

Derivation of Human Corneal Keratocytes from ReLEx SMILE Lenticules for Cell Therapy and Tissue Engineering

 , ,
, ,  , ,
, ,
Abstract
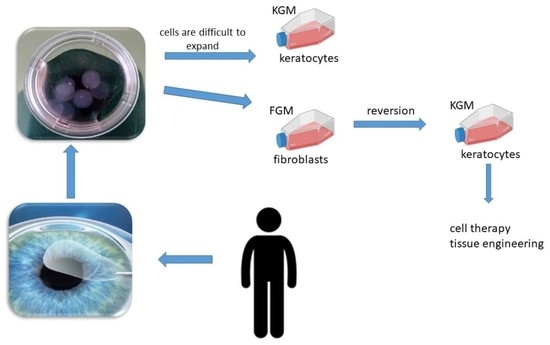
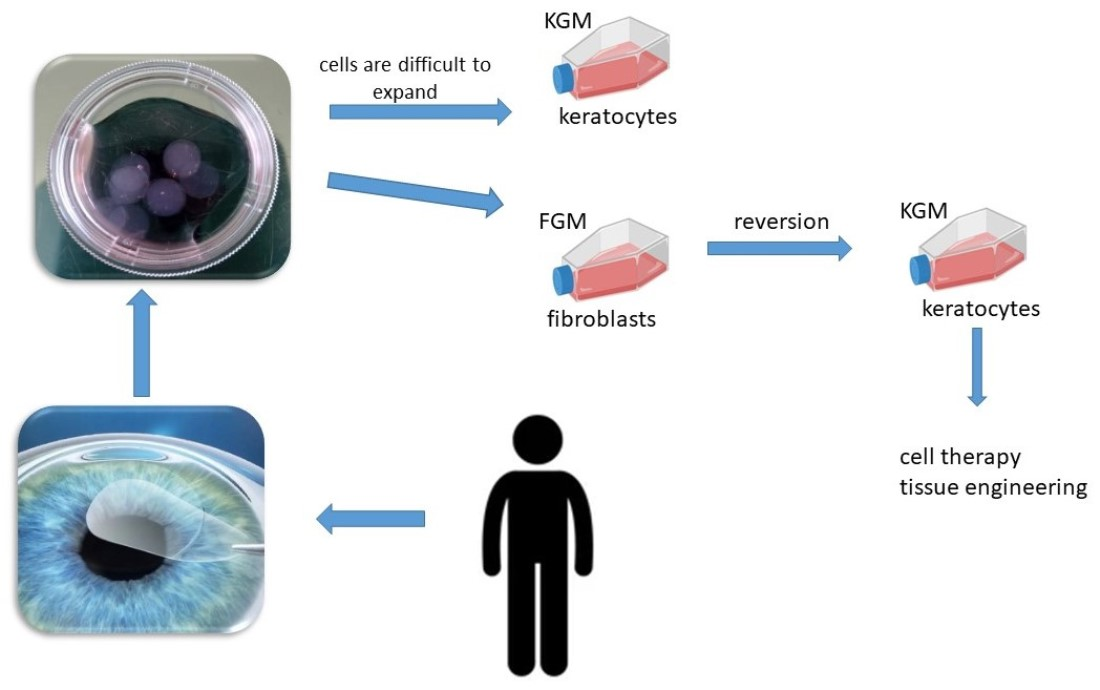{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
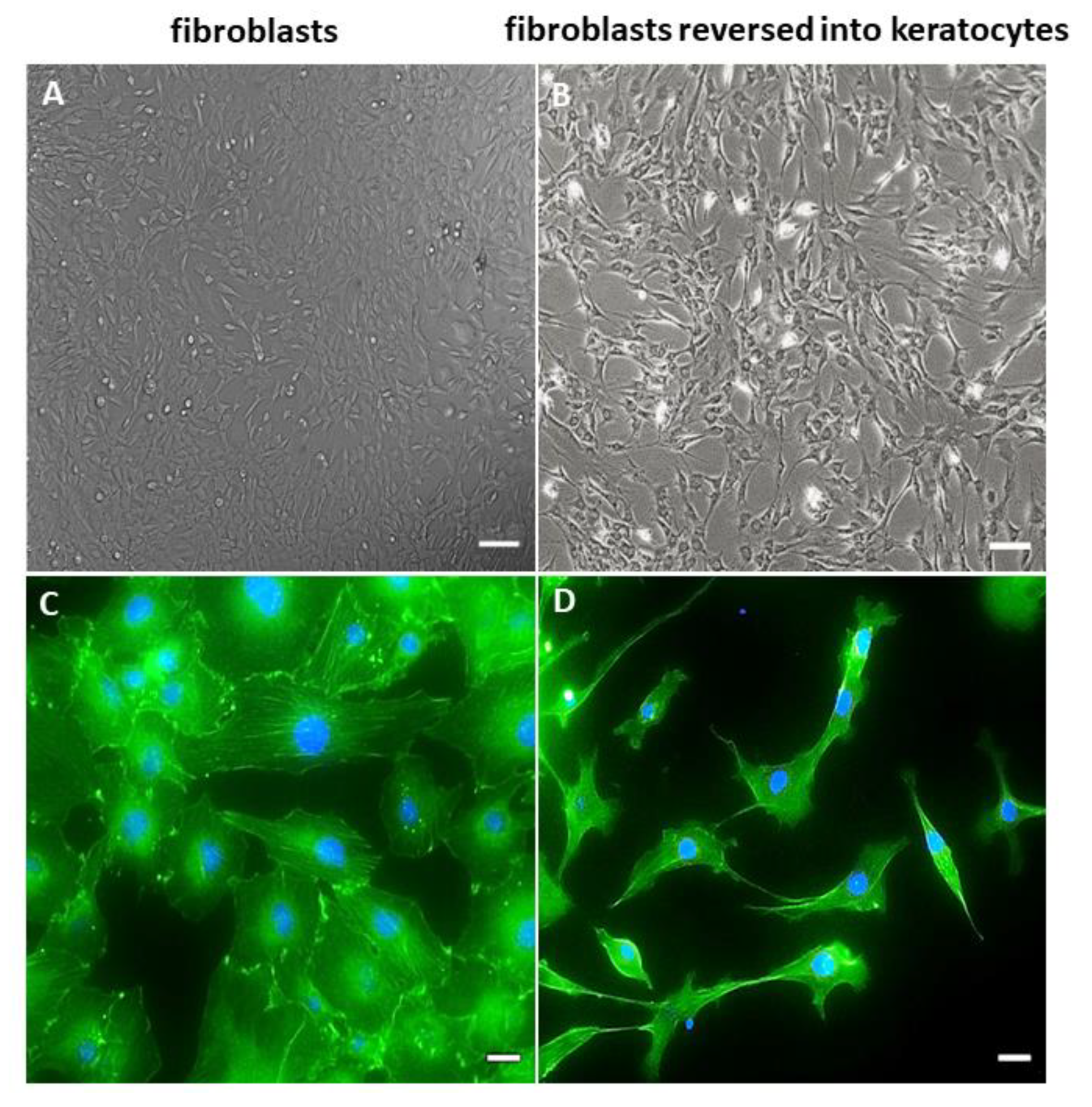
2.1. Morphology of hCSCs
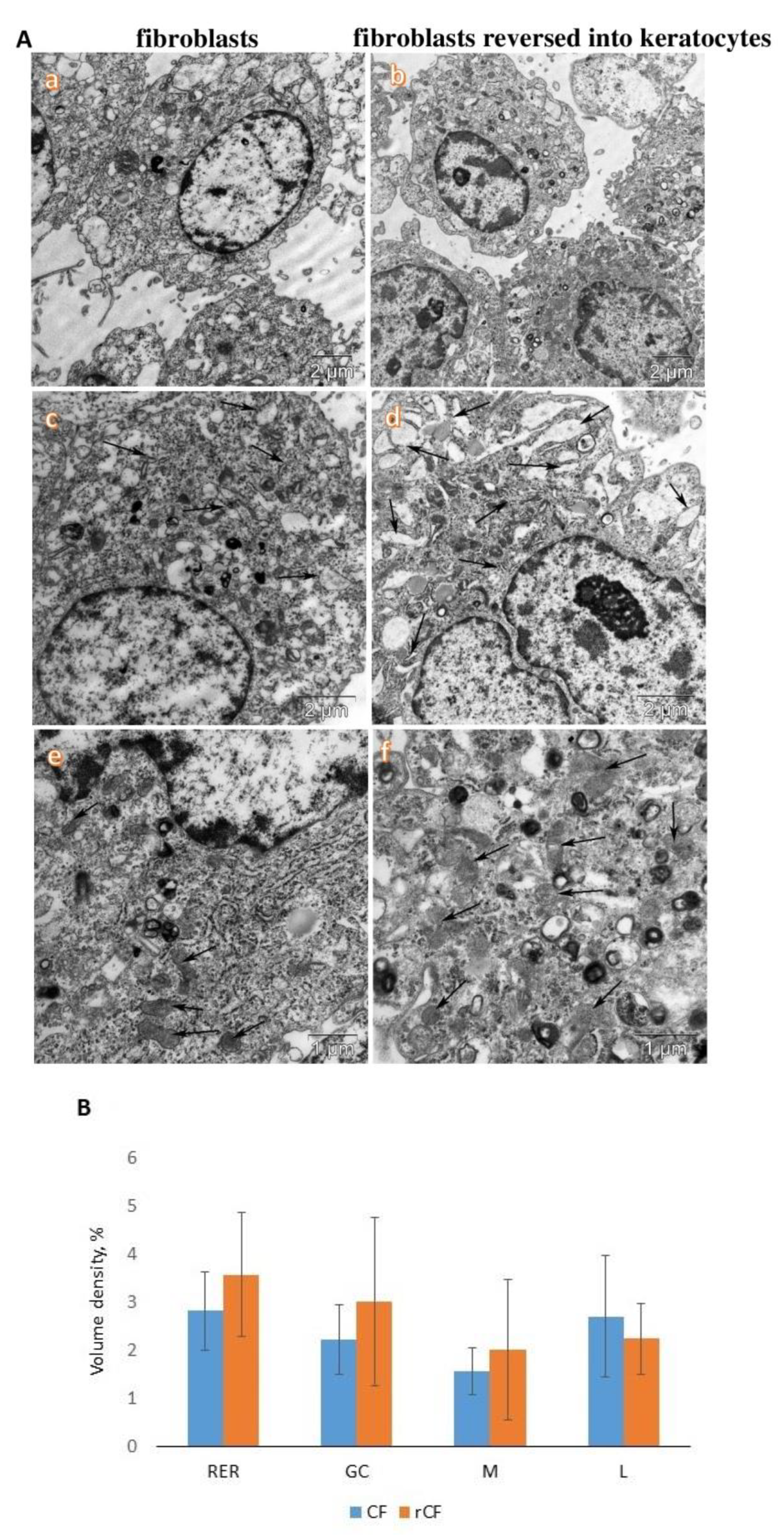
2.2. Transmission Electron Microscopy of hCSCs
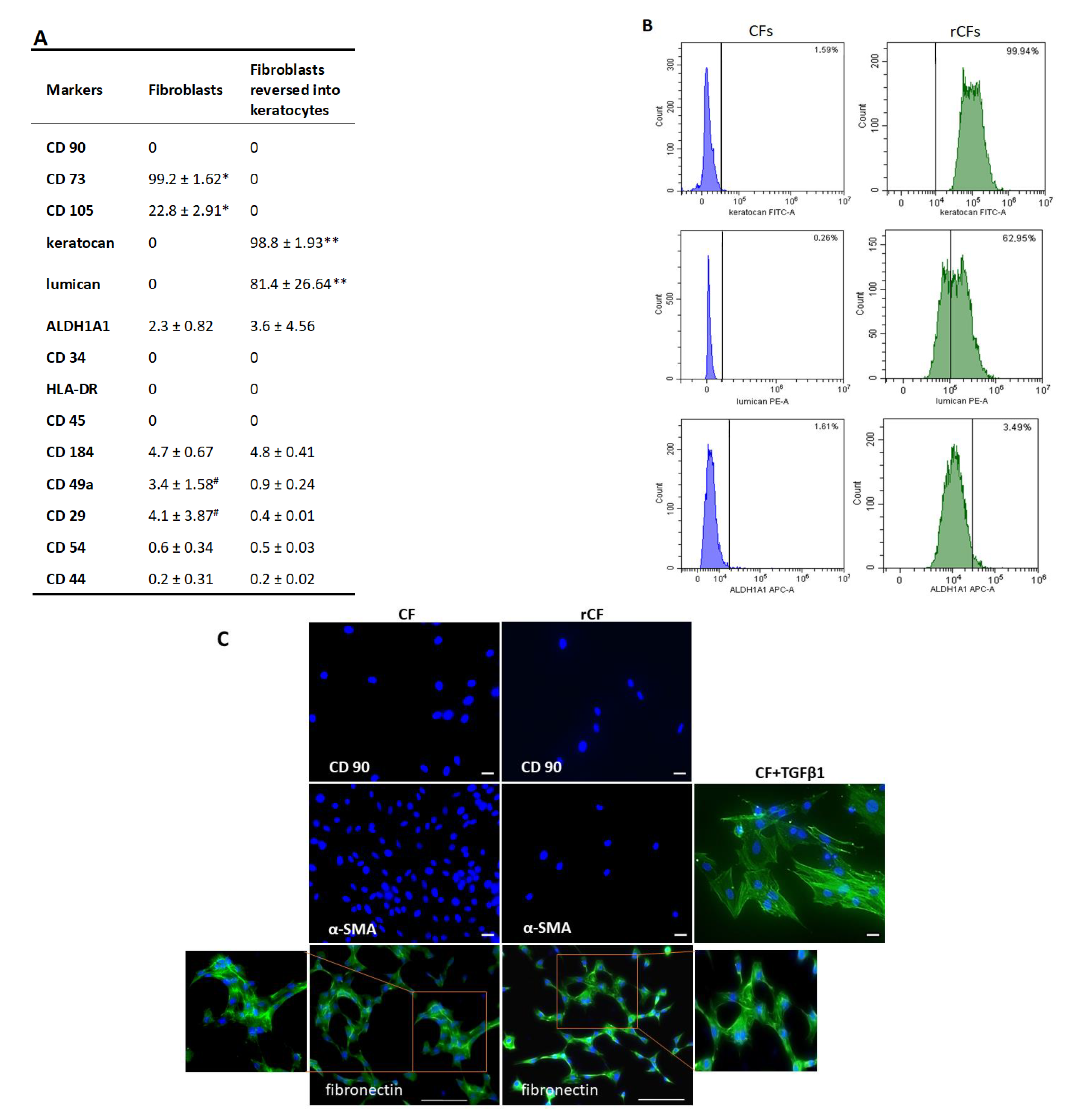
2.3. Phenotype of hCSCs
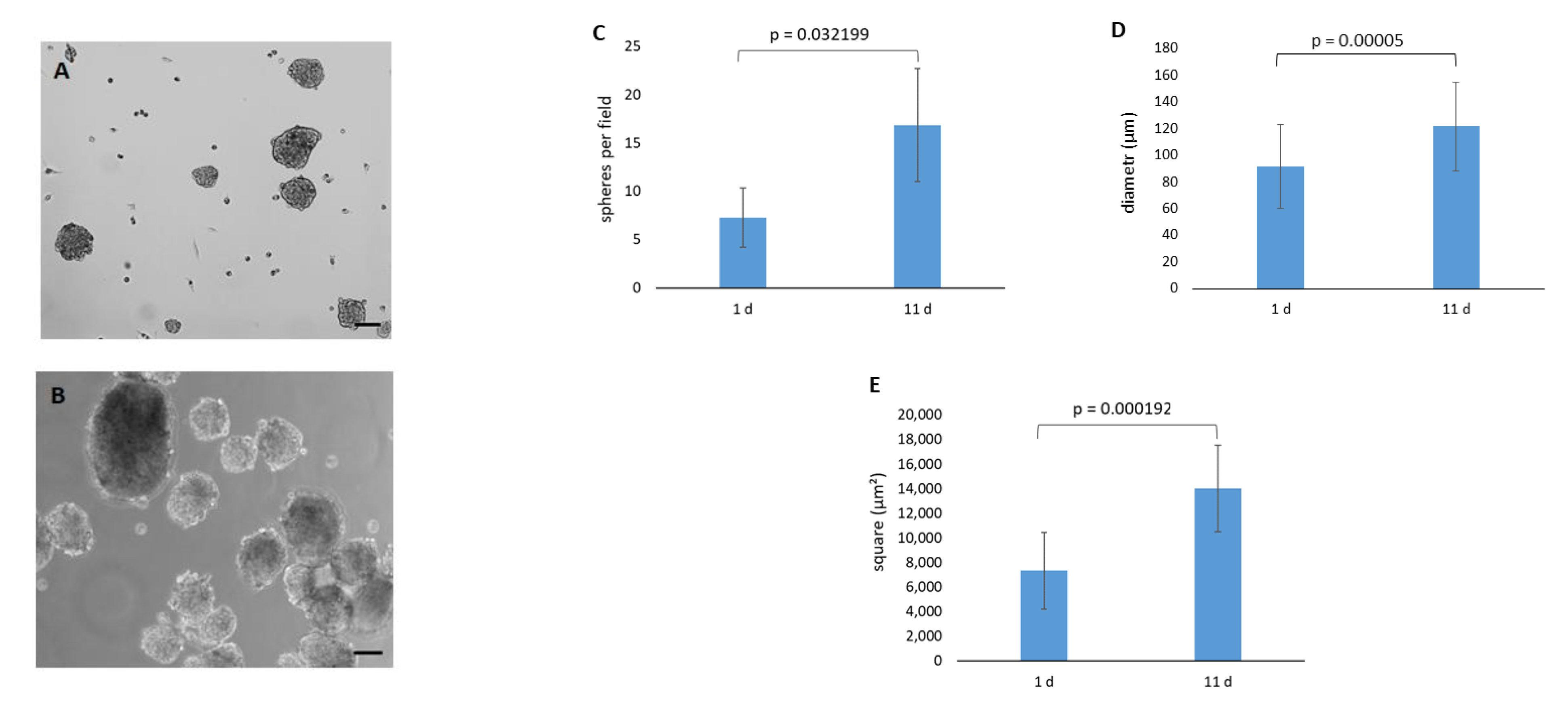
2.4. Spontaneous Sphere Formation
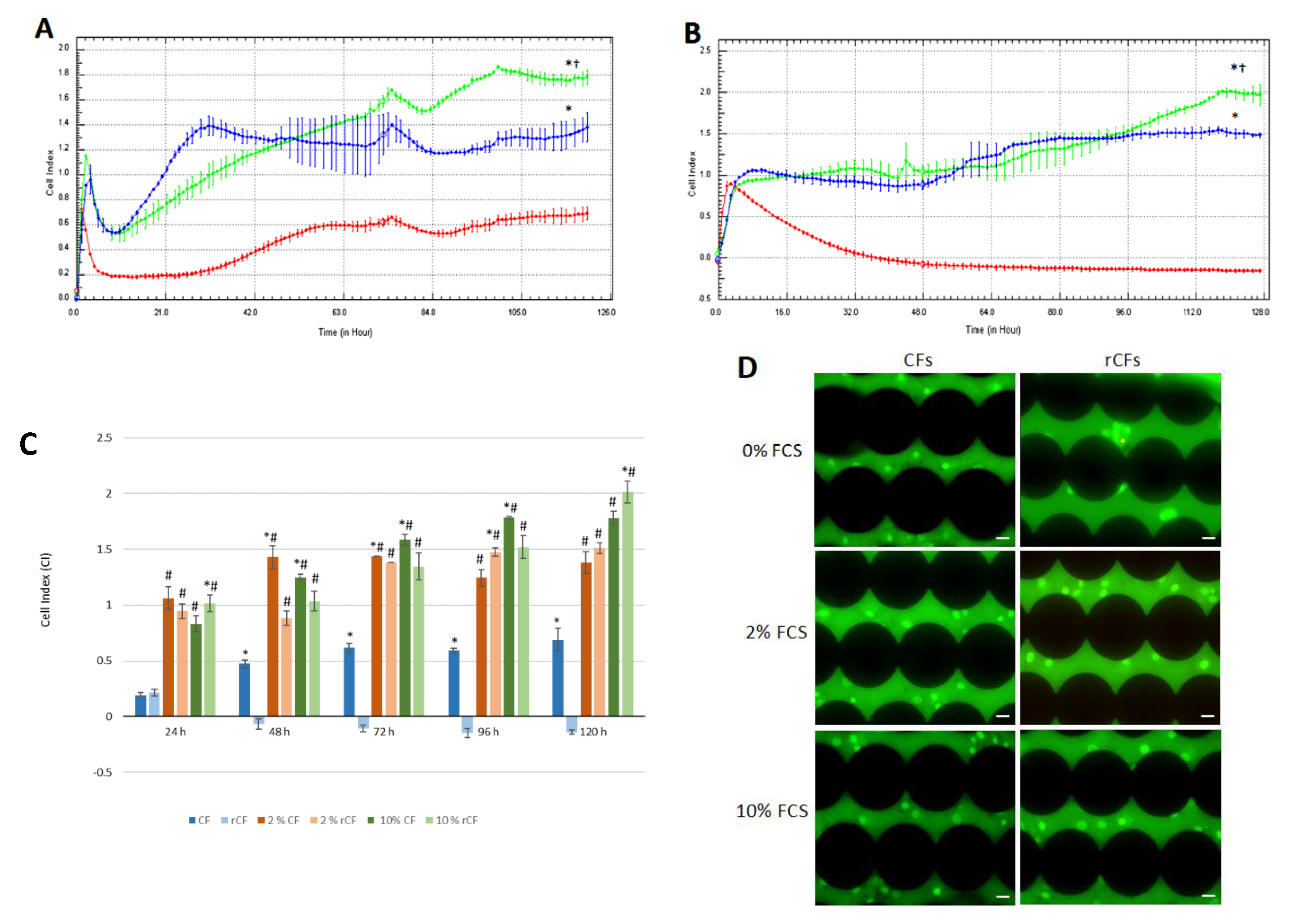
2.5. hCSC Proliferation
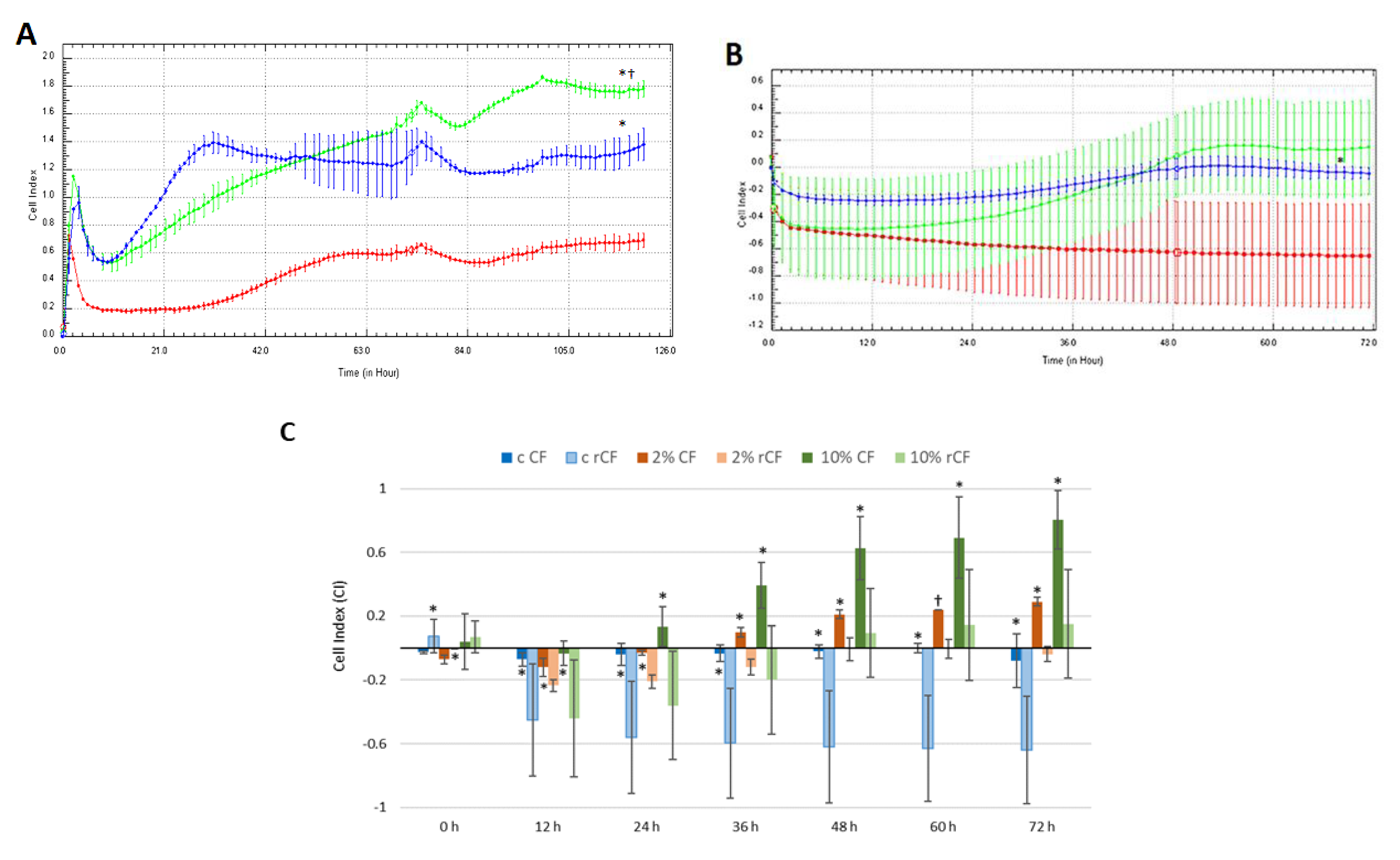
2.6. hCSC Migration
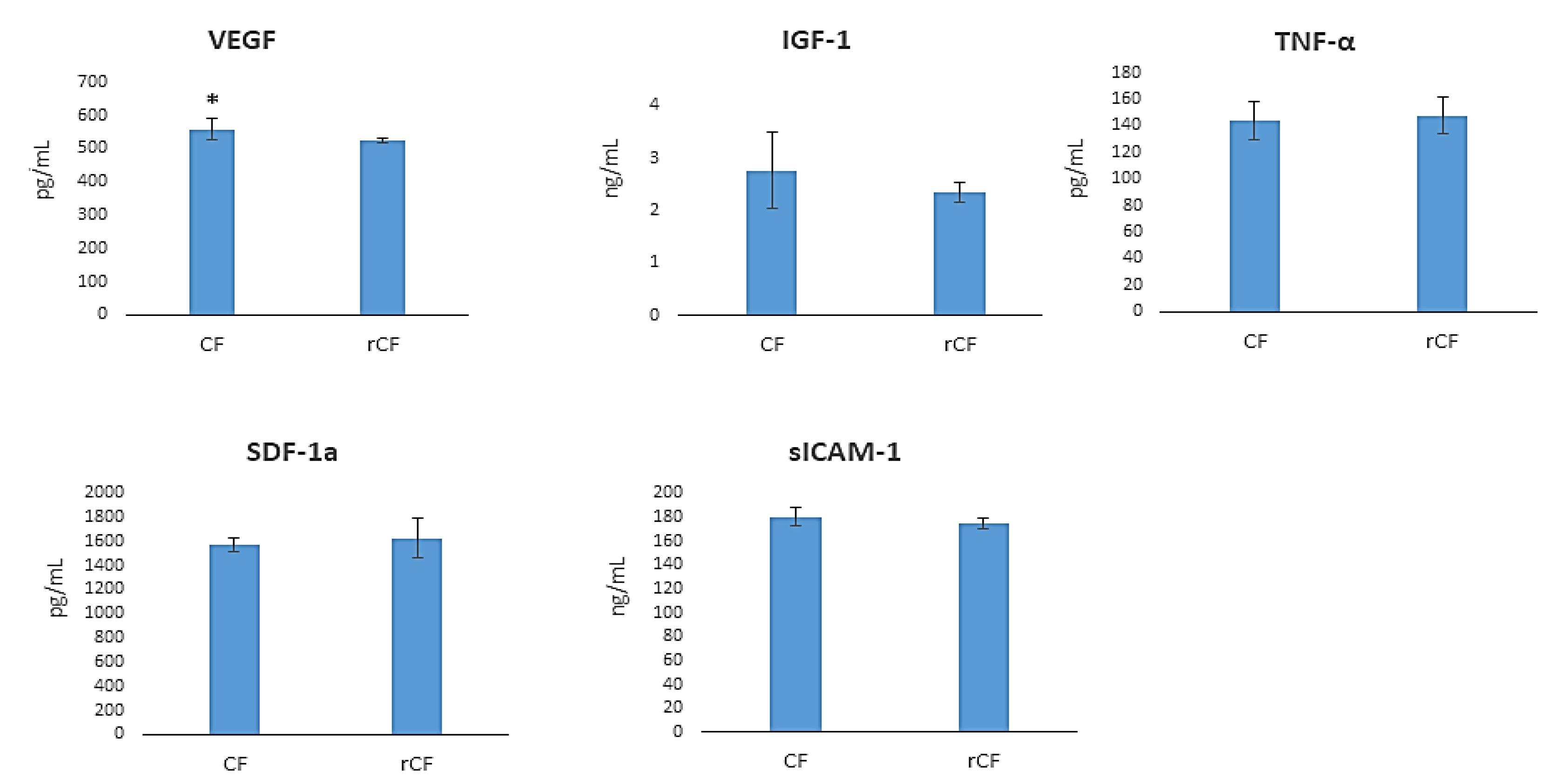
2.7. hCSC Protein Components Production
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. ReLEx SMILE
4.3. Corneal Stromal Cell Isolation and Expansion
4.4. Corneal Fibroblast Reversion into Keratocytes
4.5. Transmission Electron Microscopy
4.6. Phenotyping of Corneal Stromal Cells
4.7. Immunocytochemistry
4.8. Sphere Formation of Reversed Fibroblasts
4.9. Real-Time Cell Proliferation Monitoring
4.10. Real-Time Cell Migration Monitoring
4.11. Obtaining Conditioned Media
4.12. ELISA
4.13. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fukuda, K. Corneal fibroblasts: Function and markers. Exp. Eye Res. 2020, 200, 108229. [Google Scholar] [CrossRef] [PubMed]
- Young, R.D.; Knupp, C.; Pinali, C.; Png, K.M.; Ralphs, J.R.; Bushby, A.J.; Starborg, T.; Kadler, K.E.; Quantock, A.J. Three-dimensional aspects of matrix assembly by cells in the developing cornea. Proc. Natl. Acad. Sci. USA 2014, 111, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Lync, A.P.; O’Sullivan, F.; Ahearne, M. The effect of growth factor supplementation on corneal stromal cell phenotype in vitro using a serum-free media. Exp. Eye Res. 2016, 151, 26–37. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, K.W.; Chun, Y.S.; Kim, J.C. Human mesenchymal stem cells differentiate into keratocyte-like cells in keratocyte-conditioned medium. Exp. Eye Res. 2012, 101, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.-G.; Jun, A.S.; Chakravarti, S. Sphere formation from corneal keratocytes and phenotype specific markers. Exp. Eye Res. 2011, 93, 898–905. [Google Scholar] [CrossRef]
- Hinz, B. Myofibroblasts. Exp. Eye Res. 2016, 142, 56–70. [Google Scholar] [CrossRef]
- Zhou, X.; Li, J.; Backman, L.J.; Danielson, P. Keratocyte Differentiation Is Regulated by NF-κB and TGFβ Signaling Crosstalk. Int. J. Mol. Sci. 2022, 23, 11073. [Google Scholar] [CrossRef]
- Fini, M.E. Keratocyte and fibroblast phenotypes in the repairing cornea. Prog. Retin. Eye Res. 1999, 18, 529–551. [Google Scholar] [CrossRef]
- Ljubimov, A.V.; Saghizadeh, M. Progress in corneal wound healing. Prog. Retin. Eye Res. 2015, 49, 17–45. [Google Scholar] [CrossRef]
- Robaei, D.; Watson, S. Corneal blindness: A global problem. Clin. Exp. Ophthalmol. 2014, 42, 213–214. [Google Scholar] [CrossRef]
- Gain, P.; Jullienne, R.; He, Z.; Aldossary, M.; Acquart, S.; Cognasse, F.; Thuret, G. Global survey of corneal transplantation and eye banking. JAMA Ophthalmol. 2016, 134, 167–173. [Google Scholar] [CrossRef]
- Kureshi, A.K.; Funderburgh, J.L.; Daniels, J.T. Human corneal stromal stem cells exhibit survival capacity following isolation from stored organ-culture corneas. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7583–7588. [Google Scholar] [CrossRef]
- Nagymihaly, R.M.; Moe, M.C.; Petrovski, G. Isolation and culture of corneal stromal stem cells. Methods Mol. Biol. 2020, 2145, 1–15. [Google Scholar] [PubMed]
- Chaurasia, S.S.; Kaur, H.; de Medeiros, F.W.; Smith, S.D.; Wilson, S.E. Dynamics of the expression of intermediate filaments vimentin and desmin during myofibroblast differentiation after corneal injury. Exp. Eye Res. 2009, 89, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Karimi, A.-H.; Wong, A.; Bizheva, K. Automated detection and cell density assessment of keratocytes in the human corneal stroma from ultrahigh resolution optical coherence tomograms. Biomed. Opt. Express 2011, 2, 2905–2916. [Google Scholar] [CrossRef]
- Moshirfar, M.; Thomson, A.C.; West, W.B.; Hall, M.N.; McCabe, S.E.; Thomson, R.J.; Ronquillo, Y.C.; Hoopes, P.C. Initial single-site experience using SMILE for the treatment of astigmatism in myopic eyes and comparison of astigmatic outcomes with existing literature. Clin. Ophthalmol. 2020, 14, 3551–3562. [Google Scholar] [CrossRef]
- Santra, M.; Liu, Y.-C.; Jhanji, V.; Yam, G.H.-F. Human SMILE-Derived Stromal Lenticule Scaffold for Regenerative Therapy: Review and Perspectives. Int. J. Mol. Sci. 2022, 23, 7967. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Qiu, P.; Wu, F.; Zhang, W.; Teng, W.; Qin, Z.; Li, C.; Zhou, J.; Fang, Z.; Tang, Q.; et al. Construction of a corneal stromal equivalent with SMILE-derived lenticules and fibrin glue. Sci. Rep. 2016, 6, 33848. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Li, Y.; Liu, X.-W.; Xu, J. A novel tectonic keratoplasty with femtosecond laser intrastromal lenticule for corneal ulcer and perforation. Chin. Med. J. 2016, 129, 1817–1821. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Yang, S.; Lu, T.C. Tuck-in lamellar keratoplasty with an lenticule obtained by small incision lenticule extraction for treatment of post- LASIK ectasia. Sci. Rep. 2017, 7, 17806. [Google Scholar] [CrossRef]
- Liu, Y.C.; Teo, E.P.W.; Ang, H.P.; Seah, X.Y.; Lwin, N.C.; Yam, G.H.; Mehta, J.S. Biological corneal inlay for presbyopia derived from small incision lenticule extraction (SMILE). Sci. Rep. 2018, 8, 1831. [Google Scholar] [CrossRef]
- El Zarif, M.; Alio, J.L.; Del Barrio, J.L.A.; De Miguel, M.P.; Jawad, A.K.; Makdissy, N. Corneal stromal regeneration: A review of human clinical studies in keratoconus treatment. Front. Med. 2021, 8, 650724. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Cui, Z.; Gu, J.; Wang, Y.; Tang, S.; Chen, J. Effect of porcine corneal stromal extract on keratocytes from SMILE-derived lenticules. J. Cell. Mol. Med. 2021, 25, 1207–1220. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, S.; Tan, Y.; Wang, Y. Effects of Mechanical Compression on Cell Morphology and Function in Human Corneal Fibroblasts. Curr. Eye Res. 2021, 46, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Surovtseva, M.A.; Poveshchenko, O.V.; Krasner, K.Y.; Kim, I.I.; Lykov, A.P.; Bondarenko, N.A.; Shulmina, L.A.; Trunov, A.N.; Chernykh, V.V. Morphofunctional Properties of Corneal Stromal Cells. Bull. Exp. Biol. Med. 2021, 172, 96–99. [Google Scholar] [CrossRef] [PubMed]
- West-Mays, J.A.; Dwivedi, D.J. The keratocyte: Corneal stromal cell with variable repair phenotypes. Int. J. Biochem. Cell Biol. 2006, 38, 1625–1631. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, A.; Swioklo, S.; Connon, C.J. 3D bioprinting of a corneal stroma equivalent. Exp. Eye. Res. 2018, 173, 188–193. [Google Scholar] [CrossRef]
- Sidney, L.E.; Branch, M.J.; Dunphy, S.E.; Dua, H.S.; Hopkinson, A. Concise review: Evidence for CD34 as a common marker for diverse progenitors. Stem Cells 2014, 32, 1380–1389. [Google Scholar] [CrossRef]
- Wilson, S.L.; Yang, B.Y.; Haj, A.J. Corneal stromal cell plasticity: In vitro regulation of cell phenotype through cell-cell interactions in a three-dimensional model. Tissue Eng. 2014, 20, 225–238. [Google Scholar] [CrossRef]
- Joseph, A.; Hossain, P.; Jham, S.; Jones; R.E.; Tighe, P.; McIntosh, R.S.; Dua, H.S. Expression of CD34 and L-selectin on human corneal keratocytes. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4689–4692. [Google Scholar] [CrossRef]
- Fries, K.M.; Blieden, T.; Looney, R.J.; Sempowski, G.D.; Silvera, M.R.; Willis, R.A.; Phipps, R.P. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin. Immunol. Immunopathol. 1994, 72, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Wilson, S.E. The Yin and Yang of Mesenchymal Cells in the Corneal Stromal Fibrosis Response to Injury: The Cornea as a Model of Fibrosis in Other Organs. Biomolecules 2023, 13, 87. [Google Scholar] [CrossRef]
- Buss, D.G.; Giuliano, E.A.; Sharma, A.; Mohan, R.R. Isolation and cultivation of equine corneal keratocytes, fibroblasts and myofibroblasts. Vet. Ophthalmol. 2010, 13, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Funderburgh, M.L.; Mann, M.M.; Funderburgh, J.L. Keratocyte phenotype is enhanced in the absence of attachment to the substratum. Mol. Vis. 2008, 14, 308–317. [Google Scholar]
- Sidney, L.E.; McIntosh, O.D.; Hopkinson, A. Phenotypic change and induction of cytokeratin expression during in vitro culture of corneal stromal cells. Investig. Ophthalmol. Vis. Sci. 2015, 56, 7225–7235. [Google Scholar] [CrossRef] [PubMed]
- Miron-Mendoza, M.; Graham, E.; Manohar, S.; Petroll, W.M. Fibroblast-fibronectin patterning and network formation in 3D fibrin matrices. Matrix Biol. 2017, 64, 69–80. [Google Scholar] [CrossRef]
- Stepp, M.A.; Daley, W.P.; Bernstein, A.M.; Pal-Ghosh, S.; Tadvalkar, G.; Shashurin, A.; Palsen, S.; Jurjus, R.A.; Larsen, M. Syndecan-1 Regulates Cell Migration and Fibronectin Fibril Assembly. Exp. Cell Res. 2010, 14, 2322–2339. [Google Scholar] [CrossRef]
- Torres, P.F.; Kijlstra, A. The role of cytokines in corneal immunopathology. Ocul. Immunol. Inflamm. 2001, 9, 9–24. [Google Scholar] [CrossRef]
- Dan Cosnita, A.; Raicam, M.; Sava, M.P.; Cimpean, I.M. Gene expression profile of vascular endothelial growth factors (VEGFs) and platelet-derived growth factors (PDGFs) in the normal cornea. In Vivo 2021, 35, 805–813. [Google Scholar] [CrossRef]
- Philipp, W.; Speicher, L.; Humpel, C. Expression of vascular endothelial growth factor and its receptors in inflamed and vascularized human corneas. Investig. Ophthalmol. Vis. Sci. 2000, 41, 2514–2522. [Google Scholar]
- Mirshahi, P.; Berthaut, A.V.; Soria, J.; Mirshahi., M.; Legeais, J.-M. Igf via Ekr Pathway Signalling Induces Human Corneal Fibroblasts Network Formation in vitro. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4824. [Google Scholar]
- Stuard, W.L.; Titone, R.; Robertson, D.M. The IGF/Insulin-IGFBP Axis in Corneal Development, Wound Healing, and Disease. Front. Endocrinol. 2020, 11, 24. [Google Scholar] [CrossRef]
- Sarenac, M.; Trapecar, T.; Gradisnik, L.; Rupnik, M.S.; Pahor, D. Single-cell analysis reveals IGF-1 potentiation of inhibition of the TGF-β/Smad pathway of fibrosis in human keratocytes in vitro. Sci. Rep. 2016, 6, 34373. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.A.; Yanai, R.; Nishida, T. IGF-1 released by corneal epithelial cells induces up-regulation of N-cadherin in corneal fibroblasts. J. Cell Physiol. 2009, 221, 254–261. [Google Scholar] [CrossRef]
- Berthaut, A.; Mirshahi, P.; Benabbou, N.; Ducros, E.; Agra, A.; Therwath, A.; Legeais, J.M.; Mirshahi, M. Insulin growth factor promotes human corneal fibroblast network formation in vitro. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7647–7653. [Google Scholar] [CrossRef] [PubMed]
- Saika, S.; Ikeda, K.; Yamanaka, O.; Flanders, K.C.; Okada, Y.; Miyamoto, T.; Kitano, A.; Ooshima, A.; Nakajima, Y.; Ohnishi, Y.; et al. Loss of tumor necrosis factor potentiates transforming growth factor-mediated pathogenic tissue response during wound healing. Am. J. Pathol. 2006, 168, 1848–1860. [Google Scholar] [CrossRef] [PubMed]
- Okada, N.; Fukagawa, K.; Takano, Y. The implications of the upregulation of ICAM-1/VCAM-1 expression of corneal fibroblasts on the pathogenesis of allergic keratopathy. Investig. Ophthalmol. Vis. Sci. 2005, 46, 4512–4518. [Google Scholar] [CrossRef]
- Bourcier, T.; Berbar, T.; Paquet, S.; Rondeau, N.; Thomas, F.; Borderie, V.; Laroche, L.; Rostène, W.; Haour, F.; Lombet, A. Characterization and functionality of CXCR4 chemokine receptor and SDF-1 in human corneal fibroblasts. Mol. Vis. 2003, 9, 96–102. [Google Scholar]
- Vorkauf, W.; Vorkauf, M.; Nölle, B.; Duncker, G. Adhesion molecules in normal and pathological corneas. Graefe’s Archive for Clin. Exp. Ophthalmol. 1995, 233, 209–219. [Google Scholar] [CrossRef]
- Sekundo, W.; Kunert, K.; Russmann, C.; Gill, A.; Bissman, W.; Stobrawa, G.; Sticker, M.; Bischoff, M.; Blum, M. First efficacy and safety study of femtosecond lenticule extraction for the correction of myopia: Six-month results. J. Cataract. Refract. Surg. 2008, 34, 1513–2150. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Surovtseva, M.A.; Kim, I.I.; Bondarenko, N.A.; Lykov, A.P.; Krasner, K.Y.; Chepeleva, E.V.; Bgatova, N.P.; Trunov, A.N.; Chernykh, V.V.; Poveshchenko, O.V. Derivation of Human Corneal Keratocytes from ReLEx SMILE Lenticules for Cell Therapy and Tissue Engineering. Int. J. Mol. Sci. 2023, 24, 8828. https://doi.org/10.3390/ijms24108828
Surovtseva MA, Kim II, Bondarenko NA, Lykov AP, Krasner KY, Chepeleva EV, Bgatova NP, Trunov AN, Chernykh VV, Poveshchenko OV. Derivation of Human Corneal Keratocytes from ReLEx SMILE Lenticules for Cell Therapy and Tissue Engineering. International Journal of Molecular Sciences. 2023; 24(10):8828. https://doi.org/10.3390/ijms24108828
Chicago/Turabian StyleSurovtseva, Maria A., Irina I. Kim, Natalia A. Bondarenko, Alexander P. Lykov, Kristina Yu. Krasner, Elena V. Chepeleva, Nataliya P. Bgatova, Alexander N. Trunov, Valery V. Chernykh, and Olga V. Poveshchenko. 2023. "Derivation of Human Corneal Keratocytes from ReLEx SMILE Lenticules for Cell Therapy and Tissue Engineering" International Journal of Molecular Sciences 24, no. 10: 8828. https://doi.org/10.3390/ijms24108828
APA StyleSurovtseva, M. A., Kim, I. I., Bondarenko, N. A., Lykov, A. P., Krasner, K. Y., Chepeleva, E. V., Bgatova, N. P., Trunov, A. N., Chernykh, V. V., & Poveshchenko, O. V. (2023). Derivation of Human Corneal Keratocytes from ReLEx SMILE Lenticules for Cell Therapy and Tissue Engineering. International Journal of Molecular Sciences, 24(10), 8828. https://doi.org/10.3390/ijms24108828