic Procedures
Preparation of 2-(hydroxymethyl)-2-nitropropane-1,3-diol 1
A 1000 mL round-bottomed flask was charged with nitromethane (73.2 g, 1.2 mol), dry ethyl acetate (600 mL), and paraformaldehyde (108 g, 3.6 mol) followed by the addition of 33% aqueous potassium hydroxide (0.5 mL). The reaction mixture was heated under reflux with stirring in an oil bath at 100 °C, until all of the solids dissolved. The resulting mixture was cooled to room temperature and concentrated to 400 mL on a rotary evaporator. Following the addition of CHCl3 (1200 mL), the reaction mixture was reheated until all of the solids dissolved and then refluxed for 1 h to obtain a crystalline precipitate. The flask was sealed and placed in a freezer at −18 °C for 2 days. The precipitate was then filtered off, washed with CHCl3 (2 × 100 mL), and dried in vacuo to give 2-(hydroxymethyl)-2-nitropropane-1,3-diol (152.0 g, 84%) as a white solid.
M.p. 159 °C.
1H NMR (400 MHz, DMSO-d6) δ, ppm: 5.12 (t, J = 5.5 Hz, 3H), 3.76 (d, J = 5.5 Hz, 6H).
13C{1H} NMR (101 MHz, DMSO-d6) δ, ppm: 95.1, 58.5.
Preparation of trimethylol nitromethane trinitrate (NIBTN) 3
A 90% HNO3 solution (5 mL) was added dropwise to a mixture of acetic acid (8 mL) and acetic anhydride (10 mL) and stirred for 20 min at 0 °C. Then a solution of 2-(hydroxymethyl)-2-nitropropane-1,3-diol (1.5 g, 0.01 mol) in acetic acid (4 mL) was added dropwise and the mixture was stirred for 2 h at 0 °C and for another 2 h at 20 °C. After that, the mixture was poured into 200 mL of ice and extracted with ether (2 × 50 mL). The organic layer was washed with water (3 × 100 mL), dried over Na2SO4, and evaporated in vacuo. The residue was purified by flash chromatography on silica gel (Et2O) to give trimethylol nitromethane trinitrate as a colorless oil (2.43 g, 85%).
1H NMR (400 MHz, CDCl3) δ, ppm: 4.99 (s, 6H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 85.3, 67.4.
15N NMR (40.5 MHz, CDCl3) δ, ppm: 373.9, 328.2.
IR (KBr): ν, cm−1: 1644, 1562, 1462, 1459, 1429, 1373, 1346, 1270, 1006, 814, 746, 721, 685, 615.
Preparation of (2,2-dimethyl-5-nitro-1,3-dioxan-5-yl)methanol 8
To a mixture of tris-(hydroxymethyl)-nitromethane (15.1 g, 0.1 mol) and acetone (50 mL, 0.68 mol) phosphorus pentoxide (5 g, 0.035 mol) was added with stirring at 10 °C. The reaction mixture was stirred for 10 min, after which it was poured into 100 mL of a NaHCO3-saturated solution with ice. After the ice was completely dissolved, the product was filtered, washed with cold water, and dried in air to obtain 11.5 g (71%) of 2,2-dimethyl-5-hydroxymethyl-5-nitro-1,3-dioxane.
1H NMR (400 MHz, DMSO-d6) δ, ppm: 5.48 (t, J = 5.8 Hz, 1H), 4.35 (d, J = 13.1 Hz, 2H), 4.05 (d, J = 13.1 Hz, 2H), 3.73 (d, J = 5.8 Hz, 2H), 1.40 (s, 3H), 1.25 (s, 3H).
13C{1H} NMR (101 MHz, DMSO-d6) δ, ppm: 98.4, 87.5, 62.4, 61.2, 26.8, 20.1.
Preparation of 2,2,2′,2′-tetramethyl-5,5′-dinitro-5,5′-bi(1,3-dioxane) 9
A 1000 mL round-bottomed flask, equipped with a reflux condenser and a magnetic stir bar, was charged with (2,2-dimethyl-5-nitro-1,3-dioxan-5-yl) methanol (41.37 g, 0.216 mol) and a solution of sodium hydroxide (17.31 g, 0.432 mol) in water (542 mL) at an ambient temperature. The reaction mixture was heated at 60 °C for 1 h and then cooled to 20 °C. Portions of solid sodium peroxodisulfate (103.05 g, 0.432 mol) were added and stirred at 20 °C for 20 h. Maintaining a temperature of no higher than 25 °C, a 40% aqueous sodium hydroxide solution was added to the resulting mixture to pH > 11, and the precipitated crystals were filtered off and washed with water on the filter. The solid was then dissolved in methylene chloride (200 mL), washed with water [TLC control (CH2Cl2) until any impurities had disappeared, approximately 5 times 50 mL each], dried over anhydrous sodium sulphate, and concentrated in a rotary evaporator to a volume of 30 mL.
The product was purified by chromatography on silica gel (eluent-methylene chloride) to give 2,2,2′,2′-tetramethyl-5,5′-dinitro-5,5′-bi(1,3-dioxane) (13.9 g, 40%) as a colorless fine crystalline powder.
1H NMR (400 MHz, CDCl3) δ, ppm: 4.45 (d, J = 13.6 Hz, 4H), 4.34 (d, J = 13.5 Hz, 4H) 1.42 (s, 6H), 1.36 (s, 6H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 100.8, 90.1, 60.5, 24.1, 21.9.
Preparation of 1,4-dinitrato-2,3-dinitro-2,3-bis(nitratomethyl)butane (SMX) 2 [15]
A 50 mL round-bottomed flask, equipped with a magnetic stir bar, was charged with 98% nitric acid (33 mL, 49.5 g, 0.78 mol) and cooled to 0 °C. 2,2,2′,2′- Tetramethyl-5,5′-dinitro-5,5′-bi(1,3-dioxane) (2.4 g, 7.5 mmol) was then added at 0 °C. The reaction mixture was stirred for 3 h at 0 °C, then poured into ice water (170 mL) and stirred for an additional 15 min at room temperature to crystallize the precipitated oily liquid. The yellowish crystals were filtered off, dissolved in methylene chloride (50 mL), and the organic phase was washed with a solution of NaHCO3 (2 × 50 mL). After drying over Na2SO4, the solvent was evaporated in vacuo. The resulting product was transferred to a filter, washed with diethyl ether (2 × 4 mL), and dried in vacuo to give 1,4-dinitrato-2,3-dinitro-2,3-bis(nitratomethyl)butane (2.41 g, 76%) as a colorless fine crystalline powder.
1H NMR (400 MHz, CDCl3) δ, ppm: 5.26 (d, J = 12.2 Hz, 4H), 5.09 (d, J = 12.2 Hz, 4H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 89.0, 66.1.
IR (KBr): ν, cm−1: 3045, 3028, 2982, 2927, 1658, 1583, 1491, 1465, 1450, 1390, 1371, 1334, 1287, 1156, 1099, 1056, 1022, 995, 898, 854.
15N NMR (40.5 MHz, CDCl3) δ, ppm: 325.0, 357.5.
Preparation of 2,2-dimethyl-5,5-dinitro-1,3-dioxane 10
In a 250 mL round-bottomed flask equipped with a magnetic stir bar, 2,2-dimethyl-5-hydroxymethyl-5-nitro-1,3-dioxane (1.5 g, 0.37 mol) was dissolved in 48 mL of a 20% NaOH solution and stirred for 20 min at room temperature. Then, the solution of NaNO2 (21 g, 0.30 mol) in 24 mL of H2O was added, and the resulting homogeneous mixture was quickly poured into 250 mL of a K3Fe(CN)6 saturated solution. The reaction mixture was stirred for 4 h at room temperature and extracted with ethyl acetate (4 × 100 mL). The extract was washed with brine and water and dried over sodium sulphate. The crude product was concentrated on a rotary evaporator, and purified by preparative chromatography (SiO2, hexane/CH2Cl2 = 1/1). Furthermore, 11 g (90%) of 2,2-dimethyl-5,5-dinitro-1,3-dioxane was obtained.
1H NMR (400 MHz, CDCl3) δ, ppm: 4.67 (s, 2H), 1.47 (s, 3H).
13C{1H} NMR (101 MHz, CDCl3), δ, ppm: 101.1, 62.3, 23.1.
Preparation of 2,2-dinitropropane-1,3-diyl dinitrate (NPN) 4
2,2-Dimethyl-5,5-dinitro-1,3-dioxane (4.12 g, 0.02 mol) was added with vigorous stirring to 60 mL of 90% nitric acid cooled to −20 °C in 100 mL round-bottomed flask, equipped with a magnetic stir bar. The reaction mixture was stirred for 3 h at a temperature not exceeding 5 °C before being poured into 300 mL of ice water. The crude product was extracted with methylene chloride (4 × 75 mL), washed with water and sodium carbonate saturated solution, dried by filtration through a small plug of silica gel, and concentrated and purified by preparative chromatography (SiO2, hexane/EtOAc = 10/1) to result in 3.58 g (70%) of 2,2-dinitropropane-1,3-diyl dinitrate.
1H NMR (400 MHz, CDCl3) δ, ppm: 5.41 (s, 2H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 110.5, 66.32.
15N NMR (40.5 MHz, CDCl3) δ, ppm: 324.7, 358.6.
IR (KBr): ν, cm−1: 3685, 3606, 3340, 3029, 2978, 2940, 2896, 2653, 2553, 2441, 2114, 1960, 1877, 1674, 1582, 1454, 1425, 1387, 1347, 1365, 1281, 1208, 1180, 1129, 1026, 940, 828, 743, 675, 629, 579, 504.
Preparation of 2,2-dimethyl-5-nitro-1,3-dioxane 11
(2,2-Dimethyl-5-nitro-1,3-dioxan-5-yl)methanol (5.7 g, 0.03 mol) was dissolved in 52 mL of a 10% NaOH solution and stirred at 60 °C for 1 h in a 250 mL round-bottomed flask, equipped with a magnetic stir bar. After that, the reaction mixture was cooled to 5 °C and neutralized with 15% acetic acid (100 mL). The precipitate was filtered off and recrystallized from petroleum ether to give 2,2-dimethyl-5-nitro-1,3-dioxane in a 1.6 g (44%) yield.
1H NMR (400 MHz, CDCl3) δ, ppm: 4.48 (dd, J = 13.1, 4.1 Hz, 2H), 4.35 (p, J = 4.1 Hz, 1H), 4.24 (dd, J = 13.0, 4.0 Hz, 2H), 1.45 (s, 3H), 1.41 (s, 3H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 99.3, 77.3, 59.9, 25.5, 21.4.
Preparation of 2-nitropropane-1,3-diol 12
In a 50 mL round-bottomed flask, 2,2-dimethyl-5-nitro-1,3-dioxane (1.35 g, 0.008 mol) was stirred with 3 mL of hydrochloric acid in 30 mL of methanol at 60 °C for 24 h, then the solvent was evaporated in vacuo to dryness and the mixture was purified by flash chromatography (DCM/methanol = 1/10, then 1/5) to give 2-nitropropane-1,3-diol in 0.95 g (97%) yield.
1H NMR (400 MHz, DMSO-d6) δ, ppm: 5.26 (s, 2H), 4.70–4.60 (m, 1H), 3.82–3.66 (m, 4H).
13C{1H} NMR (101 MHz, DMSO-d6) δ, ppm: δ 92.4, 60.0.
Preparation of 2-nitro-1,3-dinitrooxypropane 5 [24]
To a mixture of acetic anhydride (7 mL) and acetic acid (5 mL), 90% nitric acid (2.8 mL) was added dropwise at 0 °C and stirred for 20 min. Then, a solution of 2-nitropropane-1,3-diol (0.95 g, 0.0078 mol) in 2 mL of acetic acid was added dropwise and stirred for another 2 h at 0 °C. The reaction mixture was poured into 25 mL of ice, the precipitated crystals were filtered, washed with water on the filter, and recrystallized from CCl4 to obtain (1.3 g, 80%) 2-nitro-1,3-dinitrooxypropane.
1H NMR (400 MHz, acetone-d6) δ, ppm: 5.60–5.52 (m, 1H), 5.34–5.22 (m, 4H).
13C{1H} NMR (101 MHz, acetone-d6) δ, ppm: 81.3, 69.6.
15N NMR (40.5 MHz, CDCl3) δ, ppm: 330.4, 375.9.
IR (KBr): ν, cm−1: 3434, 3309, 3032, 2985, 2933, 2798, 2709, 2570, 2424, 1767, 1659, 1676, 1562, 1500, 1468, 1437, 1404, 1339, 1355, 1310, 1285, 1254, 1229, 1116, 1082, 1052, 1013, 989, 911, 837, 749, 691, 627, 646, 600, 513, 465.
Preparation of (2,2-dimethyl-5-nitro-1,3-dioxan-5-yl)methyl methanesulfonate 13
A solution of (2,2-dimethyl-5-nitro-1,3-dioxan-5-yl)methanol (5.0 g, 0.026 mol) in dry methylene chloride (70 mL) was cooled to −10 °C and DIPEA (5 mL, 0.028 mol) was added in portions, then the mixture was stirred for 15 min at this temperature. Then, mesyl chloride (2.22 mL, 0.028 mol) was added dropwise and stirred for another hour at −5 °C to 5 °C. The reaction mixture was warmed to room temperature and stirred for another 2 h, then washed 3 times with 100 mL of water, dried over Na2SO4, and purified by flash chromatography on silica gel (DCM) to give (2,2-dimethyl-5-nitro-1,3-dioxan-5-yl)methyl methanesulfonate in 6 g (86%) yield.
1H NMR (400 MHz, DMSO-d6) δ, ppm: δ 5.55–5.43 (m, 1H), 4.35 (dd, J = 13.1, 1.4 Hz, 2H), 4.11–3.97 (m, 2H), 3.78–3.63 (m, 2H), 3.36 (d, J = 1.3 Hz, 3H), 1.40 (s, 3H), 1.25 (s, 3H).
13C{1H} NMR (101 MHz, DMSO-d6) δ, ppm: 98.9, 87.9, 62.8, 61.6, 27.3, 20.5.
Preparation of 5-(azidomethyl)-2,2-dimethyl-5-nitro-1,3-dioxane 14
(2,2-Dimethyl-5-nitro-1,3-dioxan-5-yl)methyl methanesulfonate (3.0 g, 0.008 mol) was stirred for 18 h with sodium azide (1.1 g, 0.016 mol) in DMF (25 mL) at 95 °C. The reaction mixture was cooled to ambient temperature, poured into cool water, extracted with ethyl acetate (3 × 100 mL), dried over Na2SO4, and the solvent was evaporated in vacuo. The residue was purified by flash chromatography on silica gel (hexane-DCM 1/1), handled with potassium permanganate (Rf 0.67 DCM) to give 5-(azidomethyl)-2,2-dimethyl-5-nitro-1,3-dioxane (1.8 g, 76%).
1H NMR (400 MHz, CDCl3) δ, ppm: 4.36 (d, J = 12.6 Hz, 2H), 4.00 (d, J = 12.6 Hz, 2H), 3.96 (s, 2H), 1.44 (s, 3H), 1.41 (s, 3H).
13C{1H} NMR (101 MHz, CDCl3), δ, ppm: 84.2, 62.1, 52.8, 24.6, 22.0.
Preparation of 2-(azidomethyl)-2-nitropropane-1,3-diol 15
5-(Azidomethyl)-2,2-dimethyl-5-nitro-1,3-dioxane (4.2 g, 0.019 mol) was stirred in a mixture of HCl (7 mL) and methanol (70 mL) at 60 °C for 17 h. Afterward, the mixture was evaporated in vacuo and the residue was purified by flash chromatography (DCM/Methanol = 20/1 then 5/1) to give 2-(azidomethyl)-2-nitropropane-1,3-diol (2.3 g, 97%).
1H NMR (400 MHz, DMSO-d6) δ, ppm: 5.56–5.42 (m, 2H), 3.91 (s, 2H), 3.82–3.68 (m, 4H).
13C{1H} NMR (101 MHz, DMSO-d6) δ, ppm: 93.9, 60.1, 48.6.
Preparation of 2-(azidomethyl)-2-nitropropane-1,3-diyl dinitrate (AMDNNM) 6
A 90% HNO3 solution (5 mL) was added dropwise to a mixture of acetic acid (9.4 mL) and acetic anhydride (15 mL) and stirred for 20 min at 0 °C. Then a solution of 2-(azidomethyl)-2-nitropropane-1,3-diol (2.27 g, 0.013 mol) in acetic acid (4 mL) was added dropwise and the mixture was stirred for 2 h at 0 °C and for another 2 h at 20 °C. After that, the mixture was poured into 200 mL of ice and extracted with ether (2 × 150 mL). The organic layer was washed with water (3 × 100 mL) and saturated with NaHCO3 solution, dried over Na2SO4, and evaporated in vacuo. The residue was purified by flash chromatography on silica gel (hexane/DCM = 2/1) to give 2-(azidomethyl)-2-nitropropane-1,3-diyl dinitrate as a colorless oil (3.12 g, 91%).
1H NMR (400 MHz, CDCl3) δ, ppm: 4.94 (s, 4H), 4.01 (s, 2H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 86.5, 67.8, 50.4.
15N NMR (40.5 MHz, CDCl3) δ, ppm: 244.4, 329.2, 376.6.
IR (KBr): ν, cm−1: 3683, 3369, 3023, 2926, 2680, 2554, 2120, 1888, 1825, 1659, 1561, 1444, 1460, 1373, 1394, 1345, 1278, 1130, 1027, 946, 838, 750, 720, 688, 633, 550.
Preparation of 2-((methylsulfonyloxy)methyl)-2-nitropropane-1,3-diyl dimethanesulfonate 16
DIPEA (50 mL) was added dropwise to a cooled mixture of 2-(hydroxymethyl)-2-nitropropane-1,3-diol (13 g, 0.086 mol) in DCM (250 mL) at −10 °C and stirred for 15 min at this temperature. Then MsCl (22 mL, 0.284 mol) was added dropwise for 1 h at −15 °C. The mixture was stirred for 1 h at −15 °C, then heated to 5 °C and filtered. The precipitate was washed with DCM, cold methanol, and the filtrate was evaporated. The precipitate was washed with ice water, filtered, and reprecipitated from acetone/methanol to obtain 2-((methylsulfonyloxy)methyl)-2-nitropropane-1,3-diyl dimethanesulfonate (27 g, 82%).
1H NMR (400 MHz, acetone-d6) δ, ppm: 4.86 (s, 1H), 3.28 (s, 2H).
13C{1H} NMR (101 MHz, acetone-d6) δ, ppm: 89.1, 65.7, 37.5.
Preparation of 1,3-diazido-2-(azidomethyl)-2-nitropropane (ANDP) 7
2-((Methylsulfonyloxy)methyl)-2-nitropropane-1,3-diyl dimethanesulfonate (12.0 g, 0.031 mol) was stirred with sodium azide (12.1 g, 0.187 mol) in DMF (100 mL) at 95 °C overnight, then poured into ice (200 g) and extracted with EtOAc (3 × 100 mL). The organic extract was washed with water, dried over sodium sulfate, and concentrated under reduced pressure to give a residue that was chromatographed to obtain pure 1,3-diazido-2-(azidomethyl)-2-nitropropane (4,99 g, 71%).
1H NMR (400 MHz, CDCl3) δ, ppm: 3.88 (s, 6H).
13C{1H} NMR (101 MHz, CDCl3) δ, ppm: 89.4, 51.1.
15N NMR (40.5 MHz, CDCl3) δ, ppm: 245.15, 382.58.
IR (KBr): ν, cm−1: 3376, 2937, 2885, 2527, 2112, 1960, 1727, 1662, 1556, 1447, 1380, 1339, 1356, 1289, 1079, 941, 963, 918, 893, 863, 743, 661, 551, 519, 488
3.3. Methods
Thermal analysis runs were carried out using the DSC 204 HP (Netzsch) apparatus. Samples of 0.3–1.0 mg mass were loaded in standard aluminum crucibles and covered with pierced lids to relieve the pressure build-up by gas decomposition products. A heating rate of 5 K min−1 and nitrogen flow within the furnace were applied to the samples. To investigate the thermal behavior of liquid compounds, the LN2 cooling unit was used, and the additional step of cooling with a 5 K min−1 rate down to −150 °C was introduced.
Electronic structure calculations were performed using the Gaussian 09 [47], Molpro 2010 [48], and ORCA 4.0 [49] program packages. The gas-phase enthalpies of formation (at p0 = 1 bar and T = 298.15 K, ΔfHm0(g)) were calculated using the explicitly correlated W1-F12 multi-level procedure and atomization energy and isodesmic reaction approaches described in detail elsewhere [50,51,52,53]. As a general rule, the explicitly correlated F12 procedure markedly accelerates the slow basis set convergence of conventional CCSD(T) techniques [54]. Note that the W1-F12 procedure employed in the present work had been slightly modified in comparison with the originally proposed technique; namely, the B3LYP-D3BJ/def2-TZVPP-optimized geometries (with a ZPE correction factor of 0.99) were used [43,55], and the diagonal Born-Oppenheimer corrections were omitted. The multireference character of the wave functions of the reagents, intermediates, and transition states considered in the present work was estimated using the T1 diagnostic for the CCSD calculation [56]. The modest T1 values obtained in all cases (<0.025) justify the reliability of the single reference-based electron correlation procedure employed in the present study. The heats of formation at 0 K for the elements in the gas phase ΔfHm0K(g) [H] = 216.04 kJ mol−1, ΔfHm0K(g) [C] = 711.19 kJ mol−1, ΔfHm0K(g) [N] = 470.82 kJ mol−1, and ΔfHm0K(g) [O] = 246.79 kJ mol−1 were taken from the NIST-JANAF tables [57].
For several isodesmic reactions (in the case of (6)), single-point electronic energies were calculated using the DLPNO-CCSD(T) methodology (the “NormalPNO” truncation thresholds were set) [55] along with the aug-cc-pVQZ basis set [58]. For the sake of brevity, it is denoted henceforth as aVQZ. The RIJK density fitting (DF) approximation [59] was used to accelerate the convergence of the SCF components of DLPNO-CCSD(T) energy. The corresponding auxiliary basis sets (aug-cc-pVQZ/JK and aug-cc-pVQZ/C in the ORCA nomenclature) [49] were used in the DF calculations of the SCF and correlation energies.
The sublimation/evaporation enthalpy was calculated using the refined Trouton-type equation suggested previously [32]. The impact and friction sensitivities for solid samples were obtained according to STANAG standards; the details can be found elsewhere [32]. For liquids, the experimental assembly with free volume was used in line with UN Recommendations on the Transport of Dangerous Goods [60]. The detonation parameters were calculated using the default set of empirical equations implemented in the web application PILEM [61].
4. Conclusions
We reported comprehensive syntheses of a bunch of useful high-energetic azidonitrate derivatives (viz., ANDP, SMX, AMDNNM, NIBTN, NPN, 2-nitro-1,3-dinitro-oxypropane). The protocols used are very promising from safety, convenience, and efficiency points of view. Nitroisobutylglycerol was chosen as a common starting reagent for all the compounds, and the procedures were improved to obtain optimal yields and better working conditions. Moreover, a comparative systematic study of their physiochemical and energetic properties was performed. All missing parameters for SMX, NPN, ANDP, AMDNNM, NIBTN, and 2-nitro-1,3-dinitrooxypropane were carefully collected and compared with each other. All compounds were recognized as promising high-energetic fillers.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24108541/s1.
Author Contributions
Conceptualization: A.F.A.; formal analysis: V.G.K.; investigation: S.A.R., M.A.T., I.N.M., V.G.K. and A.N.P.; writing—original draft: L.I.M. and S.A.R.; writing—review and editing: I.V.F. and V.G.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in Supplementary Material here.
Acknowledgments
This work was carried out by S.A.R., L.I.M., M.A.T., and A.F.A. as a part of the A. V. Topchiev Institute of Petrochemical Synthesis (TIPS) Russian Academy of Sciences (RAS) State Program. This work was performed using the equipment of the Shared Research Center “Analytical center of deep oil processing and petrochemistry of TIPS RAS”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Shukla, M.; Boddu, V.M.; Steevens, J.A.; Damavarapu, R.; Leszczynski, J. Energetic Materials. From Cradle to Grave, 1st ed.; Springer: Cham, Switzerland, 2017; p. 482. [Google Scholar]
- Charles, E.; Carraher, J. Seymour/Carraher’s Polymer Chemistry, 5th ed.; Marcel Dekker, Inc.: New York, NY, USA; Basel, Switzerland, 2000; p. 741. [Google Scholar]
- Wingborg, N.; Eldsäter, C. 2,2-Dinitro-1,3-Bis-Nitrooxy-Propane (NPN): A New Energetic Plasticizer. Propellants Explos. Pyrotech. 2002, 27, 314–319. [Google Scholar] [CrossRef]
- Akhavan, J. The Chemistry of Explosives; The Royal Society of Chemistry: London, UK, 2004. [Google Scholar]
- Badgujar, D. Review on Promising Insensitive Energetic Materials. Cent. Eur. J. Energetic Mater. 2017, 14, 821–843. [Google Scholar] [CrossRef] [PubMed]
- Zeman, S.; Jungová, M. Sensitivity and Performance of Energetic Materials. Propellants Explos. Pyrotech. 2016, 41, 426–451. [Google Scholar] [CrossRef]
- Kumari, D.; Balakshe, R.; Banerjee, S.; Singh, H. Energetic plasticizers for gun & rocket propellants. Rev. J. Chem. 2012, 2, 240–262. [Google Scholar] [CrossRef]
- Vinogradov, D.B.; Bulatov, P.V.; Petrov, E.Y.; Gribov, P.S.; Kondakova, N.N.; Il’icheva, N.N.; Stepanova, E.R.; Denisyuk, A.P.; Sizov, V.A.; Sinditskii, V.P.; et al. Promising Oxygen- and Nitrogen-Rich Azidonitramino Ether Plasticizers for Energetic Materials. Molecules 2022, 27, 7749. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, J.; Wang, B.; Qiu, L.; Xu, R.; Sheremetev, A.B. Recent synthetic efforts towards high energy density materials: How to design high-performance energetic structures? FirePhysChem 2022, 2, 83–139. [Google Scholar] [CrossRef]
- Kuchurov, I.V.; Arabadzhi, S.S.; Zharkov, M.N.; Fershtat, L.L.; Zlotin, S.G. Sustainable Synthesis of Polynitroesters in the Freon Medium and their in Vitro Evaluation as Potential Nitric Oxide Donors. ACS Sustain. Chem. Eng. 2018, 6, 2535–2540. [Google Scholar] [CrossRef]
- Fedorov, B.S.; Fadeev, M.A.; Eremenko, L.T. Tris(nitroxymethyl)methylamine and tris(nitroxymethyl)nitrosomethane. Mendeleev Commun. 1997, 7, 40. [Google Scholar] [CrossRef]
- Banert, K. Product Class 2: Nitrosoalkanes and Nitroso Acetals (N,N-Dialkoxyamines). In Category 5, Compounds with One Saturated Carbon Heteroatom Bond; Science of Synthesis; Banert, K., Ed.; Georg Thieme Verlag KG: Stuttgart, Germany, 2010; Volume 41. [Google Scholar]
- Urbanski, T. Chemistry and Technology of Explosives; Laverton, S., Ed.; Pergamon Press: Warszawa, Poland, 1967; Volume 3, p. 717. [Google Scholar]
- Legocki, H., Jr.; Hackel, J. Nitroalkyl esters of α,β-unsaturated acids. II. Synthesis of 2-nitro-2,2-bis(nitromethyl)ethanol. Przem. Chem. 1966, 45, 321–324. [Google Scholar]
- Sizov, V.; Pleshakov, D.; Andrey, A.; Topchiy, M.; Nechaev, M. Synthesis and Study of the Thermal and Ballistic Properties of SMX. Cent. Eur. J. Energetic Mater. 2018, 15, 30–46. [Google Scholar] [CrossRef]
- Oxley, J.C.; Smith, J.L.; Brady Iv, J.E.; Brown, A.C. Characterization and Analysis of Tetranitrate Esters. Propellants Explos. Pyrotech. 2012, 37, 24–39. [Google Scholar] [CrossRef]
- Chavez, D.E.; Hiskey, M.A.; Naud, D.L.; Parrish, D. Synthesis of an Energetic Nitrate Ester. Angew. Chem. Int. Ed. 2008, 47, 8307–8309. [Google Scholar] [CrossRef]
- Gankin, J.; Rjabikova, N.; Ilin, V.; Smirnov, S. Nitroisobytilglycerol Nitrate Preparation Method. RU2316538C1 RUS, 17 July 2006. [Google Scholar]
- Hofwimmer, F. Zeitschrift fur das Gesamte Schiess und Sprengstoffwesen (Journal for the Field of Gunpowder and Explosives); J. F. Lehmanns Verlag: Munchen, Germany, 1912; Volume VII, p. 43. [Google Scholar]
- Khmelnitsky, L.I. Handbook of Explosives: A Manual—Part II; F. E. Dzerzhinsky Military Engineering Academy: Moscow, Russia, 1962. [Google Scholar]
- Rudolf Meyer, J.K.; Axel Homburg, N. Explosives; Wiley-VCH Verlag GmbH & Co., KGaA: Weinheim, Germany, 2007; p. 230. [Google Scholar]
- Herman, P. 2,2-Dinitropropane-1,3-dinitrate. U.S. Patent US2978484A, 1 June 1951. [Google Scholar]
- Feuer, H.; Bachman, G.B.; Kispersky, J.P. A New Preparation of Potassium Dinitromethane and its Conversion to 2,2-Dinitro-1,3-propanediol1,2. J. Am. Chem. Soc. 1951, 73, 1360. [Google Scholar] [CrossRef]
- Breiner, M.M.; Chavez, D.E.; Parrish, D.A. 2-Nitro-1,3-dinitro-oxypropane. Acta Crystallogr. Sect. E Struct. Rep. Online 2013, 69, 384. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Su, S.; He, G.; Zhang, L.; Wang, F. A new group of energetic materials. Azidonitrates. In Proceedings of the Internationals Pyrotechnics Seminar, Internationals Pyrotechnics Seminar, Seattle, WA, USA, 8–11 October 1997; Volume 23, pp. 187–192. [Google Scholar]
- Schulze, M.C.; Chavez, D.E. Synthesis and Characterization of Energetic Plasticizer AMDNNM. J. Energetic Mater. 2016, 34, 129–137. [Google Scholar] [CrossRef]
- Ou, Y.; Chen, B.; Yan, H.; Jia, H.; Li, J.; Dong, S. Development of energetic additives for propellants in China. J. Propuls. Power 1995, 11, 838–847. [Google Scholar] [CrossRef]
- Bestuzheva, V.V.; Dushenok, S.A.; Zheltikov, F.A.; Lysov, A.N. Plasticized polyurethanes. Russ. J. Appl. Chem. 2008, 81, 1019–1022. [Google Scholar] [CrossRef]
- Dave, P.R.; Duddu, R.G.; Gelber, N.; Yang, K.; Surapaneni, C.R.; Damavarapu, R. Polyazido Compounds. U.S. Patent US6965042B1, 15 November 2003. [Google Scholar]
- Agrawal, J.P. Recent trends in high-energy materials. Prog. Energy Combust. Sci. 1998, 24, 1–30. [Google Scholar] [CrossRef]
- Talawar, M.B.; Sivabalan, R.; Anniyappan, M.; Gore, G.M.; Asthana, S.N.; Gandhe, B.R. Emerging trends in advanced high energy materials. Combust. Explos. Shock Waves 2007, 43, 62–72. [Google Scholar] [CrossRef]
- Muravyev, N.V.; Monogarov, K.A.; Melnikov, I.N.; Pivkina, A.N.; Kiselev, V.G. Learning to fly: Thermochemistry of energetic materials by modified thermogravimetric analysis and highly accurate quantum chemical calculations. Phys. Chem. Chem. Phys. 2021, 23, 15522–15542. [Google Scholar] [CrossRef]
- Romer, F. Uber Abkommlinge des Nitromethans. Angewante Chem. 1955, 67, 157. [Google Scholar] [CrossRef]
- Khisamutdinov, G.K.; Karpychev, Y.V.; Zhbanova, Y.S.; Kondyukov, I.Z.; Kashaev, V.A.; Il’in, V.P. Development of new methods for the synthesis of 2,3-bis(nitroxymethyl)-2,3-dinitrobutane-1,4-diol dinitrate and its intermediates. Russ. Chem. Bull. 2015, 64, 1967–1970. [Google Scholar] [CrossRef]
- Katorov, D.V.; Rudakov, G.F.; Zhilin, V.F. Synthesis of heterocyclic geminal nitro azides. Russ. Chem. Bull. 2009, 58, 2311–2317. [Google Scholar] [CrossRef]
- Kissinger, L.W.; Benziger, T.M.; Ungnade, H.E.; Rohwer, R.K. gem-Dinitro Esters. IV. Pyridine-Catalyzed Esterification of β-Dinitro Alcohols1. J. Org. Chem. 1963, 28, 2491–2494. [Google Scholar] [CrossRef]
- Latour, S.; Wuest, J.D. Simple Syntheses of 2-Hydroxymethyl-1,3-propanediol and Related Compounds. Synthesis 1987, 1987, 742–745. [Google Scholar] [CrossRef]
- Zhang, C.; Tang, G. Preparation of Triazo Alkane. CN1600778A CH, 30 March 2005. [Google Scholar]
- Lenz, T.; Klapötke, T.M.; Mühlemann, M.; Stierstorfer, J. About the Azido Derivatives of Pentaerythritol Tetranitrate. Propellants Explos. Pyrotech. 2021, 46, 723–731. [Google Scholar] [CrossRef]
- Manelis, G.B.; Nazin, G.M.; Rubtsov Yu, I.; Strunin, V.A. Thermal Decomposition and Combustion of Expolsives and and Propellants; Taylor & Francis: London, UK, 2003. [Google Scholar]
- Yan, Q.-L.; Künzel, M.; Zeman, S.; Svoboda, R.; Bartošková, M. The effect of molecular structure on thermal stability, decomposition kinetics and reaction models of nitric esters. Thermochim. Acta 2013, 566, 137–148. [Google Scholar] [CrossRef]
- Kanno, H. A simple derivation of the empirical rule TGTM = 23. J. Non-Cryst. Solids 1981, 44, 409–413. [Google Scholar] [CrossRef]
- Karton, A.; Schreiner, P.R.; Martin, J.M.L. Heats of formation of platonic hydrocarbon cages by means of high-level thermochemical procedures. J. Comput. Chem. 2016, 37, 49–58. [Google Scholar] [CrossRef]
- Liu, J. Multivariate Nitrates. In Nitrate Esters Chemistry and Technology; Liu, J., Ed.; Springer: Singapore, 2019; pp. 377–443. [Google Scholar]
- Song, X.; Wang, Y.; An, C. Thermochemical properties of nanometer CL-20 and PETN fabricated using a mechanical milling method. AIP Adv. 2018, 8, 065009. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Lemarchand, G.; Lenz, T.; Mühlemann, M.; Stierstorfer, J.; Weber, R. Impact and Friction Sensitivities of PETN: I. Sensitivities of the Pure and Wetted Material. Propellants Explos. Pyrotech. 2022, 47, e202200150. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, version D. 01; Gaussian Wallingford: Wallingford, CT, USA, 2009. [Google Scholar]
- Werner, H.-J.; Knowles, P.J.; Knizia, G.; Manby, F.R.; Schütz, M. Molpro: A general-purpose quantum chemistry program package. WIREs Comput. Mol. Sci. 2012, 2, 242–253. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system, version 4.0. WIREs Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]
- Karton, A.; Martin, J.M.L. Explicitly correlated Wn theory: W1-F12 and W2-F12. J. Chem. Phys. 2012, 136, 124114. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Raghavachari, K.; Redfern, P.C.; Pople, J.A. Assessment of Gaussian-2 and density functional theories for the computation of enthalpies of formation. J. Chem. Phys. 1997, 106, 1063–1079. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Gritsan, N.P. Theoretical Study of the Nitroalkane Thermolysis. 1. Computation of the Formation Enthalpy of the Nitroalkanes, Their Isomers and Radical Products. J. Phys. Chem. A 2008, 112, 4458–4464. [Google Scholar] [CrossRef]
- Kiselev, V.G.; Goldsmith, C.F. Accurate Thermochemistry of Novel Energetic Fused Tricyclic 1,2,3,4-Tetrazine Nitro Derivatives from Local Coupled Cluster Methods. J. Phys. Chem. A 2019, 123, 9818–9827. [Google Scholar] [CrossRef]
- Kong, L.; Bischoff, F.A.; Valeev, E.F. Explicitly Correlated R12/F12 Methods for Electronic Structure. Chem. Rev. 2012, 112, 75–107. [Google Scholar] [CrossRef]
- Liakos, D.G.; Sparta, M.; Kesharwani, M.K.; Martin, J.M.L.; Neese, F. Exploring the Accuracy Limits of Local Pair Natural Orbital Coupled-Cluster Theory. J. Chem. Theory Comput. 2015, 11, 1525–1539. [Google Scholar] [CrossRef]
- Lee, T.J.; Taylor, P.R. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. 1989, 36, 199–207. [Google Scholar] [CrossRef]
- Chase, M.W., Jr. NIST-JANAF Thermochemical Tables, Monograph 9, 4th ed.; American Chemical Society: Washington, DC, USA, 1998; Volume 9, pp. 1–1951. [Google Scholar]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Kossmann, S.; Neese, F. Comparison of two efficient approximate Hartee–Fock approaches. Chem. Phys. Lett. 2009, 481, 240–243. [Google Scholar] [CrossRef]
- UN Recommendations on Transport of Dangerous Goods. Environ. Conserv. 2009, 22, 370–371. [CrossRef]
- Muravyev, N.V.; Wozniak, D.R.; Piercey, D.G. Progress and performance of energetic materials: Open dataset, tool, and implications for synthesis. J. Mater. Chem. A 2022, 10, 11054–11073. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).