Abstract
Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (isoBBT) is a new electron-withdrawing building block that can be used to obtain potentially interesting compounds for the synthesis of OLEDs and organic solar cells components. The electronic structure and delocalization in benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole), 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole), and 4,8-dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) were studied using X-ray diffraction analysis and ab initio calculations by EDDB and GIMIC methods and were compared to the corresponding properties of benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (BBT). Calculations at a high level of theory showed that the electron affinity, which determines electron deficiency, of isoBBT was significantly smaller than that of BBT (1.09 vs. 1.90 eV). Incorporation of bromine atoms improves the electrical deficiency of bromobenzo-bis-thiadiazoles nearly without affecting aromaticity, which increases the reactivity of these compounds in aromatic nucleophilic substitution reactions and, on the other hand, does not reduce the ability to undergo cross-coupling reactions. 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) is an attractive object for the synthesis of monosubstituted isoBBT compounds. The goal to find conditions for the selective substitution of hydrogen or bromine atoms at position 4 in order to obtain compounds containing a (het)aryl group in this position and to use the remaining unsubstituted hydrogen or bromine atoms to obtain unsymmetrically substituted isoBBT derivatives, potentially interesting compounds for organic photovoltaic components, was not set before. Nucleophilic aromatic and cross-coupling reactions, along with palladium-catalyzed C-H direct arylation reactions for 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole), were studied and selective conditions for the synthesis of monoarylated derivatives were found. The observed features of the structure and reactivity of isoBBT derivatives may be useful for building organic semiconductor-based devices.
Keywords:
sulfur-nitrogen heterocycles; 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole); X-ray analysis; ab initio calculations; EDDB and GIMIC methods; UV-Vis spectra; aromatic nucleophilic substitution; Suzuki and Stille cross-coupling reactions; direct (het)arylation palladium-catalyzed reactions 1. Introduction
The aromaticity and reactivity of heterocyclic compounds are among the most studied problems in organic chemistry [1,2,3,4,5]. The properties of monocyclic heterocycles have been thoroughly studied for a long time, while fused heterocyclic systems often face a number of problems with their aromaticity/antiaromaticity and the consequent differences in reactivity [1,2]. Fused heterocyclic systems containing many nitrogen and chalcogen (mainly sulfur) atoms, which have pronounced acceptor properties, in the rings attracted particular interest in recent years [6]. Electron-accepting moieties are widely represented in π-conjugated organic molecules in various combinations with electron donors and π-conjugated bridges. These organic chromophores are widely used in semiconductor-based devices such as dye-sensitized solar cells (DSSCs), organic field-effect transistors (OFETs), organic light-emitting diodes (OLEDs), and electrochromic devices (ECDs) [7]. Hybridization of the energy levels between the donor and acceptor parts in molecules can decrease the difference between EHOMO and ELUMO (the energy band gap, Egap), thus improving the optoelectronic properties of the molecule [8]. The important feature of the acceptor fragment is the electron affinity (EA) that is related to the energies of the lowest unoccupied molecular orbital (ELUMO).
A variety of heterocyclic acceptors are well known and intensely studied [9,10,11]. Heterocycles with a high electron affinity as electron-acceptor blocks have received a lot of attention [12]. An exceptional place among these heterocycles is occupied by 2,1,3-benzothiadiazole (BTD) and their derivatives (Figure 1) due to their excellent properties, such as strong electron-withdrawing properties, intense light absorption, and good photochemical stability [13,14]. Nevertheless, there is a strong demand to create electron-deficient heterocyclic systems with a stronger accepting ability. One of the options is to annulate the BTD cycle with another thiadiazole ring in order to build benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (benzo-bis-thiadiazole, BBT), a sulfur–nitrogen heterocycle with the lowest LUMO energy [15]. In fact, BBT derivatives are being actively explored for application in pharmacology and in various optoelectronic devices [16]. On the other hand, replacement of the 1,2,5-thiadiazole ring in 2,1,3-benzothiadiazole (BTD) by the 1,2,3-thiadiazole ring results in benzo[d][1,2,3]thiadiazole (isoBTD) compounds with properties similar to those of BTD but with a higher ELUMO and band gap (Eg) [17] values. It was recently found that a BBT isomer, benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (isoBTD), also had promising electron-accepting properties, and its 4,8-dibromo derivative could be successfully involved in aromatic nucleophilic substitution reactions and in Suzuki–Miyaura and Stille palladium-catalyzed cross-coupling reactions with selective formation of mono- and bis-arylated heterocycles [18]. Many sulfur-containing heterocycles are known to possess anticancer activity [19] due to low-lying C-S σ* or C-N σ* orbitals, which are responsible for drug–target interactions [20]. Therefore, isoBBT derivatives are of additional interest in terms of potential biological activity.
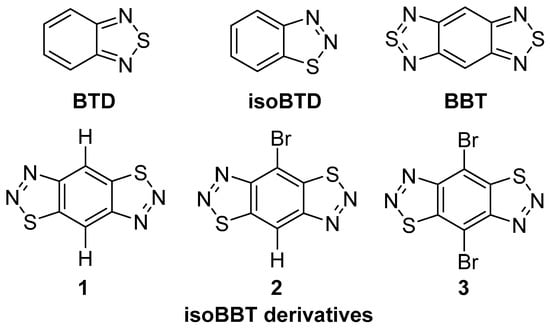
Figure 1.
Structures of 2,1,3-benzothiadiazole (BTD), benzo-[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (BBT), and benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (isoBBT) derivatives.
In order to reveal the electronic structure of isoBBT derivatives and find a possibility to obtain mono-arylated isoBBT derivatives selectively, we describe a study of the electronic structure and electron delocalization in benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole), its 4-bromo and 4,8-dibromo derivatives by X-ray analysis and ab initio calculations using the electron density of delocalized bonds (EDDB) [21,22] and the gauge-including magnetically induced currents (GIMIC) [23,24] methods, as well as palladium-catalyzed C-H direct arylation reactions, and C-Br aromatic nucleophilic and cross-coupling reactions for 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) with selective formation of mono-arylated derivatives.
2. Results and Discussion
2.1. Structure and Electronic Percularities of Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) and Its Bromo Derivatives
According to X-ray diffraction studies, the conformations of 1–3 in the crystal are planar (Figure 2), and the lengths of all chemical bonds fall in the range typical of thiadiazole derivatives. The molecules of 1 and 3 occupy the positions at the crystallographic center of inversion, so the unique parts of their unit cells are two halves of the corresponding molecules. The small number or absence of hydrogen atoms in 1–3 allow one to assume that the contribution of hydrogen bonds to the respective lattice energies are low. Indeed, all N...H and S...H distances in 1 and 2 exceed the sums of the Van der Waals radii of these elements. Thus, other types of intermolecular interactions are responsible for the stabilization of the crystal structures of 1–3. The planar molecules of 1–3 are assembled into infinite stacks. Each crystallographically independent molecule in 1 and 3 forms separate stacks (Figure 3 and Figure 4). The stacks are interlinked by weak N...S and N...Br interactions.
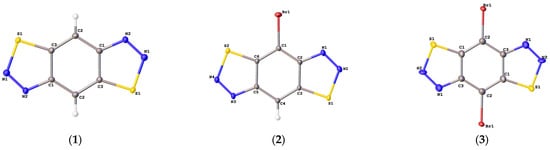
Figure 2.
Molecular structure of 1–3 presented in ADP ellipsoids (probability is equal to 50%).
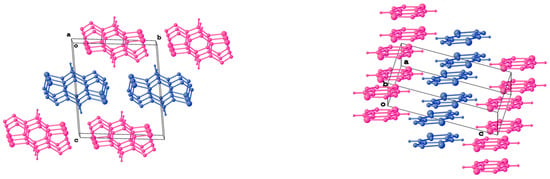
Figure 3.
Mutual orientation of stacks formed by two crystallographically independent molecules in crystal packing of 1. Magenta and blue colors are used to highlight the stacks formed by a particular type of independent molecules.
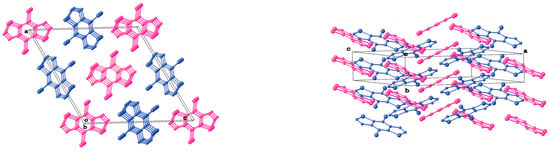
Figure 4.
Mutual orientation of stacks formed by two crystallographically independent molecules in crystal packing of 3. Magenta and blue colors are used to highlight the stacks formed by a particular type of independent molecules.
The distribution of electrostatic potential mapped on the molecular Hirshfeld surfaces [25,26] of 1–3 indicates that the N...S and N...Br interactions are complementary and maintain the crystal packing stability (Figure 5). Two-dimensional fingerprint plots (Figures S2–S4) for 1–3 indicate that they made a relatively large contribution to the intermolecular contacts with de = di ≈ 2 Å, which is characteristic for N…S, N…Br and stacking interactions. In turn, the nature of stacking interactions can be described as the interaction of the N=N bond with the central benzene rings (positive and negative regions of electrostatic potential).
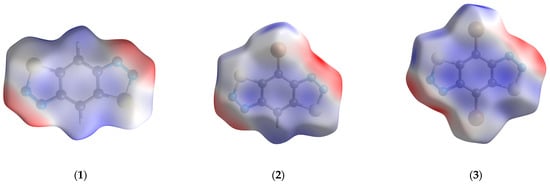
Figure 5.
The distribution of electrostatic potential mapped on molecular Hirshfeld surfaces of 1–3. The positive values are blue, while negative ones are red. The ranges of mapped functions are −0.065–0.087, −0.061–0.087, and −0.061–0.069 a.u. for 1–3, respectively.
To compare the electron-deficiency of compounds 1–3 and BBT, the EA values were calculated at the MP2 and bt-STEOM-CCSD levels (for details, see Section 3.3. Calculations Details) (Table 1, Table S3). Although EA values calculated at the bt-STEOM-CCSD level should be more accurate, the EA MP2 values for 1–3 differ by no more than 0.1 eV. The difference between the adiabatic and vertical EA values is not large (up to ~0.15 eV), which indicates a weak structural rearrangement upon electron attachment. The adiabatic EA value of isoBBT (1) is 1.09 eV, which is significantly lower than that of BBT (1.90 eV). Thus, the desymmetrization of BBT leads to a strong decrease in its electron deficiency. In series 1–3, the Br substituent exhibits electron acceptor properties and significantly increases the EA values of each Br substituent by ~0.2 eV. The HOMO–LUMO gap (Egap) variation in series 1–3 and BBT is opposite to the EA variation, which is not surprising for aromatic polycyclic molecules.

Table 1.
The electronic characteristics (eV) for 1–3 and BBT.
The UV-Vis spectra of 1–3 (Figure 6) show the longest wavelength band with an ill-defined vibrational resolution in the region of 375–425 nm, which corresponds to the π-π* HOMO–LUMO transition (the orbital shapes are given in Table S1 in ESI). This band, which corresponds to Egap, gradually red-shifts from 1 to 2 and 3 and increases in intensity, indicating a conjugation of the Br substituent with the π-orbital system [27].
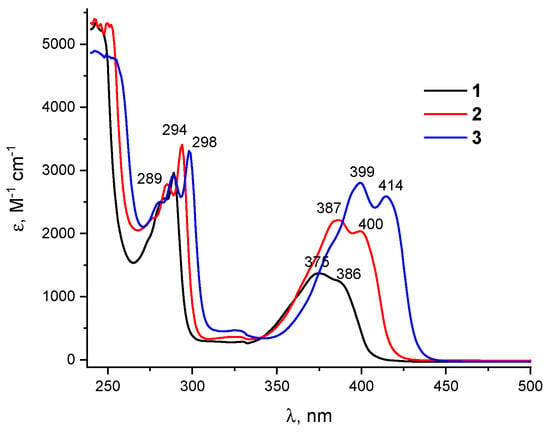
Figure 6.
UV-vis spectra for 1–3 in DMF solution.
Two modern criteria, EDDB [21,22] and GIMIC [23,24], at the MP2 level of theory have been applied to estimate the conjugation and aromaticity in molecules 1–3 (Table 2, Figure S1). The π-conjugation system of BBT and isoBBT consists of 14 ē on 7 orbitals (see Table S1 in ESI), which obey the Hückel rule. The BBT and isoBBT molecules are π-aromatic, as confirmed by EDDB and GIMIC results. The IC distributions in 1–3 (Figure 7) are typical of aromatic rings, but the IC density is distributed unevenly. The current strength (IRCS value) for the six-membered ring (~15 nA/T) is greater than that for the five-membered ring (~11 nA/T) despite the different ring size and the presence of three types of ring currents: local (for each ring), semilocal (over two adjacent rings), and global (over three rings) [28]. The difference in IRCS values indicates a significantly greater degree of delocalization in the six-membered formally benzene ring compared to the heterocyclic five-membered ring. This also leads to the manifestation of a local diamagnetic current of the six-membered ring, which is well distinguishable in the ring annulation region (Figure 7), which is usually non-observable.
Table 2.
The GIMIC and EDDB results at the MP2/cc-pVTZ level for aromaticity evaluation in 1–3 and related molecules.
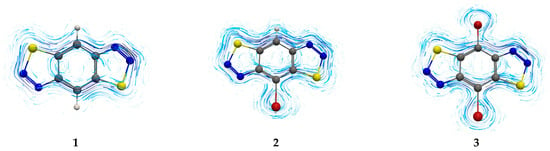
Figure 7.
IC streamlines located above 0.5 Å on the molecular plane. The presented IC streamlines are clockwise, which, in the used coordinate system, corresponds to diatropic currents.
Numerical analysis of the degree of delocalization by the EDDB method revealed that the total number of effective delocalized π-electrons (π-EDDBH value in Table 2) increased slightly from 1 to 2 and 3, which agrees with the UV-vis spectra. The low sensitivity of the EDDB method compared to optical spectra is worthy of note [28]. Analysis of the local conjugation effects by comparing the EDDB values for the C6 and C6Br fragments (the π-EDDBF values in Table 2) showed that the participation of a lone pair of the Br atoms in the conjugation was 0.39–0.48 ē, which corresponds to ~8% of the delocalization degree in the central ring. A comparison of the π-EDDBF values for the five- and six-membered rings indicates that the delocalization in the thiadiazole ring is smaller by 6–8%, which qualitatively agrees with the GIMIC data. Upon Br substitution, the small decrease of aromaticity degree in the six-membered ring (π-EDDBF in the C6 ring changes by 0.06 ē) is compensated by a small aromaticity increase in thiadiazole rings (π-EDDBF in the C2N2S ring changes by 0.1 ē). Thus, the overall delocalization increase from 1 to 2 and 3 is caused by the participation of lone pairs of Br in conjugation, while the change in π-aromaticity is small.
It is worthy of note that the global aromaticity degree of 1–3 is significantly weaker than that of BBT, as evidenced by both π-EDDBH and IRCS values (Table 2). Moreover, the delocalization in BBT is stronger in five-membered rings in contrast to isoBBT. The same trend is observed for isoBTD and BTD (see Table S2 in ESI). This difference in electron delocalization is caused by the lower degree of aromaticity of the 1,2,3-thiadazole ring compared to that of the 1,2,5-thiadazole ring being fused with the benzene ring. Apparently, the significantly lower EA value for isoBBT as compared to BBT is due to the difference in delocalization.
2.2. Aromatic Nucleophilic Substitution of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2
The main goal of this stage of the work was to develop the optimal conditions for obtaining the products of nucleophilic aromatic substitution reactions (SNAr) in 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 using various aromatic and aliphatic O-, S-, and N-nucleophiles and to compare these conditions with the reactions of dibromo derivative 3 [18].
We have studied the reactions of replacement of the bromine atom in the benzene ring of 2 for amino groups in order to obtain substitution products. It was shown that the reaction of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with two equivalents of morpholine in DCM at room temperature for 12 h gave monoamine derivative 4a in trace amounts (Table 3, entry 1). To increase the yield of compound 4a, we studied various conditions for this chemical reaction. It was found that the nature of the solvent significantly affected the yield of the final product by changing the rate of the reaction. Using TLC analysis, we showed that morpholine nearly did not react with monobromo derivative 2 in MeCN at room temperature within 12 h (Table 3, entry 2), compared to DMF (Table 3, entry 3), and gave mono-substitution product 4a in 6 and 15% yields, respectively. Refluxing the reaction mixture in MeCN for 24 h with two equivalents of morpholine gave mono-substitution product 4a in 85% yield. Through heating in DMF at 80 °C, a complete conversion of the initial dibromide was observed within 12 h with the formation of mono-substitution product 4a in 65% yield (Table 3, entry 5). Thus, the optimal conditions for the synthesis of unsymmetrical compound 4a that was involved the treatment of bromo derivative 2 with two equivalents of morpholine in refluxing MeCN (Table 3, entry 4) was extended to other primary and secondary amines. It was found that piperidine 5b and pyrrolidine 5c reacted with bromide 2 to form substitution products 4 in high yields (Table 3, entries 6–7). It should be noted that attempts to perform the reaction with cyclopentaindole 5d failed due to the decomposition of monobromide 2 into a mixture of unidentifiable compounds (Table 3, entry 8). Bromide 2 reacted with primary amines, for example, with aniline 5e, on heating at 130 °C in DMF to form substitution product 4e in moderate yield (Table 3, entry 9). However, with aliphatic primary amines, such as cyclohexylamine 5f and tert-butylamine 5g, the reaction resulted in partial decomposition of the starting bromide 2, even on heating to 80 °C in DMF (Table 3, entries 10–13).
Table 3.
The nucleophilic substitution of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with aromatic and aliphatic amines.
Thus, we have shown that nucleophilic substitution reactions of monobromide 2 occurred more slowly than those of dibromide 3. For example, complete conversion of dibromide 3 by treatment with morpholine occurred within 18 h [18], while monobromide 2 reacted in 24 h; the same trend was observed for piperidine and pyrrolidine. However, in the case of aniline, it was necessary to heat the reaction mixture in DMF to high temperatures. Monobromide 2 reacted only when the reaction mixture was heated to 130 °C for 24 h to give a monosubstitution product in 40% yield, while dibromide 3 reacted at 100 °C in 18 h to give a mono-substitution product with a higher yield of 50%. It should be noted that upon incorporation of an amine into the molecule of dibromide 3, the rate of the substitution reaction of the second bromine atom sharply decreased, which required the use of more drastic conditions, namely, prolonged heating in DMF at 130 °C [18]. Based on this, we can conclude that the reactivity of monobromide 2 is between those of dibromide 3 and mono-substituted amino derivatives. In addition, monobromide 2 did not react with cyclopentaindoline 5d since complete decomposition of the starting bromide 2 into a mixture of unidentifiable compounds was observed, while dibromide 3 reacted with it to give only the mono-substitution product [18].
A study of the reactions of monobromide 2 with such S-nucleophiles as thiophenol, hexynethiol, and dodecanethiol showed that they occurred similarly to the reactions of these nucleophiles with dibromide 3 [18] in the presence of sodium hydride in tetrahydrofuran at room temperature to give monomercapto derivatives 6 in high yields (Scheme 1).
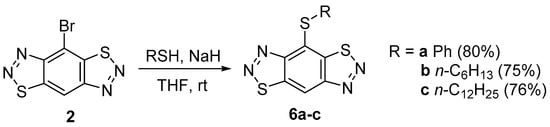
Scheme 1.
Reaction of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with S-nucleophiles.
4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2, like 4,8-dibromo derivative 3, was found to be resistant to various O-nucleophiles, such as water, methanol, ethanol, phenol, and the corresponding sodium alcoholates. We have shown that monobromide 2 did not react with water upon heating in either THF or DMSO at 80 °C. It was shown that in all cases 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 did not react with them, either in THF or in DMF. Heating of the reaction mixtures did not result in the nucleophilic substitution of bromine atoms but, rather, resulted only in partial decomposition of the starting compound 2.
2.3. Cross-Coupling Reactions of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2
We studied the Suzuki–Miyaura reaction of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with various aromatic and heteroaromatic pinacolate esters of boronic acids 7. The selection of conditions for the Suzuki reaction was based on the example of the reaction with thiophene pinacolate ester 7a. We have shown that the nature of the reagents, solvents, and the temperature of the reaction medium significantly affect the course of the reactions. The tetrakis(triphenylphosphine)–palladium complex (Pd(PPh3)4) was used, as it is the most widely used catalyst in these reactions, and potassium carbonate was used as the base. It was shown that when the reaction was carried out at 110 °C in toluene for 24 h, mono-coupling product 8a was isolated in 60% yield. (Table 4, entry 1). At the same time, the addition of water increased the yield of product 8a to 70%, which is apparently due to the solubility of the base (K2CO3) in water (Table 4, entry 2). Replacement of toluene with dioxane or xylene did not increase the yield of the cross-coupling product 8a (Table 4, entries 3–4). Thus, the highest yield of the mono-coupling product was achieved in the toluene–water medium. These conditions were extended to other organoboron esters 7; the yields of mono-coupling products 8 (Table 4, entries 5–11) varied from 64% to 70%.
Table 4.
Suzuki–Miyaura cross-coupling of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with pinacolate esters 7.
Thus, it was shown that the Suzuki reaction of 4-bromobenzo[1,2d:4,5d′]bis([1,2,3]thiadiazole) 2 and boronic esters 7 gave the best results with a toluene/water mixture due to the fact that monobromo derivative 2 is much more hydrolytically stable than dibromo derivative 3, while water promotes the cross-coupling reaction by dissolving the inorganic base. A comparison of the Suzuki cross-coupling times of dibromide 3 [18] and monobromide 2 showed that the reactions for dibromide 3 occurred better and slightly faster under anhydrous conditions than in the presence of small amounts of water. In contrast, owing to the high hydrolytic stability of monobromide 2, the Suzuki reactions involving the latter occurred under aqueous conditions rather than under anhydrous conditions. In addition, to replace both bromine atoms in the molecule of dibromide 3, more drastic conditions were required, i.e., heating the reaction mixture in xylene at 130 °C, which, in turn, was also due to a decrease in the reactivity of monobromo derivatives upon incorporation of thienyl or phenyl substituents.
The Stille reaction of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 was studied with various aromatic and heteroaromatic stannyl derivatives 9a–h. The optimal conditions for the reactions were found using the example of a reaction with thienyltributyl stannane 9a in the presence of PdCl2(PPh3)2, a catalyst that is widely used in these reactions. The reaction with reflux in toluene in the presence of 1.2 equivalents of stannane 9a gave monoaryl derivative 8a in 75% yield (Table 5, entry 1). On replacement of toluene with THF or dioxane, a decrease in the yield of the target product 8a was observed (Table 5, entries 2,3). The best conditions were applied to other aryl (hetaryl) stannanes 9. As a result, we obtained a number of mono-coupling products 8a–h in good yields (Table 5, entries 4–10).
Table 5.
Stille cross-coupling of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with aryl and heteroaryl tributylstannanes 9.
It was previously found that in the Stille reaction of dibromide 3 and stannyl derivatives 9 under mild conditions (heating in toluene at 60 °C), only one bromine atom was replaced, whereas to replace the bromine atom in the molecule of monobromide 2, the reaction mixture had to be heated in toluene at 110 °C. Similar conditions were also required for incorporation of both thienyl and phenyl substituents into the molecule of dibromide 3. It should be noted that the yields of the Stille reaction products were similar for both monobromide 2 and dibromide 3 and varied from 50% to 73%.
2.4. Palladium-Catalyzed C-H (Het)arilation Reactions of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2
Direct C–H arylation reactions are a modern, environmentally attractive method for building a C–C bond with two aromatic and/or heteroaromatic compounds, which allows the number of steps in this process to be reduced, avoiding the use of toxic (e.g., organotin) and flammable (e.g., butyl lithium) derivatives that are used, for example, in the Stille or Suzuki reactions [29,30,31]. For BTD heterocyclic systems, three methods are known for the synthesis of 4,7-disubstituted BTD: (1) the reaction of 4,7-dibromobenzo[c][1,2,5]thiadiazoles with arenes and heteroarenes; (2) the reaction of 4,7-unsubstituted benzo[c][1,2,5]thiadiazoles with halogeno (bromo- or iodo-) arenes and heteroarenes; and (3) oxidative direct arylation of 4,7-unsubstituted benzo[c][1,2,5]thiadiazoles with arenes and heteroarenes. All of the above reactions are catalyzed by palladium compounds. The application of the direct C–H arylation method for 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 could selectively produce monoaryl derivatives and further a wide range of unsymmetrical 4,7-disubstituted isoBBT derivatives (especially push–pull compounds), which are in great demand as components of various optoelectronic devices [9,32]. The synthesis of monoaryl derivatives from dibromoBTD using the Suzuki and Stille reactions is often difficult due to the formation of hard-to-separate mixtures of the starting compound with mono- and bis-arylation products [33]. Only a few examples of direct C–H het-arylation reactions have been described for 4-bromobenzo[c][1,2,5]thiadiazole 10 by method (1) with thiophene [34] and furan derivatives (Scheme 2) [35].
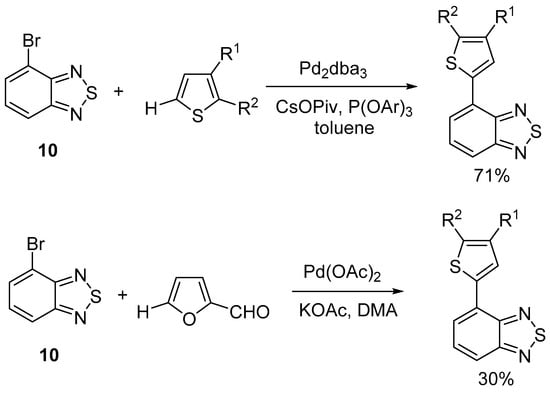
Scheme 2.
Direct C–H (het-)arylation of 4-bromobenzo[c][1,2,5]thiadiazole 10.
Only one example of direct C–H hetarylation of tricyclic benzo-bis-thiadiazoles is described in the literature: the reaction of benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1 with 2-bromothiophene 11 in the presence of palladium(II) acetate, potassium pivalate, and di-tert-butyl(methyl) salt of phosphonium tetrafluoroborate (PBut2Me HBF4) in toluene at 120 °C to give 4,8-bis(5-(triisopropylsilyl)thiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 12 in a low yield (Scheme 3) [36].
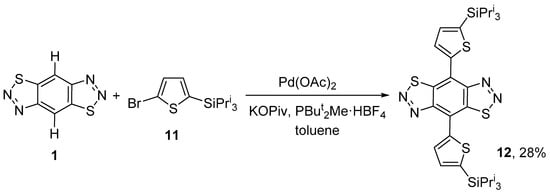
Scheme 3.
Direct C–H hetarylation of benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 1.
Considering the small number of published articles in the field of direct C–H arylation of heterocyclic systems based on BTD, we decided to use all the three methods for the isoBBT systems, namely, to study the replacement of bromine and hydrogen atoms in monobromo BBT 2 and the oxidative C–H arylation of 2 with the purpose of synthesizing mono-substituted BBT derivatives as starting compounds to obtain asymmetric diaryl derivatives of the BBT heterocyclic system.
2.4.1. Palladium-Catalyzed C–H Activation Reactions of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with Haloaromatic and Heteroaromatic Compounds (Replacement of a Hydrogen Atom in 2)
We studied the feasibility of the addition of aromatic and heteroaromatic halogen derivatives to 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 under the conditions of the C–H activation reaction to selectively obtain mono-coupling products 14. The development of the optimal reaction conditions was performed using the reaction with 2-bromo-5-(2-ethylhexyl)thiophene 13d in the presence of various palladium catalysts and organic ligands as an example. It was shown that the nature of the palladium catalyst, ligand, solvent, and temperature of the reaction significantly affected the results of the reactions (Table 6). Refluxing in toluene in the presence of palladium acetate (Pd(OAc)2) and potassium pivalate (KOPiv) resulted in partial decomposition of the starting tricycle 2 without formation of the target product 14d (Table 6, entry 1). Incorporation of ligands, such as tri-tert-butylphosphine (But3P) or bis(diphenylphosphino)ferrocene (dppf), also did not trigger the cross-coupling reaction (Table 6, entries 3,4). In contrast, incorporation of XPhos led to the formation of a mono-coupling product 14d in 7% yield (Table 6, entry 2). The addition of catalysts such as tetrakis(triphenylphosphine)palladium (Pd(PPh3)4), tris(dibenzylideneacetone)dipalladium (Pd2(dba)3), and bis(triphenylphosphine)palladium chloride (PdCl2(PPh3)2) also did not favor the reactions (Table 6, entries 5, 6, and 8). The use of a catalytic system based on (Pd(OAc)2) and PBut2Me·HBF4 made it possible to increase the yield of target C–H activation product 14d considerably. In fact, the reaction in refluxing toluene gave mono-coupling product 14d in a good yield of 65% (Table 6, entry 10). Replacement of toluene with xylene at 140 °C did not increase the yield of compound 14d (Table 6, entry 11). We have shown that the C–H activation reaction did not affect the carbon atom bound to the bromine atom, which, in turn, makes it possible to obtain monobromo derivatives in moderate yields. The optimal conditions for the cross-coupling reaction developed by us were extended to aromatic and heteroaromatic derivatives 14b–h. While the C–H activation reactions with bromothiophene compounds 13a–d occurred selectively and in moderate yields (Table 6, entries 13–16), the reactions with aryl bromides occurred with much greater difficulty. The replacement of aryl bromides with aryl iodides increased the yields of mono-coupling products 14e h significantly: the use of the Pd(OAc)2 and PBut2Me·HBF4 catalytic system in refluxing toluene gave the target products 14 in moderate yields (Table 6, entries 13, 18–22).
Table 6.
Palladium-catalyzed C–H direct arylation reactions of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with aryl and thienyl halogenides.
2.4.2. Palladium-Catalyzed C-H Activation Reactions of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with Aromatic and Heteroaromatic Compounds (Replacement of Bromine Atom in 2)
We began to study the reaction of tricycle 2 with thiophene using the conditions developed for 4-bromobenzo[c][1,2,5]thiadiazole 10 (see Scheme 2). First of all, we found that Pd2dba3 did not catalyze the reaction of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with (2-ethylhexyl)thiophene 15d in the presence of bases, such as cesium and potassium pivalates, and various phosphine ligands in toluene; the starting heterocycle was isolated from the reaction mixtures in high yields. Therefore, the main attention was further paid to the catalysis of this reaction with palladium acetate (Table 7). It was shown that the use of a catalytic system of palladium acetate (Pd(OAc)2) and potassium pivalate in the reaction of tricycle 2 and 15d gave product 8d in a moderate yield (Table 7, entry 1). The incorporation of ligands such as tri-tert-butylphosphine (But3P), bis(diphenylphosphino)ferrocene (dppf), and XPhos did not trigger the cross-coupling reaction (Table 7, entries 2–4). The use of a catalytic system based on Pd(OAc)2 and PBut2Me·HBF4 in toluene also did not result in the formation of target product 8d (Table 7, entry 5). Nevertheless, an increase in the temperature of the reaction mixture to 130 °C led to the formation of product 8d in a moderate yield (Table 7, entry 6). Unexpectedly, the reaction performed in DMA (see ref. [36]) resulted only in the decomposition of the starting dibromide 2 (Table 7, entry 7). The reaction conditions that we developed were extended to other thiophene derivatives 8a–c (Table 7, entries 9–11). Attempts to carry out the C–H arylation reaction with aromatic compounds, such as toluene or xylene using various catalytic systems, failed: the starting tricycle 2 was isolated in high yields.
Table 7.
Palladium-catalyzed C-H activation reactions of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with 2-unsubstituted thiophenes.
2.4.3. Palladium-Catalyzed Oxidative C-H Thienylation Reactions of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with Thiophenes
We studied the reactions of oxidative C–H (het)arylation of monobromide 2 with (2-ethylhexyl)thiophene 15d using silver oxide (Ag2O) as the oxidizing agent in anhydrous DMSO under the conditions reported for 2,1,3-benzothiadiazole [37,38]. It was shown that when palladium trifluoroacetate was used as the catalyst, the C–H activation reaction did not occur (Table 8, entry 1); after stirring for 24 h the starting compound 2 was isolated in high yield. Replacing palladium trifluoroacetate with palladium acetate at 110 °C resulted in successful activation of the reaction that produced bromoaryl derivative 14d in 68% yield (Table 8, entry 2). The use of such silver salts, such as silver acetate (AgOAc), silver nitrate (AgNO3), silver tetrafluoroborate (AgBF4), silver perchlorate (AgClO4), and Ag2O in anhydrous DMSO gave no results (Table 8, entries 4–6). An increase in the temperature of the reaction mixture to 120 °C did not increase the yield of target product 14d (Table 8, entry 3). The conditions we found were extended to other thiophene derivatives 15a,b,c to obtain the corresponding thienylated products 14 in low to moderate yields (Table 8, entries 7–9).
Table 8.
Palladium-catalyzed C-H oxidative thienylation reactions of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2.
Thus, depending on the chosen catalytic system, it is possible to perform the selective syntheses of mono-derivatives 8 and 14, both with a C–H component and with a bromine atom for further transformations.
3. Materials and Methods
3.1. Materials and Reagents
The chemicals were purchased from the commercial sources (Sigma-Aldrich, St. Louis, MO, USA) and used as received. 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 [39], thiophene pinacolate esters 7a–d [40], tributyl-2-thienylstannanes 9a–d [41], and tributylarylstannanes 9e–h [42] were prepared according to the published methods and characterized by NMR spectra. All synthetic operations were performed under a dry argon atmosphere. The solvents were purified by distillation over the appropriate drying agents.
3.2. Analytical Instruments
The melting points were determined on a Kofler hot-stage apparatus and were uncorrected. 1H and 13C NMR spectra were recorded on a Bruker AM-300 instrument (Bruker Ltd., Moscow, Russia) with TMS as the standard. J values are given in Hz. MS spectra (EI, 70 eV) were obtained with a Finnigan MAT INCOS 50 instrument (Thermo Finnigan LLC, San Jose, CA, USA). High-resolution MS spectra were measured on a Bruker micrOTOF II instrument using electrospray ionization (ESI). IR spectra were measured with a Bruker “Alpha-T” instrument (Bruker, Billerica, MA, USA) in KBr pellets. UV-vis spectra in the region 200–900 nm were registered for DMF solutions of 1–3 C= 10–4 M in the standard 10 mm quartz cell using a Carl Zeiss Specord M400 spectrophotometer.
3.3. Calculations Details
Geometry optimization, calculation of NAO and DMNAO matrices for EDDB analysis [21,22], calculation of magnetic shielding matrices for GIMIC analysis [23,24] at MP2(fc) theory level with the cc-pVTZ basis set were performed in the Gaussian program [43]. Additionally, an unrestricted MP2-level geometry optimization of anion radicals was performed to calculate adiabatic electron affinity (EA) values. The vertical and adiabatic EA values were also calculated at the bt-STEOM-CCSD level using TightPNO and RIJCOSX approximations using the ORCA [44] program for molecules with optimized geometry at the MP2(fc) level. The ZPVE correction for EA values is calculated at the TPSS-D4/cc-pVTZ level using the ORCA program. The positive EA values given in Table 1 correspond to the profitability of electron attachment to 1–5. The RunEDDB [21] script was used for EDDB analysis, the π-EDDBH value corresponds to a whole molecule, and π-EDDBF is of a selected fragment. The calculation of ring currents (IC) by the GIMIC method was performed using the GIMIC 2.0 program [23,45]. The ring current strength (IRCS) values were calculated by integrating with respect to the N–S and C–C bonds for 5- and 6-membered cycles by Scheme S1 in ESI, according to the recommendations [45]. The orbitals were visualized using the Avogadro program [46], the π-EDDB isosurfaces and IC distribution maps were constructed using the ParaView program [47]. The IC located below 0.5 Å on the molecular plane (Figure 2) are omitted for clarity of diatropic ICs from the π-cloud contribution.
3.4. X-ray Crystallography
X-ray diffraction data for 1 were collected with Bruker Quest diffractometer while the data for 2 and 3 were collected at 100 K on a four-circle Rigaku Synergy S diffractometer using graphite Mo Kα-radiation. The intensity data were integrated and corrected for absorption and decay by APEX 3 (Bruker QUEST) and CrysAlisPro software (Austin, TX, USA, accessed on 1 September 2022) [48]. The structures were solved by dual-space algorithm using SHELXT and refined on F2 using SHELXL-2018 [49] in anisotropic approximation [50] for non-hydrogen atoms. All hydrogen atoms were placed in ideal calculated positions and refined as riding atoms with relative isotropic displacement parameters. A rotating group model was applied for methyl groups. The structure 1 was refined as two component non-merohedral twin with TWINABS program implemented in APEX3 software. The scale factors for twin components are equal to 0.6673(15) and 0.3327(15). The twinning for 3 was established with Olex2 software, the scale factors for components are 0.521(3) and 0.479(3). The Cambridge Crystallographic Data Centre contains all crystallographic data for this paper (deposition numbers: 2255420, 2256390, and 2210625). These data can be obtained free of charge via http://www.ccdc.cam.ac.uk/conts/retrieving.html (accessed on 10 April 2023) (or from the CCDC, 12 Union Road, Cambridge CB2 1EZ, UK; Fax: +44-1223-336033; E-mail: deposit@ccdc.cam.ac.uk). Detailed information related to the X-ray diffraction studies of 1–3 is summarized in Table S4.
3.5. Synthesis of Compounds
3.5.1. General Procedure for the Preparation of Aminated Products 4a–c
Amine 5a–c (0.36 mmol) was added to a solution of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol) in dry MeCN (10 mL) at room temperature, and the mixture was stirred at reflux for 24 h, poured into water (20 mL), and extracted with CH2Cl2 (3 × 35 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel Merck 60).
4-(benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole)-4-yl)morpholine (4a)
Orange solid, 39 mg (85%), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.2 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 195–197 °C. IR νmax (KBr, cm–1): 2961, 2918, 1851, 1563, 1537, 1441, 1388, 1339, 1290, 1262, 1243, 1108, 1067, 1025, 980, 925, 848,835, 809, 680, 611, 514. 1H NMR (300 MHz, CDCl3): δ 8.72 (s, 1H), 4.06–3.96 (m, 8H). 13C NMR (100 MHz, CDCl3): δ 160.2, 150.3, 142.3, 139.6, 129.2, 104.4, 67.4, 52.2. HRMS (ESI-TOF), m/z: calcd for C10H9N5OS2 [M]+, 279.0243; found, 279.0240. MS (EI, 70 eV), m/z (I, %): 279 ([M]+, 6), 251 (20), 165 (55), 93 (4), 69 (80), 28 (100).
4-(Piperidin-1-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (4b)
Orange solid, 39 mg (80%), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.5 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 155–157 °C. IR νmax (KBr, cm–1): 2928, 2847, 2809, 1565, 1534, 1443, 1387, 1341, 1288, 1243, 1222, 1123, 1086, 975, 844, 810, 678, 609, 625, 609, 543. 1H NMR (300 MHz, CDCl3): δ 8.59 (s, 1H), 3.98–3.88 (m, 4H), 1.96–1.98 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 160.2, 149.8, 142.4, 140.7, 129.0, 102.6, 53.4, 26.8, 24.4. HRMS (ESI-TOF), m/z: calcd for C11H11N5S2 [M]+, 277.0450; found, 277.0444. MS (EI, 70 eV), m/z (I, %): 277 ([M]+, 45), 249 (90), 220 (6), 192 (46), 165 (30), 96 (40), 69 (100), 41 (95), 27 (80).
4-(Pyrrolidin-1-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (4c)
Orange solid, 37 mg (79%), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.4 (CH2Cl2/Hexane, 1:1, (ν/ν)). Mp = 196 –198 °C. IR νmax (KBr, cm–1): 2969, 2950, 2875, 2856, 1635, 1555, 1538, 1450, 1388, 1337, 1315, 1278, 1194, 1037, 982, 844, 807, 669, 640, 528. 1H NMR (300 MHz, CDCl3): δ 8.19 (s, 1H), 4.39–4.28 (m, 4H), 2.23–2.14 (m, 4H). 13C NMR (100 MHz, CDCl3): δ 160.5, 145.4, 143.3, 138.8, 124.8, 97.5, 53.6, 26.1. HRMS (ESI-TOF), m/z: calcd for C10H9N5S2 [M]+, 263.0294; found, 263.0295. MS (EI, 70 eV), m/z (I, %): 263 ([M]+, 8), 235 (15), 206 (5), 192 (3), 178 (6), 96 (13), 69 (100), 41 (50), 27 (55), 18 (10).
N-Phenylbenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole)-4-amine (4e)
Aniline 5e (33 mg, 0.36 mmol) was added to a solution of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol) in dry DMF (10 mL), and the mixture was stirred at 130 °C for 24 h, poured into water, and extracted with CH2Cl2 (3 × 35 mL). The combined organic layers were washed with water, brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (silica gel Merck 60). Red solid, yield 20 mg (40%), eluent–CH2Cl2/hexane, 1:1 (v/v)). Rf = 0.4 (CH2Cl2/hexane, 1:1 (v/v)). Mp = 157–160 °C. IR νmax (KBr, cm–1): 3344, 1581, 1546, 1495, 1449, 1429, 1385, 1343, 1298, 1255, 1072, 854, 816, 764, 702, 677, 538. 1H NMR (300 MHz, CDCl3): δ 8.59 (s, 1H), 8.33 (br. s, 1H), 7.56–7.45 (m, 3H), 7.31 (d, J = 7.1, 2H). 13C NMR (100 MHz, CDCl3): δ 161.3, 146.7, 141.3, 137.2, 134.7, 129.8, 127.8, 126.9, 121.4, 101.7. HRMS (ESI-TOF), m/z: calcd for C12H8N5S2 [M + H]+, 286.0216; found, 286.0221. MS (EI, 70 eV), m/z (I, %): 285 ([M]+, 4), 257 (55), 228 (4), 196 (5), 185 (30), 160 (12), 153 (13), 125 (10), 93 (14), 77 (60), 69 (100), 51 (58).
3.5.2. General Procedure for the Reaction of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 with Thiols
Sodium hydride (7.2 mg, 0.18 mmol) was added to a solution of thiol (0.18 mmol) in dry THF (15 mL) at 0 °C with stirring. The reaction mixture was stirred at 0 °C for 30 min, then 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol) was added. The mixture was stirred for 6 h at room temperature. On completion (monitored by TLC), the mixture was poured into water (20 mL) and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography.
4-(Phenylthio)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (6a)
Yellow solid, 43 mg (80%), Mp = 140–142 °C, eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.5 (CH2Cl2/hexane, 1:1 (v/v)). IR νmax (KBr, cm–1): 1638, 1578, 1475, 1438, 1388, 1330, 1286, 1196, 1075, 1019, 901, 866, 832, 800, 734, 686, 660, 541, 515, 470. 1H NMR (300 MHz, CDCl3): δ 9.11 (s, 1H), 7.65 (d, J = 7.4, 2H), 7.60–7.54 (m, 1H), 7.46 (t, J = 7.4, 2H). 13C NMR (75 MHz, CDCl3): δ 158.6, 155.5, 140.2, 139.0, 135.7, 130.6, 129.9, 129.2, 127.4, 111.3. HRMS (ESI-TOF), m/z: calcd for C12H6N4S3Ag [M + Ag]+, 408.8800; found, 408.8790. MS (EI, 70 eV), m/z (I, %): 304 ([M + 2]+, 2), 303 ([M + 1]+, 3), 302 ([M + 1]+, 35), 274 (70), 245 (8), 214 (14), 201 (100), 177 (90), 170 (65), 158 (85), 145 (50), 133 (53), 124 (80), 109 (90), 100 (92), 93(95), 76 (94), 70 (95), 65 (94), 52 (94), 45 (96), 39 (95), 27 (55).
4-(Hexylthio)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (6b)
Yellow solid, 41 mg (75%), eluent–CH2Cl2/hexane, 1:4 (v/v). Rf = 0.7 (CH2Cl2/hexane, 1:1 (v/v)). Mp = 112–115 °C. IR νmax (KBr, cm–1): 2956, 2924, 2853, 1615, 1510, 1456, 1391, 1334, 1285, 1196, 1104, 901, 868, 798, 752, 721, 658, 543. 1H NMR (300 MHz, CDCl3): δ 9.12 (s, 1H), 3.68 (t, J = 7.3, 2H), 1.67 (p, J = 7.3, 2H), 1.49–1.40 (m, 2H), 1.27–1.21 (m, 4H), 0.84 (t, J = 6.7, 3H). 13C NMR (75 MHz, CDCl3): δ 157.1, 156.4, 140.8, 126.5, 125.8, 111.5, 36.8, 31.2, 30.0, 28.2, 22.4, 13.9. HRMS (ESI-TOF), m/z: calcd for C12H14N4S3Ag [M + Ag]+, 416.9426; found, 416.9425. MS (EI, 70 eV), m/z (I, %): 311 ([M + 1]+, 10), 310 ([M]+, 66), 311 ([M − 1]+, 15), 281 (60), 225 (55), 197 (35), 183 (65), 125 (36), 93 (58), 69 (100), 41 (80), 29 (79).
4-(Dodecylthio)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (6c)
Green solid, 53 mg (76%), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.8 (CH2Cl2/hexane, 1:1 (v/v). Mp = 31–33 °C. IR νmax (KBr, cm–1): 2954, 2920, 2849, 1653, 1469, 1390, 1287, 1195, 903, 871, 833, 798, 718, 656, 542. 1H NMR (300 MHz, CDCl3): δ 9.11 (s, 1H), 3.67 (t, J = 7.3, 2H), 1.67 (p, J = 7.3, 2H), 1.48–1.36 (m, 2H), 1.29–1.16 (m, 16H), 0.86 (t, J = 6.6, 3H). 13C NMR (75 MHz, CDCl3): δ 157.1, 156.4, 143.0, 140.8, 125.8, 111.4, 36.8, 31.9, 30.0, 29.6, 29.59, 29.55, 29.5, 29.4, 29.0, 28.5, 22.7, 14.1. HRMS (ESI-TOF), m/z: calcd for C18H26N4S3Na [M + Na]+, 417.1212; found, 417.1212. MS (EI, 70 eV), m/z (I, %): 396 ([M + 2]+, 5), 395 ([M + 1]+, 10), 394 ([M]+, 46), 355 (100), 341 (70), 295 (44), 281 (90), 221 (85), 207 (49), 69 (50), 55 (40).
3.5.3. General Procedure for the Preparation of Arylated Benzo-bis-thiadiazoles 8 under Suzuki Coupling Conditions (Procedure A)
A mixture of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol), boronic ether 16a–h (0.18 mmol), K2CO3 (24 mg, 0.18 mmol), 1 mL (H2O), and Pd(PPh3)4 (20 mg, 10% mmol) in dry toluene (8 mL) was degassed by argon and heated at 110 °C in a sealed vial. On completion (monitored by TLC), the mixture was poured into water and extracted with CH2Cl2 (3 × 35 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated in vacuo. The crude product was purified by column chromatography.
3.5.4. General Procedure for the Preparation of Arylated Benzo-bis-thiadiazoles 8 under Stille Coupling Conditions (Procedure B)
PdCl2(PPh3)2 (12 mg, 10% mmol) and stannane 20a–h (0.21 mmol) were added to a solution of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol) in anhydrous toluene (4 mL) The resulting cloudy, yellow mixture was stirred and degassed by argon in a sealed vial. The resulting yellow mixture was then stirred at 60 °C for the desired time. On completion (monitored by TLC), the mixture was washed with water and the organic layer was extracted with CH2Cl2 (3 × 35 mL), dried over MgSO4, and then concentrated in vacuo. The crude product was purified by column chromatography.
3.5.5. General Procedure for the Synthesis of Arylated Benzo-bis-thiadiazoles 8 by Palladium-catalyzed C-H Direct Arylation Reaction of 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (Procedure C)
A mixture of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol), thiophene 3a–e (0.20 mmol), Pd(OAc)2 (6 mg, 15% mmol), pivalic acid (20 mg, 0.20 mmol), and K2CO3 (27 mg, 0.20 mmol) were added to an air-free flask, which was then purged in dry toluene (8 mL), degassed by argon, and heated at 110 °C in a sealed vial. On completion (monitored by TLC), the mixture was poured into water and extracted with CH2Cl2 (3 × 35 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography.
4-(Thiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8a)
Orange solid, 34 mg (70%, Procedure A), 34 mg (70%, Procedure B), or 18 mg (37%, Procedure C), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.3 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 173–150 °C. IR νmax (KBr, cm–1): 1636, 1532, 1437, 1432, 1393, 1328, 1286, 1258, 1142, 858, 812, 715, 666, 544. 1H NMR (300 MHz, CDCl3): δ 9.19 (s, 1H), 8.25 (d, J = 3.7, 1H), 7.75 (d, J = 5.0, 1H), 7.43–7.32 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 158.4, 154.0, 140.9, 138.9, 137.6, 131.6, 130.5, 129.8, 128.6, 112.1. HRMS (ESI-TOF), m/z: calcd for C10H5N4S3 [M + H]+, 276.9671; found, 276.9663. MS (EI, 70 eV), m/z (I, %): 276 ([M]+, 6), 248 (75), 220 (10), 176 (11), 151 (100), 93 (25), 69 (95), 45 (12), 28 (5).
4-(4-Hexylthiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8b)
Yellow solid, 44 mg (69%, Procedure A), 46 mg (71%, Procedure B), or 23 mg (37%, Procedure C), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.4 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 65–68 °C. IR νmax (KBr, cm–1): 2956, 2924, 2853, 1640, 1540, 1513, 1494, 1451, 1398, 13754, 1333, 1287, 1249, 1188, 1081, 967, 854, 815, 775, 725, 661, 615, 522. 1H NMR (300 MHz, CDCl3): δ 9.16 (s, 1H), 8.14 (s, 1H), 7.34 (s, 1H), 3.18–2.61 (m, 2H), 1.79–1.70 (m, 2H), 1.40–1.30 (m, 6H), 0.91 (t, J = 8.0, 3H). 13C NMR (100 MHz, CDCl3): δ 157.2, 152.8, 144.1, 138.9, 136.2, 136.1, 132.2, 124.3, 122.5, 110.6, 30.7, 29.6, 29.5, 28.0, 21.6, 13.1 HRMS (ESI-TOF), m/z: calcd for C16H17N4S3 [M + H]+, 361.0610; found, 361.0606. MS (EI, 70 eV), m/z (I, %): 362 ([M + 2]+, 3), 361 ([M + 1]+, 6), 360 ([M]+, 50), 332 (100), 248 (20), 235 (19), 220 (12), 165 (18), 120 (13), 69 (60), 43 (57), 29 (48).
4-(5′-(Trimethylsilyl)-[2,2′-bithiophen]-5-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8c)
Red solid, 54 mg (70%, Procedure A), 52 mg (68%, Procedure B), or 31 mg (40%, Procedure C), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.3 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 155–157 °C. IR νmax (KBr, cm–1): 2958, 2924, 2853, 1724, 1641, 1494, 1464, 1364, 1279, 1263, 1187, 1081, 968, 892, 818, 725, 486. 1H NMR (300 MHz, CDCl3): δ 9.15 (s, 1H), 8.15 (d, J = 4.0, 1H), 7.44 (d, J = 3.5, 1H), 7.40 (d, J = 4.0, 1H), 7.22 (d, J = 3.5, 1H), 0.37 (s, 9H). 13C NMR (100 MHz, CDCl3): δ 158.6, 153.6, 143.1, 142.2, 141.2, 141.05, 136.8, 135.9, 135.2, 132.6, 126.5, 124.9, 123.1, 111.7, 0.00(TMS). HRMS (ESI-TOF), m/z: calcd for C17H15N4S4Si [M + H]+, 430.9943; found, 430.9928. MS (EI, 70 eV), m/z (I, %): 432 ([M + 2]+, 1), 431 ([M + 1]+, 2), 430 ([M]+, 8), 402 (7), 305 (6), 200 (10), 175 (12), 93 (45), 69 (100), 45 (30).
4-(5-(2-Ethylhexyl)thiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8d)
Orange solid, 47 mg (68%, Procedure A), 48 mg (70%, Procedure B), or 38 mg (55%, Procedure C), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.4 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 55–57 °C. IR νmax (KBr, cm–1): 2958, 2923, 2855, 1618, 1507, 1457, 1389, 1324, 1282, 1262, 1144, 1078, 1032, 881, 861, 847, 812, 786, 739, 618, 547. 1H NMR (300 MHz, CDCl3): δ 9.10 (s, 1H), 8.09 (d, J = 3.7, 1H), 7.01 (d, J = 3.7, 1H), 2.91 (d, J = 6.8, 2H), 1.78–1.68 (m, 1H), 1.48–1.29 (m, 8H), 0.98–0.89 (m, 6H). 13C NMR (100 MHz, CDCl3): δ 158.3, 151.1, 140.8, 136.7, 135.1, 131.8, 127.0, 126.5, 123.6, 111.1, 41.6, 34.4, 32.5, 28.9, 25.7, 23.0, 14.1, 10.9. HRMS (ESI-TOF), m/z: calcd for C18H21N4S3 [M + H]+, 389.0923; found, 389.0921. MS (EI, 70 eV), m/z (I, %): 390 ([M + 2]+, 3), 389 ([M + 1]+, 6), 388 ([M]+, 35), 360 (80), 332 (15), 261 (18), 248 (38), 233 (100), 69 (28), 57 (60), 41 (45), 29 (37).
4-Phenylbenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8e)
Yellow solid, 33 mg (68%, Procedure A), 30 mg (63%, Procedure B), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.3 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 203–205 °C. IR νmax (KBr, cm–1): 1637, 1492, 1431, 1386, 1277, 1148, 1075, 893, 862, 813, 745, 696, 673, 623, 545, 523. 1H NMR (300 MHz, CDCl3): δ 9.28 (s, 1H), 7.99 (d, J = 6.7, 2H), 7.69–7.59 (m, 3H). 13C NMR (100 MHz, CDCl3): δ 157.9, 155.6, 140.8, 140.1, 136.9, 130.3, 129.9, 129.7, 129.3, 112.7. HRMS (ESI-TOF), m/z: calcd for C12H7N4S2 [M + H]+, 271.0107/ found, 271.0109. MS (EI, 70 eV), m/z (I, %): 270 ([M+]+, 3), 242 (58), 214 (26), 170 (23), 145 (90), 93 (20), 69 (100), 28 (40).
4-(p-Tolyl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8f)
Green solid, 32 mg (64%, Procedure A), 34 mg (68%, Procedure B), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.3 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 229–232 °C. IR νmax (KBr, cm–1): 2925, 1639, 1609, 1507, 1427, 1379, 1331, 1317, 1291, 1275, 1192, 1147, 1120, 895, 865, 828, 804, 763, 716, 670, 609, 556, 536, 488. 1H NMR (300 MHz, CDCl3): δ 9.24 (s, 1H), 7.89 (d, J = 7.9, 2H), 7.46 (d, J = 7.9, 2H), 2.51 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 157.8, 155.5, 140.6, 140.4, 139.8, 134.0, 130.0, 129.8, 129.4, 112.1, 21.3. HRMS (ESI-TOF), m/z: calcd for C13H8BrN4S2 [M + H]+, 285.0263; found, 285.0266. MS (EI, 70 eV), m/z (I, %): 284 ([M]+, 3), 256 (8), 227 (5), 159 (25), 139 (5), 93 (7), 69 (100), 63 (7), 51 (10), 39 (30), 28 (45), 18 (70).
4-(4-Methoxyphenyl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (8g)
Orange solid, 37 mg (69%, Procedure A), 37 mg (70%, Procedure B), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.2 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 198–201 °C. IR νmax (KBr, cm–1): 3076, 1609, 1509, 1457, 1430, 1383, 1300, 1279, 1262, 1178, 1150, 1116, 1030, 896, 863, 835, 806, 670, 540. 1H NMR (300 MHz, CDCl3): δ 9.22 (s, 1H), 7.97 (d, J = 8.8, 2H), 7.17 (d, J = 8.8, 2H), 3.95 (s, 3H). 13C NMR (100 MHz, CDCl3): δ 161.2, 158.0, 155.6, 140.8, 139.4, 131.2, 129.9, 129.2, 114.8, 112.0, 55.6. HRMS (ESI-TOF), m/z: calcd for C13H9N4OS2 [M + H]+, 301.0212; found, 301.0215. MS (EI, 70 eV), m/z (I, %): 302 ([M + 2]+, 3), 301 ([M + 1]+, 4), 300 ([M]+, 30), 272(50), 229 (45), 201 (25), 175 (80), 132 (65), 93 (35), 69 (100), 28 (30).
4-(benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole)-4-yl)-N,N-diphenylaniline (8h)
Orange solid, 55 mg (70%, Procedure A), 50 mg (64%, Procedure B), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.25 (CH2Cl2/hexane, 1:1, (ν/ν)). Mp = 213–215 °C. IR νmax (KBr, cm–1): 1727, 1590, 1487, 1428, 1321, 1276, 1195, 1125, 1073, 894, 865, 835, 808, 748, 696, 624, 512. 1H NMR (300 MHz, CDCl3): δ 9.18 (s, 1H), 7.88 (d, J = 8.8, 2H), 7.35 (t, J = 7.8 Hz, 3H), 7.28–7.12 (m, 9H). 13C NMR (100 MHz, CDCl3): δ 158.0, 155.3, 150.0, 146.9, 140.8, 139.3, 130.6, 129.8, 129.6, 129.0, 125.7, 124.3, 121.4, 111.5. HRMS (ESI-TOF), m/z: calcd for C24H15N5S2 [M]+, 437.0763; found, 437.0757. MS (EI, 70 eV), m/z (I, %): 438 ([M + 1]+, 8), 437 ([M]+, 55), 409 (6), 381 (4), 312 (12), 168 (3), 69 (15), 18 (100).
3.5.6. General Procedure for the Preparation of Arylated Benzo-bis-thiadiazoles 14 from 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (Procedure D)
A mixture of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol), bromide or iodide 13a–d,f–j (1.03 mmol), Pd(OAc)2 (17 mg, 0.076 mmol), (P(But)2Me·HBF4) (41 mg, 0.18 mmol), pivalic acid (105 mg, 1.03 mmol), and K2CO3 (142 mg, 1.03 mmol) were added to an air-free flask, which was then purged in dry toluene (8 mL), degassed by argon, and heated at 110 °C in a sealed vial. On completion (monitored by TLC), the mixture was poured into water and extracted with CH2Cl2 (3 × 35 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography.
3.5.7. General Procedure for the Preparation of Arylated Benzo-bis-thiadiazoles 14 under C-H Oxidative Coupling Conditions (Procedure E)
A mixture of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) 2 (50 mg, 0.18 mmol), thiophene (0.36 mmol), Ag2O (83 mg, 0.36 mmol), and Pd(OAc)2 (6 mg, 0.027 mmol), were added to an air-free flask which, was then purged in dry DMSO (5 mL), degassed by argon, and heated at 90 °C in a sealed vial. On completion (monitored by TLC), the mixture was poured into water and extracted with CH2Cl2 (3 × 35 mL). The combined organic layers were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography.
4-Bromo-8-(thiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14a)
Yellow solid, 20 mg (32%, Procedure D), 19 mg (30%, Procedure E), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.4 (CH2Cl2). Mp = 198–200 °C (lit. mp 198–200 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-Bromo-8-(4-hexylthiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14b)
Yellow solid, 31 mg (40%, Procedure D), 30 mg (38%, Procedure E), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.6 (CH2Cl2/hexane, 1:1 (v/v)). Mp = 67–69 °C (lit. mp 67–69 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-([2,20-Bithiophen]-5-yl)-8-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14c)
Red solid, 39 mg (50%, Procedure D), 37 mg (48%, Procedure E), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.4 (CH2Cl2/hexane, 1:1 (v/v)). Mp = 130–132 °C (lit. mp 130–132 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-Bromo-8-(5-(2-ethylhexyl)thiophen-2-yl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14d)
Yellow solid, 54 mg (65%, Procedure D), 57 mg (68%, Procedure E), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.6 (CH2Cl2). Mp = 57–60 °C (lit. mp 57–60 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-Bromo-8-phenylbenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14e)
Yellow solid, 32 mg (52%, procedure D), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.4 (CH2Cl2). Mp = 163–165 °C (lit. mp 163–165 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-Bromo-8-(p-tolyl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14f)
Yellow solid, 44 mg (68%, procedure D), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.5 (CH2Cl2). Mp = 135–137 °C (lit. mp 135–137 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-Bromo-8-(4-methoxyphenyl)benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (14g)
Yellow solid, 41 mg (60%, procedure D), eluent–CH2Cl2/hexane, 1:1 (v/v). Rf = 0.2 (CH2Cl2). Mp = 125–127 °C (lit. mp 125–127 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4-(8-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole)-4-yl)-N,N-diphenylaniline (14h)
Red solid, 53 mg (58%, procedure D), eluent–CH2Cl2/hexane, 1:2 (v/v). Rf = 0.5 (CH2Cl2/hexane, 1:1 (v/v)). Mp = 165–168 °C. (lit. mp 165–168 °C [18]). The data of the 1H and 13C NMR spectra correspond to the literature data [18].
4. Conclusions
The aromaticity of benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (isoBBT) and benzo[1,2-c:4,5-c′]bis[1,2,5]thiadiazole (BBT) heterocycles was confirmed by two modern criteria, EDDB and GIMIC, at the MP2 level of theory and additionally by UV-vis spectroscopy. The aromaticity degree of isoBBT derivatives is significantly weaker than that of BBT as evidenced by both π-EDDBH and IRCS values, and the delocalization in BBT is stronger in five-membered rings in contrast to isoBBT. The incorporation of bromine substituents into the isoBBT molecule enhances the electron-withdrawing properties that may increase its ability participate in nucleophilic aromatic substitution reactions. At the same time, the aromaticity of isoBBT derivatives practically does not change, and their reactivity to cross-coupling reactions is preserved, which is confirmed by the data of 4-bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (4-bromoisoBBT) reactions. The study of aromatic nucleophilic substitution of 4-bromoisoBBT showed that its reactivity is close to that of 4,8-dibromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole; a series of 4-amino and 4-thio-substituted derivatives were obtained in high yields. A number of mono(het-)aryl-substituted isoBBT derivatives have been successfully synthesized from 4-bromoisoBBT. 4-ArylisoBBT derivatives, not available by other methods, were prepared by several variants of palladium-catalyzed cross-coupling reactions, namely, Suzuki, Stille, and direct C–H arylation with 2-unsubstituted thiophenes. Selective methods have been developed for the synthesis of 4-bromo-8-arylisoBBT derivatives by cross-coupling reactions of 4-bromoisoBBT with (het)aryl halides and an oxidative direct C–H arylation with thiophenes.
Supplementary Materials
The supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/ijms24108835/s1. Calculations data, characterization data including 1H and 13C NMR spectra for novel compounds, crystallographic data for compounds 1–3.
Author Contributions
Conceptualization, O.A.R.; methodology, O.A.R. and R.R.A.; software, R.R.A.; validation, O.A.R. and R.R.A.; formal analysis, T.N.C.; investigation, T.N.C., T.A.K., D.A.A. and A.A.K.; resources, O.A.R.; data curation, T.N.C.; writing—original draft preparation, O.A.R. and R.R.A.; writing—review and editing, O.A.R.; visualization, T.N.C.; supervision, O.A.R.; project administration, O.A.R.; funding acquisition, O.A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation (RSF Grant no. 22-23-00252). The X-ray studies and quantum chemical calculations was supported by the Ministry of Science and Higher Education of the Russian Federation (Contract No. 075-03-2023-642) and was performed employing the equipment of Center for Molecular Composition Studies of INEOS RAS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article or Supplementary Material.
Acknowledgments
Crystal structure determination of compounds 2 and 3 was performed in the Department of Structural Studies of N. D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Krygowski, T.M.; Cyrański, M.K. (Eds.) Aromaticity in Heterocyclic Compounds; Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2009; Volume 19, ISBN 978-3-540-68329-2. [Google Scholar]
- Solà, M.; Boldyrev, A.I.; Cyrański, M.K.; Krygowski, T.M.; Merino, G. Aromaticity and Antiaromaticity; Wiley: Hoboken, NJ, USA, 2022; ISBN 9781119085898. [Google Scholar]
- von Ragué Schleyer, P. Introduction: Aromaticity. Chem. Rev. 2001, 101, 1115–1118. [Google Scholar] [CrossRef]
- von Ragué Schleyer, P. Introduction: Delocalization–Pi and Sigma. Chem. Rev. 2005, 105, 3433–3435. [Google Scholar] [CrossRef]
- Martín, N.; Scott, L.T. Challenges in aromaticity: 150 years after Kekulé’s benzene. Chem. Soc. Rev. 2015, 44, 6397–6400. [Google Scholar] [CrossRef] [PubMed]
- Rakitin, O.A.; Zibarev, A.V. Synthesis and Applications of 5-Membered Chalcogen-Nitrogen π-Heterocycles with Three Heteroatoms. Asian J. Org. Chem. 2018, 7, 2397–2416. [Google Scholar] [CrossRef]
- Iftikhar, R.; Khan, F.Z.; Naeem, N. Recent synthetic strategies of small heterocyclic organic molecules with optoelectronic applications: A review. Mol. Divers. 2023. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, L.; Gong, S.; Zhong, C.; Xie, G.; Mao, E.; Fang, J.; Ma, D.; Yang, C. A Red Fluorescent Emitter with a Simultaneous Hybrid Local and Charge Transfer Excited State and Aggregation-Induced Emission for High-Efficiency, Low Efficiency Roll-Off OLEDs. Adv. Opt. Mater. 2017, 5, 1700145. [Google Scholar] [CrossRef]
- Zhang, Y.; Song, J.; Qu, J.; Qian, P.-C.; Wong, W.-Y. Recent progress of electronic materials based on 2,1,3-benzothiadiazole and its derivatives: Synthesis and their application in organic light-emitting diodes. Sci. China Chem. 2021, 64, 341–357. [Google Scholar] [CrossRef]
- Li, Y. Molecular Design of Photovoltaic Materials for Polymer Solar Cells: Toward Suitable Electronic Energy Levels and Broad Absorption. Acc. Chem. Res. 2012, 45, 723–733. [Google Scholar] [CrossRef]
- Zhou, P.; Zhang, Z.-G.; Li, Y.; Chen, X.; Qin, J. Thiophene-Fused Benzothiadiazole: A Strong Electron-Acceptor Unit to Build D–A Copolymer for Highly Efficient Polymer Solar Cells. Chem. Mater. 2014, 26, 3495–3501. [Google Scholar] [CrossRef]
- Wu, Y.; Zhu, W.-H.; Zakeeruddin, S.M.; Grätzel, M. Insight into D–A−π–A Structured Sensitizers: A Promising Route to Highly Efficient and Stable Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 9307–9318. [Google Scholar] [CrossRef]
- Parker, T.C.; Patel, D.G.; Moudgil, K.; Barlow, S.; Risko, C.; Brédas, J.-L.; Reynolds, J.R.; Marder, S.R. Heteroannulated acceptors based on benzothiadiazole. Mater. Horiz. 2015, 2, 22–36. [Google Scholar] [CrossRef]
- Rakitin, O.A. Recent Developments in the Synthesis of 1,2,5-Thiadiazoles and 2,1,3-Benzothiadiazoles. Synthesis 2019, 51, 4338–4347. [Google Scholar] [CrossRef]
- Chmovzh, T.N.; Knyazeva, E.A.; Mikhalchenko, L.V.; Golovanov, I.S.; Amelichev, S.A.; Rakitin, O.A. Synthesis of the 4,7-Dibromo Derivative of Highly Electron-Deficient [1,2,5]Thiadiazolo[3,4-d ]pyridazine and Its Cross-Coupling Reactions. Eur. J. Org. Chem. 2018, 2018, 5668–5677. [Google Scholar] [CrossRef]
- Chmovzh, T.N.; Rakitin, O.A. Benzobischalcogenadiazoles: Synthesis and applications (microreview). Chem. Heterocycl. Compd. 2022, 58, 307–309. [Google Scholar] [CrossRef]
- Gudim, N.S.; Knyazeva, E.A.; Mikhalchenko, L.V.; Golovanov, I.S.; Popov, V.V.; Obruchnikova, N.V.; Rakitin, O.A. Benzothiadiazole vs. iso-Benzothiadiazole: Synthesis, Electrochemical and Optical Properties of D–A–D Conjugated Molecules Based on Them. Molecules 2021, 26, 4931. [Google Scholar] [CrossRef] [PubMed]
- Chmovzh, T.N.; Alekhina, D.A.; Kudryashev, T.A.; Rakitin, O.A. Efficient Synthesis of 4,8-Dibromo Derivative of Strong Electron-Deficient Benzo[1,2-d:4,5-d’]bis([1,2,3]thiadiazole) and Its SNAr and Cross-Coupling Reactions. Molecules 2022, 27, 7372. [Google Scholar] [CrossRef]
- Cascioferro, S.; Petri, G.L.; Parrino, B.; Carbone, D.; Funel, N.; Bergonzini, C.; Mantini, G.; Dekker, H.; Geerke, D.; Peters, G.J.; et al. Imidazo[2,1-b] [1,3,4]thiadiazoles with antiproliferative activity against primary and gemcitabine-resistant pancreatic cancer cells. Eur. J. Med. Chem. 2020, 189, 112088. [Google Scholar] [CrossRef]
- Beno, B.R.; Yeung, K.-S.; Bartberger, M.D.; Pennington, L.D.; Meanwell, N.A. A Survey of the Role of Noncovalent Sulfur Interactions in Drug Design. J. Med. Chem. 2015, 58, 4383–4438. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, D.W.; Andrzejak, M.; Dominikowska, J.; Pawełek, B.; Krygowski, T.M.; Szatylowicz, H.; Solà, M. The electron density of delocalized bonds (EDDB) applied for quantifying aromaticity. Phys. Chem. Chem. Phys. 2017, 19, 28970–28981. [Google Scholar] [CrossRef]
- Szczepanik, D.W.; Solà, M. The electron density of delocalized bonds (EDDBs) as a measure of local and global aromaticity. In Aromaticity; Elsevier: Amsterdam, The Netherlands, 2021; pp. 259–284. [Google Scholar]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of magnetically induced current densities: Theory and applications. WIREs Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Dimitrova, M.; Sundholm, D. Current density, current-density pathways, and molecular aromaticity. In Aromaticity; Elsevier: Amsterdam, The Netherlands, 2021; pp. 155–194. [Google Scholar]
- Spackman, M.A.; Byrom, P.G. A novel definition of a molecule in a crystal. Chem. Phys. Lett. 1997, 267, 215–220. [Google Scholar] [CrossRef]
- Spackman, P.R.; Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer: A program for Hirshfeld surface analysis, visualization and quantitative analysis of molecular crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Shorygin, P.P.; Burshtein, K.Y. Conjugation and the periodic system of the elements. Russ. Chem. Rev. 1991, 60, 1–24. [Google Scholar] [CrossRef]
- Aysin, R.R.; Bukalov, S.S. Modern Criteria of Aromaticity for Organometallic Compounds. INEOS OPEN 2021, 4, 154–175. [Google Scholar] [CrossRef]
- Bohra, H.; Wang, M. Direct C–H arylation: A “Greener” approach towards facile synthesis of organic semiconducting molecules and polymers. J. Mater. Chem. A 2017, 5, 11550–11571. [Google Scholar] [CrossRef]
- Mainville, M.; Leclerc, M. Direct (Hetero)arylation: A Tool for Low-Cost and Eco-Friendly Organic Photovoltaics. ACS Appl. Polym. Mater. 2021, 3, 2–13. [Google Scholar] [CrossRef]
- Albano, G.; Punzi, A.; Capozzi, M.A.M.; Farinola, G.M. Sustainable protocols for direct C–H bond arylation of (hetero)arenes. Green Chem. 2022, 24, 1809–1894. [Google Scholar] [CrossRef]
- Yen, Y.-S.; Chou, H.-H.; Chen, Y.-C.; Hsu, C.-Y.; Lin, J.T. Recent developments in molecule-based organic materials for dye-sensitized solar cells. J. Mater. Chem. 2012, 22, 8734–8747. [Google Scholar] [CrossRef]
- Konstantinova, L.S.; Knyazeva, E.A.; Rakitin, O.A. Recent Developments in the Synthesis and Applications of 1,2,5-Thia- and Selenadiazoles. A Review. Org. Prep. Proced. Int. 2014, 46, 475–544. [Google Scholar] [CrossRef]
- Moser, M.; Thorley, K.J.; Moruzzi, F.; Ponder, J.F.; Maria, I.P.; Giovannitti, A.; Inal, S.; McCulloch, I. Highly selective chromoionophores for ratiometric Na + sensing based on an oligoethyleneglycol bridged bithiophene detection unit. J. Mater. Chem. C 2019, 7, 5359–5365. [Google Scholar] [CrossRef]
- Pandey, V.; Raza, M.K.; Sonowal, M.; Gupta, I. BODIPY based red emitters: Synthesis, computational and biological studies. Bioorg. Chem. 2021, 106, 104467. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, L.; Zhang, X.; Chen, Z.; Chen, P.; Zhou, X.; Tang, Y.; Liu, B.; Guo, X.; Facchetti, A. New Benzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole) (iso-BBT)-Based Polymers for Application in Transistors and Solar Cells. Chem. Mater. 2019, 31, 6519–6529. [Google Scholar] [CrossRef]
- He, C.-Y.; Wu, C.-Z.; Zhu, Y.-L.; Zhang, X. Selective Thienylation of Fluorinated Benzothiadiazoles and Benzotriazoles for Organic Photovoltaics. Chem. Sci. 2014, 5, 1317–1321. [Google Scholar] [CrossRef]
- Hu, H.; Jiang, K.; Yang, G.; Liu, J.; Li, Z.; Lin, H.; Liu, Y.; Zhao, J.; Zhang, J.; Huang, F.; et al. Terthiophene-Based D–A Polymer with an Asymmetric Arrangement of Alkyl Chains That Enables Efficient Polymer Solar Cells. J. Am. Chem. Soc. 2015, 137, 14149–14157. [Google Scholar] [CrossRef] [PubMed]
- Chmovzh, T.N.; Alekhina, D.A.; Kudryashev, T.A.; Rakitin, O.A. 4-Bromobenzo[1,2-d:4,5-d′]bis([1,2,3]thiadiazole). Molbank 2022, 2022, M1362. [Google Scholar] [CrossRef]
- Trippé-Allard, G.; Lacroix, J.-C. Synthesis of nitro- and amino-functionalized π-conjugated oligomers incorporating 3,4-ethylenedioxythiophene (EDOT) units. Tetrahedron 2013, 69, 861–866. [Google Scholar] [CrossRef]
- Kuo, C.-Y.; Huang, Y.-C.; Hsiow, C.-Y.; Yang, Y.-W.; Huang, C.-I.; Rwei, S.-P.; Wang, H.-L.; Wang, L. Effect of Side-Chain Architecture on the Optical and Crystalline Properties of Two-Dimensional Polythiophenes. Macromolecules 2013, 46, 5985–5997. [Google Scholar] [CrossRef]
- Vegiraju, S.; Hsieh, C.-M.; Huang, D.-Y.; Chen, Y.-C.; Priyanka, P.; Ni, J.-S.; Esya, F.A.; Kim, C.; Yau, S.L.; Chen, C.-P.; et al. Synthesis and characterization of solution-processable diketopyrrolopyrrole (DPP) and tetrathienothiophene (TTA)-based small molecules for organic thin film transistors and organic photovoltaic cells. Dye. Pigment. 2016, 133, 280–291. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 16, Revision A.03; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef]
- Fliegl, H.; Jusélius, J.; Sundholm, D. Gauge-Origin Independent Calculations of the Anisotropy of the Magnetically Induced Current Densities. J. Phys. Chem. A 2016, 120, 5658–5664. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef]
- Henderson, A. ParaView Guide: A Parallel Visualization Application; Kitware Inc.: Clifton Park, NY, USA, 2007; Available online: https://www.paraview.org/ (accessed on 15 March 2023).
- CrysAlisPro, version 1.171.41.106a. User Inspired Software for Single Crystal X-Ray Diffractometers. Rigaku Oxford Diffraction: Warriewood, Australia, 2021. Available online: https://www.rigaku.com/products/crystallography/chrysalis (accessed on 15 March 2023).
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).