
Halogen Bond to Experimentally Significant N-Heterocyclic Carbenes (I, IMe2, IiPr2, ItBu2, IPh2, IMes2, IDipp2, IAd2; I = Imidazol-2-ylidene)


Abstract
:
1. Introduction
2. Results and Discussion
2.1. Nucleophilicity of Carbenes
2.2. Characteristics of IRXCCN and IRXCN Dimers
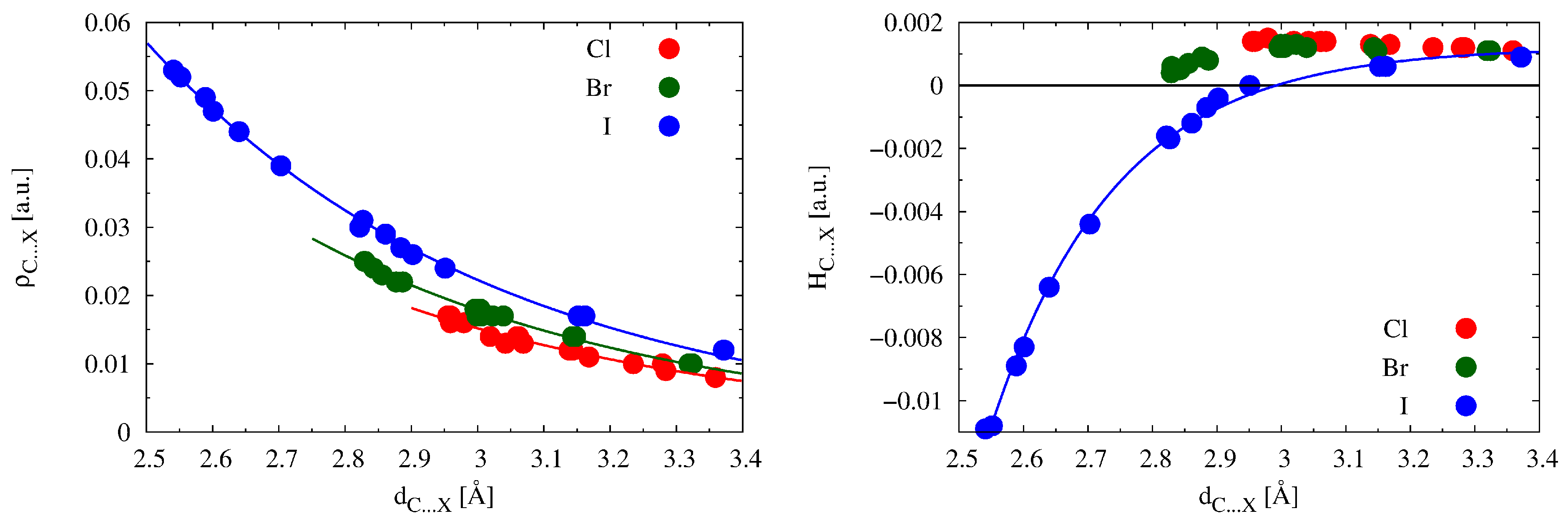
2.3. Relationships between Physical Quantities
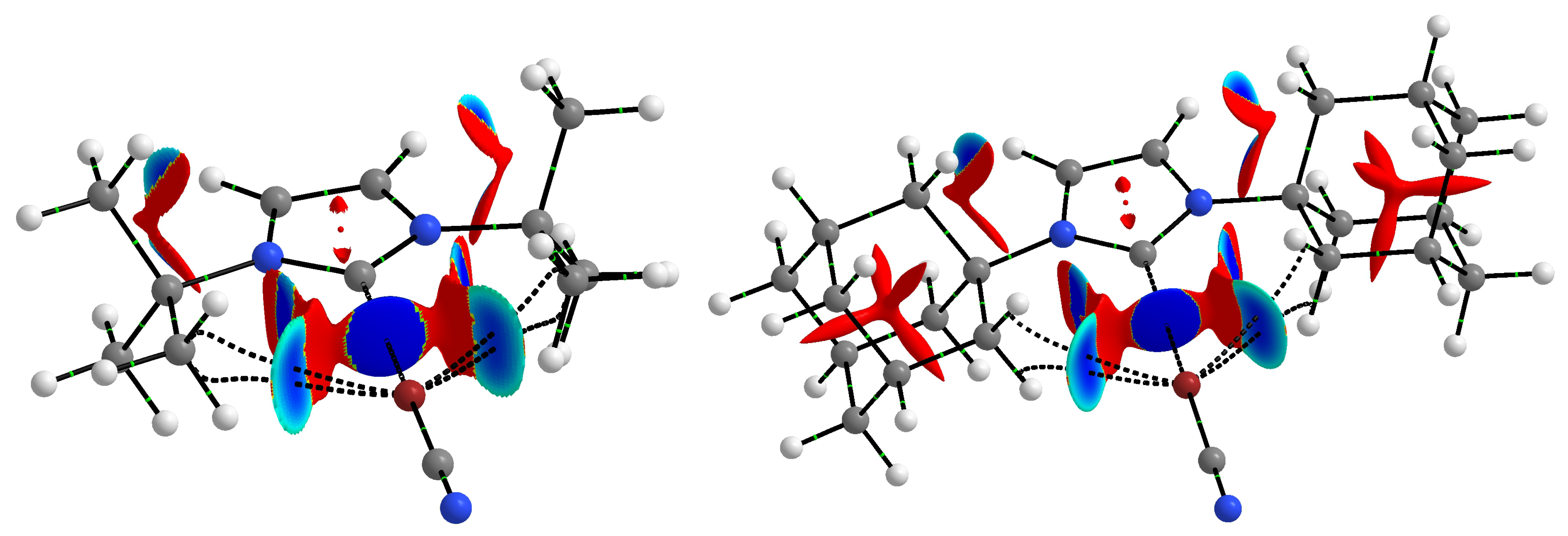
2.4. Halogen Bond vs. Hydrogen Bond
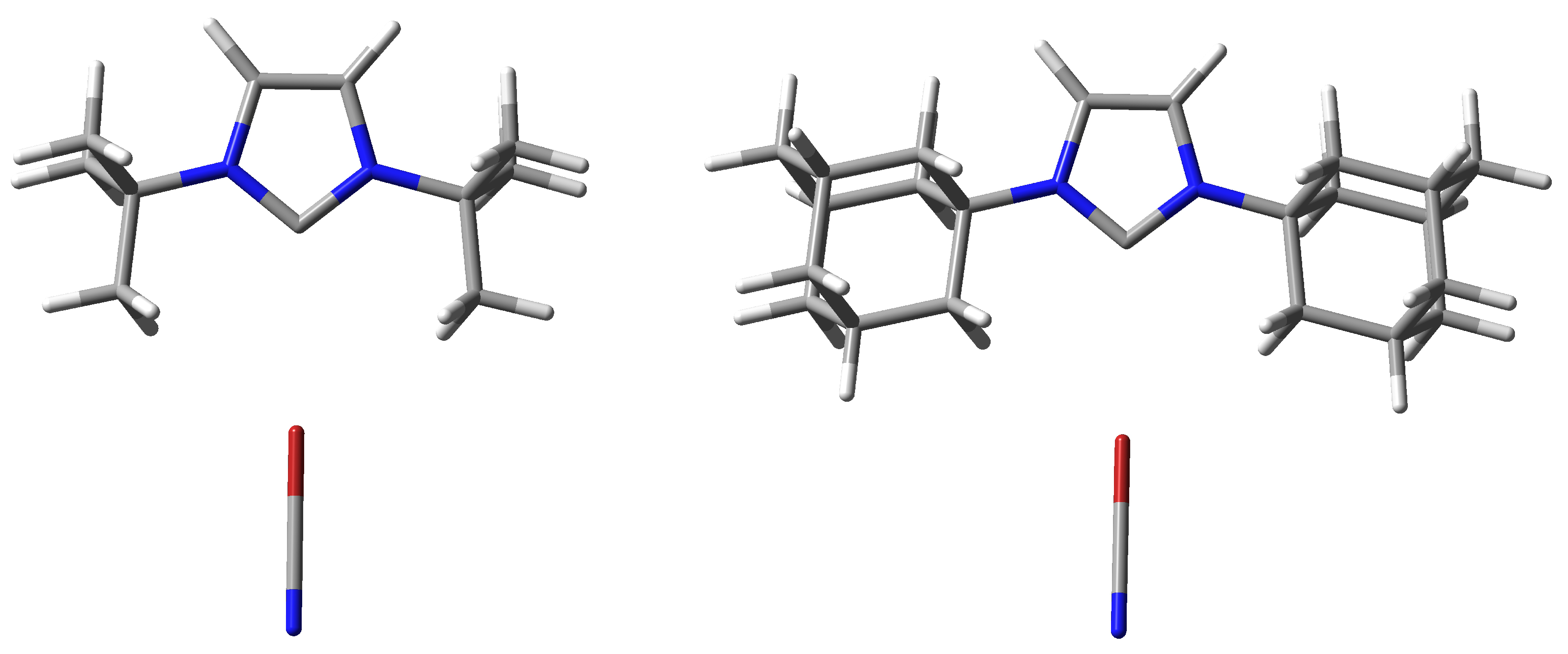
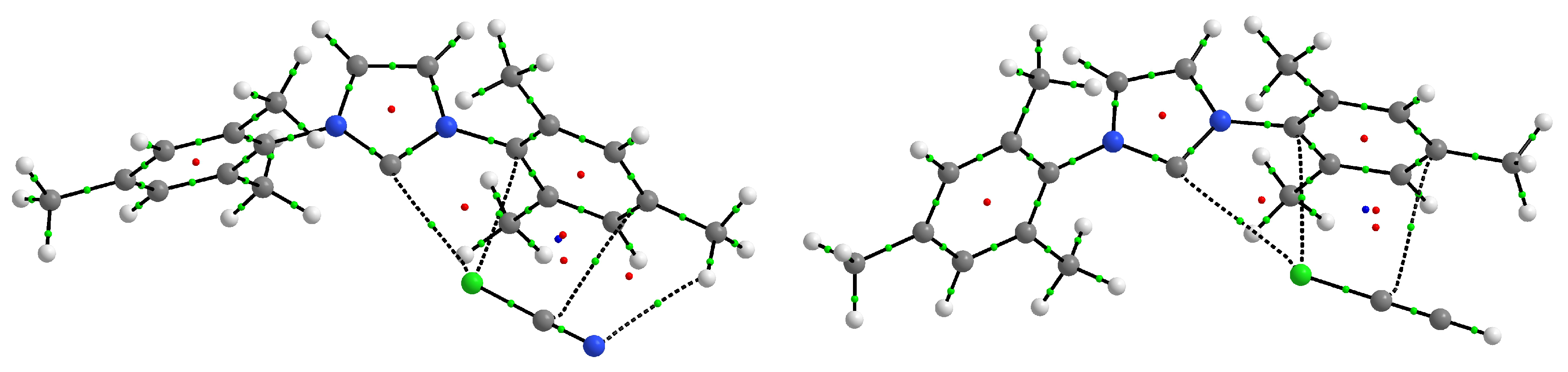
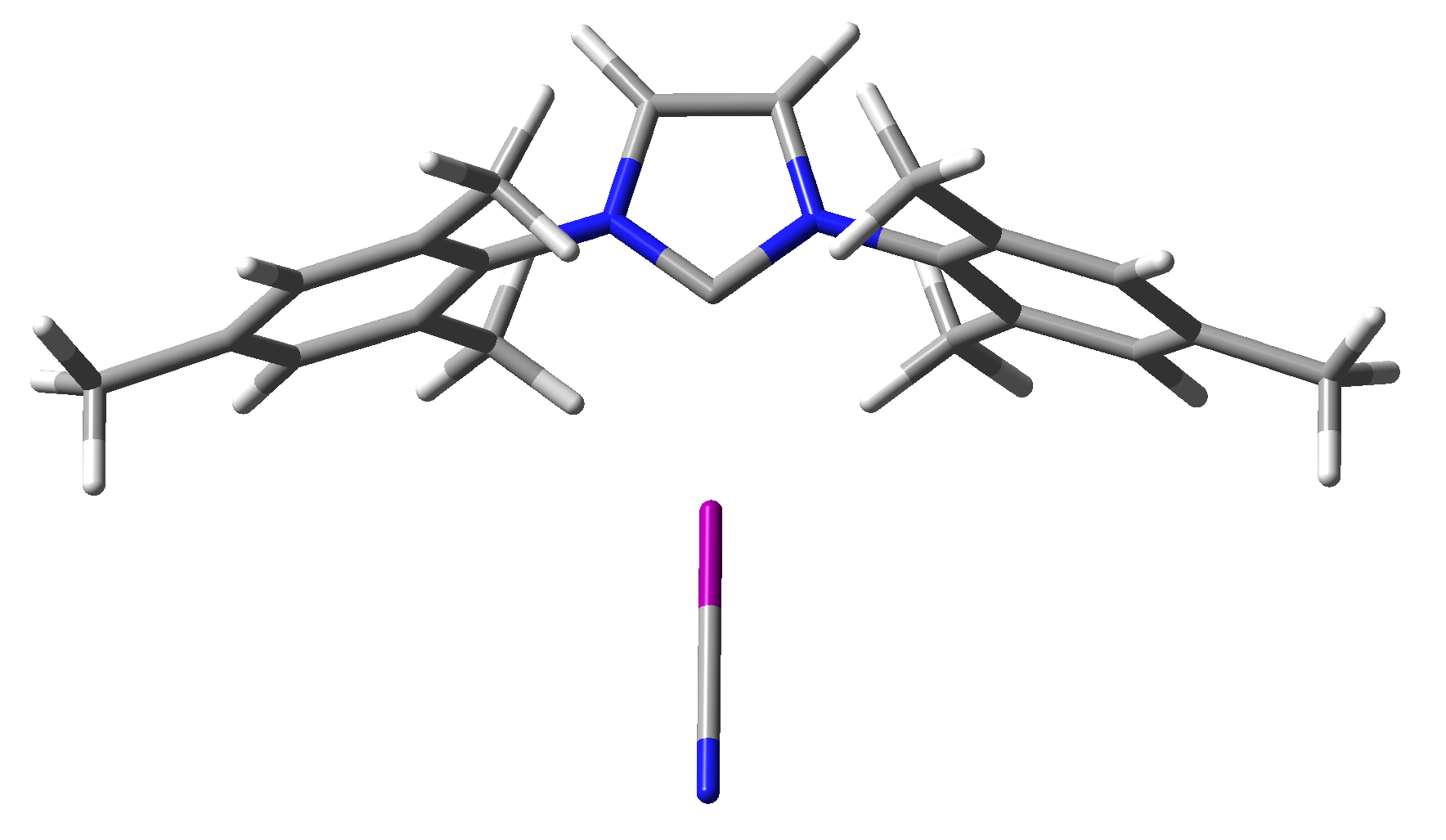
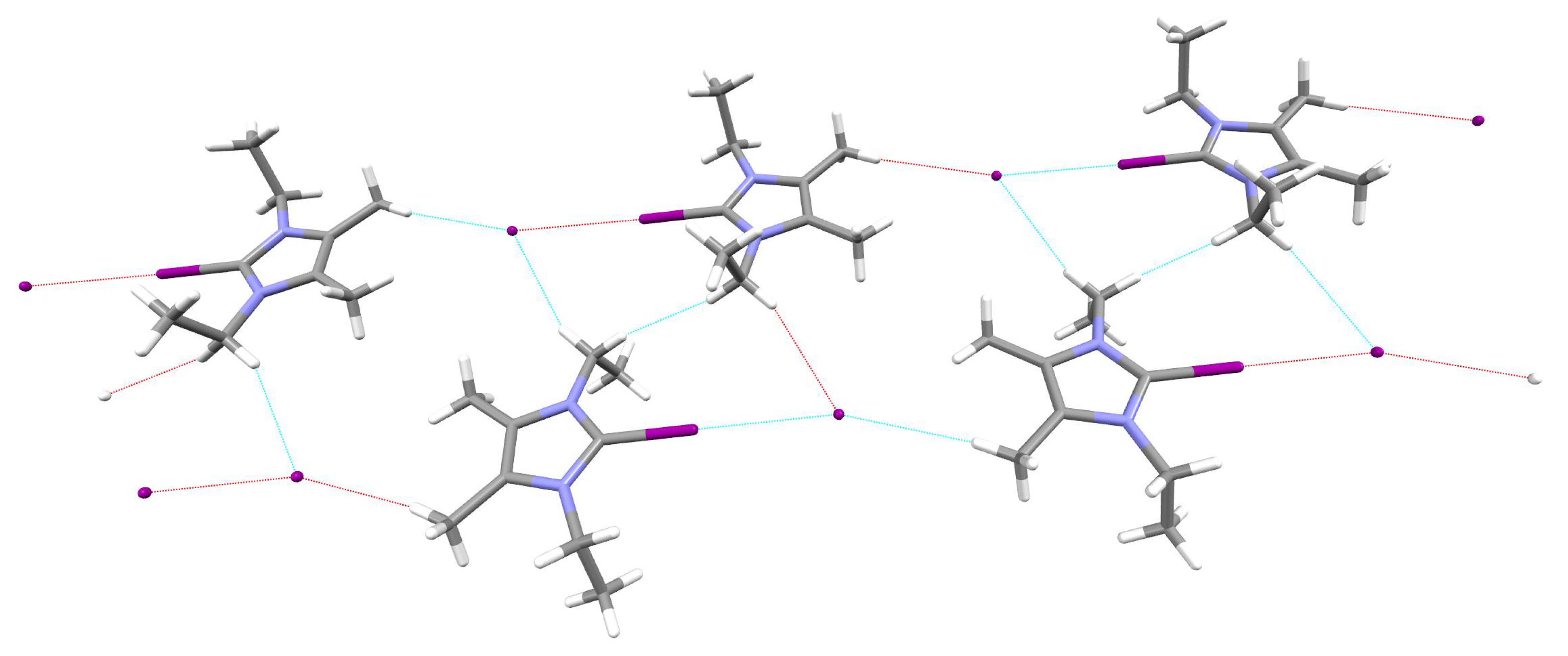
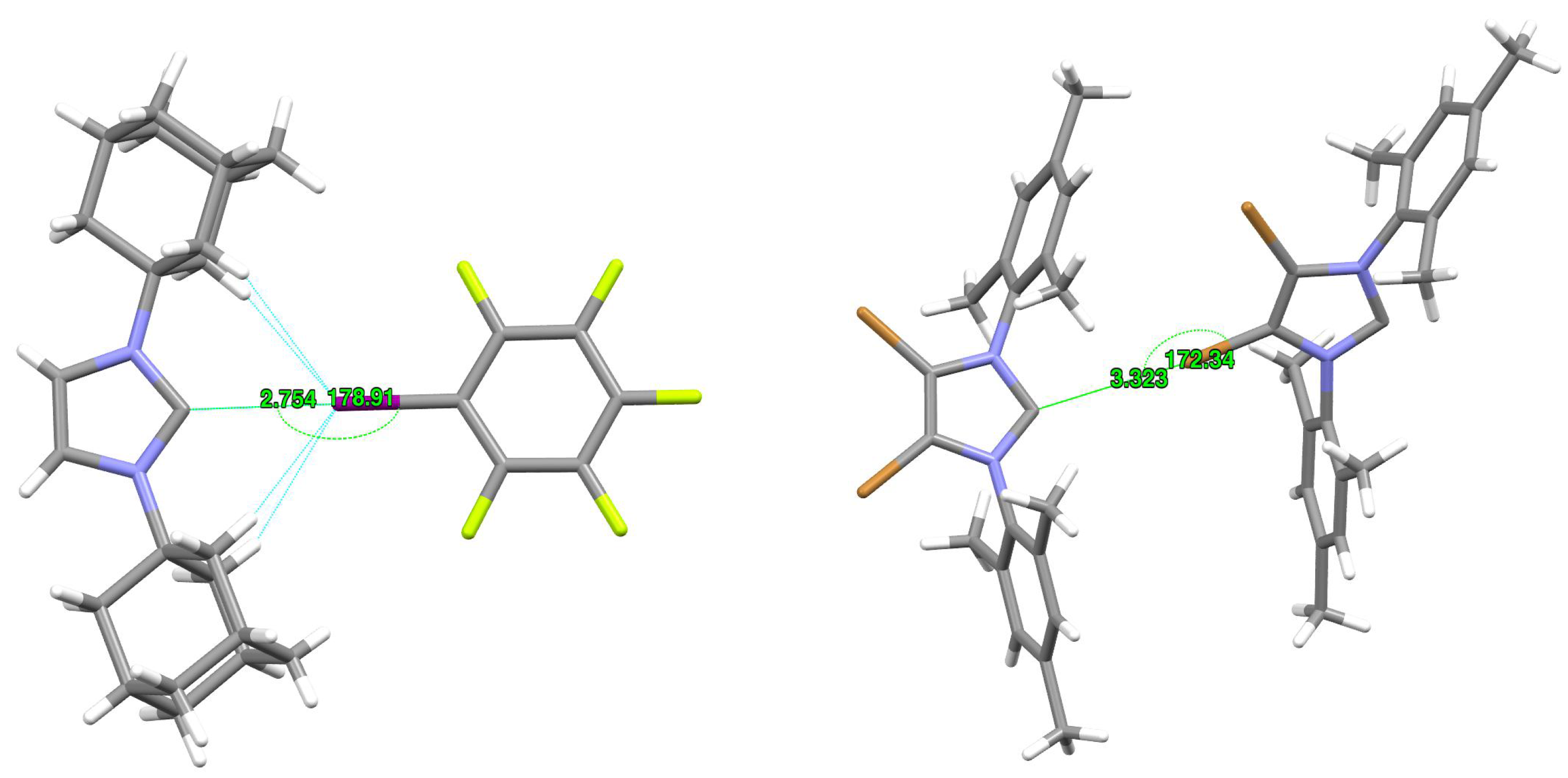
2.5. Crystal Structures
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| XB | halogen bond |
| XD | halogen-donor |
| NHC | N-heterocyclic-carbenes |
| I | imidazol-2-ylidene |
| IR | R-substituted imidazol-2-ylidene |
| Me | methyl group |
| Pr | isopropyl group |
| Bu | tert-butyl group |
| Ph | phenyl group |
| Mes | mesityl group |
| Dipp | 2,6-diisopropylphenyl group |
| Ad | adamantyl group |
| DFT | Density Functional Theory |
| QTAIM | Quantum Theory of Atoms in Molecules |
| BCP | bond critical point |
| RCP | ring critical point |
| NCI | Noncovalent Interaction (index) |
References
- Wanzlick, H.W. Aspects of Nucleophilic Carbene Chemistry. Angew. Chem. Int. Ed. Engl. 1962, 1, 75–80. [Google Scholar] [CrossRef]
- Kirmse, W. Carbene Chemistry; Academic Press: Cambridge, MA, USA, 1964. [Google Scholar]
- Hubert, A.J. Catalysis in C1 Chemistry; Springer: Dordrecht, The Netherlands, 1983. [Google Scholar]
- Schubert, U. Advances in Metal Carbene Chemistry; Springer: Dordrecht, The Netherlands, 1989. [Google Scholar]
- Bourissou, D.; Guerret, O.; Gabbaï, F.P.; Bertrand, G. Stable Carbenes. Chem. Rev. 2000, 100, 39–91. [Google Scholar] [CrossRef] [PubMed]
- Bertrande, G. Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents; FontisMedia S.A.: Lausanne, Switzerland; Marcel Dekker, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Moss, R.A.; Platz, M.S.; Jones, M., Jr. (Eds.) Reactive Intermediate Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2005. [Google Scholar]
- Carey, F.A.; Sundberg, R.J. Carbenes, Part B: Reactions and Synthesis. Advanced Organic Chemistry; Springer: New York, NY, USA, 2007. [Google Scholar]
- de Frémont, P.; Marion, N.; Nolan, S.P. Carbenes: Synthesis, properties, and organometallic chemistry. Coord. Chem. Rev. 2009, 253, 862–892. [Google Scholar] [CrossRef]
- Moss, R.A.; Doyle, M.P. Contemporary Carbene Chemistry; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013. [Google Scholar]
- Herrmann, W.A.; Köcher, C. N-Heterocyclic Carbenes. Angew. Chem. Int. Ed. Engl. 1997, 36, 2162–2187. [Google Scholar] [CrossRef]
- Nemcsok, D.; Wichmann, K.; Frenking, G. The Significance of π Interactions in Group 11 Complexes with N-Heterocyclic Carbenes. Organometallics 2004, 23, 3640–3646. [Google Scholar] [CrossRef]
- Scott, N.M.; Nolan, S.P. Stabilization of Organometallic Species Achieved by the Use of N-Heterocyclic Carbene (NHC) Ligands. Eur. J. Inorg Chem. 2005, 1815–1828. [Google Scholar] [CrossRef]
- Nolan, S.P. N-Heterocyclic Carbenes in Synthesis; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Díez-González, S.; Nolan, S.P. Stereoelectronic parameters associated with N-heterocyclic carbene (NHC) ligands: A quest for understanding. Coord. Chem. Rev. 2007, 251, 874–883. [Google Scholar] [CrossRef]
- Tonner, R.; Heydenrych, G.; Frenking, G. Bonding Analysis of N-Heterocyclic Carbene Tautomers and Phosphine Ligands in Transition-Metal Complexes: A Theoretical Study. Chem. Asian J. 2007, 2, 1555–1567. [Google Scholar] [CrossRef]
- Jacobsen, H.; Correa, A.; Poater, A.; Costabile, C.; Cavallo, L. Understanding the M–(NHC) (NHC = N-heterocyclic carbene) bond. Coord. Chem. Rev. 2009, 253, 687–703. [Google Scholar] [CrossRef]
- Srebro, M.; Michalak, A. Theoretical Analysis of Bonding in N-Heterocyclic Carbene–Rhodium Complexes. Inorg. Chem. 2009, 48, 5361–5369. [Google Scholar] [CrossRef]
- Nelson, D.J.; Nolan, S.P. Quantifying and understanding the electronic properties of N-heterocyclic carbenes. Chem. Soc. Rev. 2013, 42, 6723–6753. [Google Scholar] [CrossRef] [PubMed]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef]
- Bellemin-Laponnaz, S.; Dagorne, S. Group 1 and 2 and Early Transition Metal Complexes Bearing N-Heterocyclic Carbene Ligands: Coordination Chemistry, Reactivity, and Applications. Chem. Rev. 2014, 114, 8747–8774. [Google Scholar] [CrossRef] [PubMed]
- Andrada, D.M.; Holzmann, N.; Hamadi, T.; Frenking, G. Direct estimate of the internal π-donation to the carbene centre within N-heterocyclic carbenes and related molecules. Beilstein J. Org. Chem. 2015, 11, 2727–2736. [Google Scholar] [CrossRef] [PubMed]
- Nesterov, V.; Reiter, D.; Bag, P.; Frisch, P.; Holzner, R.; Porzelt, A.; Inoue, S. NHCs in Main Group Chemistry. Chem. Rev. 2018, 118, 9678–9842. [Google Scholar] [CrossRef]
- Santoro, O.; Nahra, F.; Cordes, D.B.; Slawin, A.M.Z.; Nolan, S.P.; Cazin, C.S.J. Synthesis, characterization and catalytic activity of stable [(NHC)H][ZnXY2] (NHC = N-Heterocyclic carbene, X, Y = Cl, Br) species. J. Mol. Catal. 2016, 423, 85–91. [Google Scholar] [CrossRef]
- Dagorne, S. Recent Developments on N-Heterocyclic Carbene Supported Zinc Complexes: Synthesis and Use in Catalysis. Synthesis 2018, 50, 3662–3670. [Google Scholar] [CrossRef]
- Procter, R.J.; Uzelac, M.; Cid, J.; Rushworth, P.J.; Ingleson, M.J. Low-Coordinate NHC–Zinc Hydride Complexes Catalyze Alkyne C–H Borylation and Hydroboration Using Pinacolborane. ACS Catal. 2019, 9, 5760–5771. [Google Scholar] [CrossRef]
- Dzieszkowski, K.; Barańska, I.; Mroczyńska, K.; Słotwiński, M.; Rafinski, Z. Organocatalytic Name Reactions Enabled by NHCs. Materials 2020, 13, 3574. [Google Scholar] [CrossRef]
- Specklin, D.; Fliedel, C.; Dagorne, S. Recent Representative Advances on the Synthesis and Reactivity of N-Heterocyclic-Carbene-Supported Zinc Complexes. Chem. Rec. 2021, 21, 1130–1143. [Google Scholar] [CrossRef]
- Huang, J.; Stevens, E.D.; Nolan, S.P.; Petersen, J.L. Olefin Metathesis-Active Ruthenium Complexes Bearing a Nucleophilic Carbene Ligand. J. Am. Chem. Soc. 1999, 121, 2674–2678. [Google Scholar] [CrossRef]
- Scholl, M.; Trnka, T.M.; Morgan, J.P.; Grubbs, R.H. Increased Ring Closing Metathesis Activity of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with Imidazolin-2-ylidene Ligands. Tetrahedron Lett. 1999, 40, 2247–2250. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef]
- Ackermann, L.; Fürstner, A.; Weskamp, T.; Kohl, F.J.; Herrmann, W.A. Ruthenium Carbene Complexes with Imidazolin-2-ylidene Ligands Allow the Formation of Tetrasubstituted Cycloalkenes by RCM. Tetrahedron Lett. 1999, 40, 4787–4790. [Google Scholar] [CrossRef]
- Kingsbury, J.S.; Harrity, J.P.A.; Bonitatebus, P.J., Jr.; Hoveyda, A.H. A Recyclable Ru-Based Metathesis Catalyst. J. Am. Chem. Soc. 1999, 121, 791–799. [Google Scholar] [CrossRef]
- Walsh, D.J.; Lau, S.H.; Hyatt, M.G.; Guironnet, D. Kinetic Study of Living Ring-Opening Metathesis Polymerization with Third-Generation Grubbs Catalysts. J. Am. Chem. Soc. 2017, 139, 13644–13647. [Google Scholar] [CrossRef]
- Leitgeb, A.; Wappel, J.; Slugovc, C. The ROMP toolbox upgraded. Polymer 2010, 51, 2927–2946. [Google Scholar] [CrossRef]
- Mercs, L.; Albrecht, M. Beyond catalysis: N-heterocyclic carbene complexes as components for medicinal, luminescent, and functional materials applications. Chem. Soc. Rev. 2010, 39, 1903–1912. [Google Scholar] [CrossRef]
- Visbal, R.; Concepción Gimeno, M. N-heterocyclic carbene metal complexes: Photoluminescence and applications. Chem. Soc. Rev. 2014, 43, 3551–3574. [Google Scholar] [CrossRef]
- Garrison, J.C.; Youngs, W.J. Ag(I) N-Heterocyclic Carbene Complexes: Synthesis, Structure, and Application. Chem. Rev. 2005, 105, 3978–4008. [Google Scholar] [CrossRef]
- Hindi, K.M.; Panzner, M.J.; Tessier, C.A.; Cannon, C.L.; Youngs, W.J. The Medicinal Applications of Imidazolium Carbene—Metal Complexes. Chem. Rev. 2009, 109, 3859–3884. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Davidson, F.; Krafczyk, R.; Marshall, W.J.; Tamm, M. Adducts of Carbenes with Group II and XII Metallocenes. Organometallics 1998, 17, 3375–3382. [Google Scholar] [CrossRef]
- Cases, M.; Frenking, G.; Duran, M.; Solà, M. Molecular Structure and Bond Characterization of the Fischer-Type Chromium–Carbene Complexes (CO)5Cr = C(X)R (X = H, OH, OCH3, NH2, NHCH3 and R = H, CH3, CH = CH2, Ph, C ≡ CH). Organometallics 2002, 21, 4182–4191. [Google Scholar] [CrossRef]
- Abernethy, C.D.; Baker, R.J.; Cole, M.L.; Davies, A.J.; Jones, C. Reactions of a carbene stabilised indium trihydride complex, [InH3{CN(Mes)-C2H2N(Mes)}] Mes = mesityl, with transition metal complexes. Trans. Metal. Chem. 2003, 28, 296–299. [Google Scholar] [CrossRef]
- Wang, D.; Wurst, K.; Buchmeiser, M.R. N-heterocyclic carbene complexes of Zn(II): Synthesis, X-ray structures and reactivity. J. Organomet. Chem. 2004, 689, 2123–2130. [Google Scholar] [CrossRef]
- Frenking, G.; Solà, M.; Vyboishchikov, S.F. Chemical bonding in transition metal carbene complexes. J. Organomet. Chem. 2005, 690, 6178–6204. [Google Scholar] [CrossRef]
- Jensen, T.R.; Schaller, C.P.; Hillmyer, M.A.; Tolman, W.B. Zinc N-heterocyclic carbene complexes and their polymerization of D,L-lactide. J. Organomet. Chem. 2005, 690, 5881–5891. [Google Scholar] [CrossRef]
- Anantharaman, G.; Elango, K. Synthesis and Characterization of NHC-Stabilized Zinc Aryloxide and Zinc Hydroxyaryloxide. Organometallics 2007, 26, 1089–1092. [Google Scholar] [CrossRef]
- Radius, U.; Bickelhaupt, F.M. Bonding of Imidazol-2-ylidene Ligands in Nickel Complexes. Organometallics 2008, 27, 3410–3414. [Google Scholar] [CrossRef]
- Budagumpi, S.; Endud, S. Group XII Metal–N-Heterocyclic Carbene Complexes: Synthesis, Structural Diversity, Intramolecular Interactions, and Applications. Organometallics 2013, 32, 1537–1562. [Google Scholar] [CrossRef]
- Schnee, G.; Fliedel, C.; Avilés, T.; Dagorne, S. Neutral and Cationic N-Heterocyclic Carbene Zinc Adducts and the BnOH/Zn(C6F5)2 Binary Mixture—Characterization and Use in the Ring-Opening Polymerization of β-Butyrolactone, Lactide, and Trimethylene Carbonate. Eur. J. Inorg. Chem. 2013, 3699–3709. [Google Scholar] [CrossRef]
- Fliedel, C.; Vila-Viçosa, D.; Calhorda, M.J.; Dagorne, S.; Avilés, T. Dinuclear Zinc–N-Heterocyclic Carbene Complexes for Either the Controlled Ring-Opening Polymerization of Lactide or the Controlled Degradation of Polylactide Under Mild Conditions. ChemCatChem 2014, 6, 1357–1367. [Google Scholar] [CrossRef]
- Collins, L.R.; Moffat, L.A.; Mahon, M.F.; Jones, M.D.; Whittlesey, M.K. Lactide polymerisation by ring-expanded NHC complexes of zinc. Polyhedron 2016, 103, 121–125. [Google Scholar] [CrossRef]
- Tian, J.; Chen, Y.; Vayer, M.; Djurovic, A.; Guillot, R.; Guermazi, R.; Dagorne, S.; Bour, C.; Gandon, V. Exploring the Limits of π-Acid Catalysis Using Strongly Electrophilic Main Group Metal Complexes: The Case of Zinc and Aluminium. Chem. Eur. J. 2020, 26, 12831–12838. [Google Scholar] [CrossRef]
- Roy, M.M.D.; Baird, S.R.; Ferguson, M.J.; Rivard, E. Toward N-heterocyclic carbene stabilized zinc sulfides. Mendeleev Commun. 2021, 31, 173–175. [Google Scholar] [CrossRef]
- Jabłoński, M. Study of Beryllium, Magnesium, and Spodium Bonds to Carbenes and Carbodiphosphoranes. Molecules 2021, 26, 2275. [Google Scholar] [CrossRef] [PubMed]
- Jabłoński, M. Theoretical Study of N-Heterocyclic-Carbene–ZnX2 (X = H, Me, Et) Complexes. Materials 2021, 14, 6147. [Google Scholar] [CrossRef]
- Sagan, F.; Mitoraj, M.; Jabłoński, M. Nature of Beryllium, Magnesium, and Zinc Bonds in Carbene⋯MX2 (M = Be, Mg, Zn; X = H, Br) Dimers Revealed by the IQA, ETS-NOCV and LED Methods. Int. J. Mol. Sci. 2022, 23, 14668. [Google Scholar] [CrossRef]
- Pople, J.A.; Raghavachari, K.; Frisch, M.J.; Binkley, J.S.; Schleyer, P.v.R. Comprehensive Theoretical Study of Isomers and Rearrangement Barriers of Even-Electron Polyatomic Molecules HmABHn (A, B = C, N, O, and F). J. Am. Chem. Soc. 1983, 105, 6389–6398. [Google Scholar] [CrossRef]
- Pople, J.A. A Theoretical Search for the Methylenefluoronium Ylide. Chem. Phys. Lett. 1986, 132, 144–146. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Gamper, S.F.; Tamm, M.; Calabrese, J.C.; Davidson, F.; Craig, H.A. A Bis(carbene)–Proton Complex: Structure of a C–H–C Hydrogen Bond. J. Am. Chem. Soc. 1995, 117, 572–573. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. Carbenes and Silylenes as Hydrogen Bond Acceptors. J. Phys. Chem. 1996, 100, 19367–19370. [Google Scholar] [CrossRef]
- Jabłoński, M.; Palusiak, M. Divalent carbon atom as the proton acceptor in hydrogen bonding. Phys. Chem. Chem. Phys. 2009, 11, 5711–5719. [Google Scholar] [CrossRef] [PubMed]
- Giffin, N.A.; Makramalla, M.; Hendsbee, A.D.; Robertson, K.N.; Sherren, C.; Pye, C.C.; Masuda, J.D.; Clyburne, J.A.C. Anhydrous TEMPO-H: Reactions of a good hydrogen atom donor with low-valent carbon centres. Org. Biomol. Chem. 2011, 9, 3672–3680. [Google Scholar] [CrossRef]
- Gerbig, D.; Ley, D. Computational methods for contemporary carbene chemistry. WIREs Comput. Mol. Sci. 2013, 3, 242–272. [Google Scholar] [CrossRef]
- Samanta, R.C.; De Sarkar, S.; Fröhlich, R.; Grimme, S.; Studer, A. N-Heterocyclic carbene (NHC) catalyzed chemoselective acylation of alcohols in the presence of amines with various acylating reagents. Chem. Sci. 2013, 4, 2177–2184. [Google Scholar] [CrossRef]
- Jabłoński, M. On the Coexistence of the Carbene⋯H-D Hydrogen Bond and Other Accompanying Interactions in Forty Dimers of N-Heterocyclic-Carbenes (I, IMe2, IiPr2, ItBu2, IMes2, IDipp2, IAd2; I = imidazol-2-ylidene) and Some Fundamental Proton Donors (HF, HCN, H2O, MeOH, NH3). Molecules 2022, 27, 5712. [Google Scholar] [CrossRef]
- Li, Q.; Wang, H.; Liu, Z.; Li, W.; Cheng, J.; Gong, B.; Sun, J. Ab Initio Study of Lithium-Bonded Complexes with Carbene as an Electron Donor. J. Phys. Chem. A 2009, 113, 14156–14160. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Y.; Abraham, M.Y.; Wei, P.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. A Viable Anionic N-Heterocyclic Dicarbene. J. Am. Chem. Soc. 2010, 132, 14370–14372. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.F.; Sheng, Y.; Li, H.X. Theoretical prediction characters of unconventional weak bond with carbene as electron donors and Li–Y (Y = OH, H, F, NC and CN) as electron acceptors. J. Mol. Struct. THEOCHEM 2010, 952, 56–60. [Google Scholar]
- Herrmann, W.A.; Runte, O.; Artus, G. Synthesis and structure of an ionic beryllium—“Carbene” complex. J. Organomet. Chem. 1995, 501, C1–C4. [Google Scholar] [CrossRef]
- Gilliard, R.J., Jr.; Abraham, M.Y.; Wang, Y.; Wei, P.; Xie, Y.; Quillian, B.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. Carbene-Stabilized Beryllium Borohydride. J. Am. Chem. Soc. 2012, 134, 9953–9955. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; Kociok-Köhn, G.; MacDougall, D.J.; Mahon, M.F. Beryllium-Induced C–N Bond Activation and Ring Opening of an N-Heterocyclic Carbene. Angew. Chem. Int. Ed. 2012, 51, 2098–2100. [Google Scholar] [CrossRef]
- Walley, J.E.; Wong, Y.-O.; Freeman, L.A.; Dickie, D.A.; Gilliard, R.J., Jr. N-Heterocyclic Carbene-Supported Aryl- and Alk- oxides of Beryllium and Magnesium. Catalysts 2019, 9, 934. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Dias, H.V.R.; Davidson, F.; Harlow, R.L. Carbene adducts of magnesium and zinc. J. Organomet. Chem. 1993, 462, 13–18. [Google Scholar] [CrossRef]
- Arrowsmith, M.; Hill, M.S.; MacDougall, D.J.; Mahon, M.F. A Hydride-Rich Magnesium Cluster. Angew. Chem. Int. Ed. 2009, 48, 4013–4016. [Google Scholar] [CrossRef] [PubMed]
- Arnold, P.L.; Casely, I.J.; Turner, Z.R.; Bellabarba, R.; Tooze, R.B. Magnesium and zinc complexes of functionalised, saturated N-heterocyclic carbene ligands: Carbene lability and functionalisation, and lactide polymerisation catalysis. Dalton Trans. 2009, 7236–7247. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Dias, H.V.R.; Calabrese, J.C.; Davidson, F. A Stable Carbene–Alane Adduct. J. Am. Chem. Soc. 1992, 114, 9724–9725. [Google Scholar] [CrossRef]
- Li, X.-W.; Su, J.; Robinson, G.H. Syntheses and molecular structure of organo-group 13 metal carbene complexes. Chem. Commun. 1996, 23, 2683–2684. [Google Scholar] [CrossRef]
- Hibbs, D.E.; Hursthouse, M.B.; Jones, C.; Smithies, N.A. Synthesis, crystal and molecular structure of the first indium trihydride complex, [InH3{CN(Pri)C2Me2N(Pri)}]. Chem. Commun. 1998, 8, 869–870. [Google Scholar] [CrossRef]
- Merceron, N.; Miqueu, K.; Baceiredo, A.; Bertrand, G. Stable (Amino)(phosphino)carbenes: Difunctional Molecules. J. Am. Chem. Soc. 2002, 124, 6806–6807. [Google Scholar] [CrossRef]
- Wang, Y.; Robinson, G.H. Unique homonuclear multiple bonding in main group compounds. Chem. Commun. 2009, 5201–5213. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Carbon–Carbon Bonding between Nitrogen Heterocyclic Carbenes and CO2. J. Phys. Chem. A 2017, 121, 8136–8146. [Google Scholar] [CrossRef]
- Liu, M.; Li, Q.; Li, W.; Cheng, J. Carbene tetrel-bonded complexes. Struct. Chem. 2017, 28, 823–831. [Google Scholar] [CrossRef]
- Wang, Y.; Xie, Y.; Abraham, M.Y.; Gilliard, R.J., Jr.; Wei, P.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. Carbene-Stabilized Parent Phosphinidene. Organometallics 2010, 29, 4778–4780. [Google Scholar] [CrossRef]
- Abraham, M.Y.; Wang, Y.; Xie, Y.; Wei, P.; Schaefer, H.F., III; Schleyer, P.v.R.; Robinson, G.H. Carbene Stabilization of Diarsenic: From Hypervalency to Allotropy. Chem. Eur. J. 2010, 16, 432–435. [Google Scholar] [CrossRef]
- Patel, D.S.; Bharatam, P.V. Divalent N(I) Compounds with Two Lone Pairs on Nitrogen. J. Phys. Chem. A 2011, 115, 7645–7655. [Google Scholar] [CrossRef]
- Zhao, Q.; Feng, D.; Sun, Y.; Hao, J.; Cai, Z. Theoretical Investigations on the Weak Nonbonded C=S⋯CH2 Interactions: Chalcogen-Bonded Complexes with Singlet Carbene as an Electron Donor. Int. J. Quant. Chem. 2011, 111, 3881–3887. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Kline, M.; Calabrese, J.C.; Davidson, F. Synthesis of a Reverse Ylide from a Nucleophilic Carbene. J. Am. Chem. Soc. 1991, 113, 9704–9705. [Google Scholar] [CrossRef]
- Kuhn, N.; Kratz, T.; Henkel, G. A Stable Carbene Iodine Adduct: Secondary Bonding in 1,3-Diethyl-2-iodo-4,5-dimethylimidazolium Iodide. J. Chem. Soc. Chem. Commun. 1993, 1778–1779. [Google Scholar] [CrossRef]
- Li, Q.; Wang, Y.; Liu, Z.; Li, W.; Cheng, J.; Gong, B.; Sun, J. An unconventional halogen bond with carbene as an electron donor: An ab initio study. Chem. Phys. Lett. 2009, 469, 48–51. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Mohammadirad, N. Insights into the strength and nature of carbene⋯halogen bond interactions: A theoretical perspective. J. Mol. Model. 2013, 19, 2559–2566. [Google Scholar] [CrossRef] [PubMed]
- Esrafili, M.D.; Mohammdain-Sabet, F.; Esmailpour, P. Theoretical study on cooperative effects between X⋯N and X⋯Carbene halogen bonds (X = F, Cl, Br and I). J. Mol. Model. 2013, 19, 4797–4804. [Google Scholar] [CrossRef] [PubMed]
- Donoso-Tauda, O.; Jaque, P.; Elguero, J.; Alkorta, I. Traditional and Ion-Pair Halogen-Bonded Complexes Between Chlorine and Bromine Derivatives and a Nitrogen-Heterocyclic Carbene. J. Phys. Chem. A 2014, 118, 9552–9560. [Google Scholar] [CrossRef]
- Lv, H.; Zhuo, H.-Y.; Li, Q.-Z.; Yang, X.; Li, W.-Z.; Cheng, J.-B. Halogen bonds with N-heterocyclic carbenes as halogen acceptors: A partially covalent character. Mol. Phys. 2014, 112, 3024–3032. [Google Scholar] [CrossRef]
- Del Bene, J.E.; Alkorta, I.; Elguero, J. Halogen bonding with carbene bases. Chem. Phys. Lett. 2017, 685, 338–343. [Google Scholar] [CrossRef]
- Grabowski, S.J. Halogen bonds with carbenes acting as Lewis base units: Complexes of imidazol-2-ylidene: Theoretical analysis and experimental evidence. Phys. Chem. Chem. Phys. 2023, 25, 9636–9647. [Google Scholar] [CrossRef]
- Esrafili, M.D.; Sabouri, A. Carbene–aerogen bonds: An ab initio study. Mol. Phys. 2017, 115, 971–980. [Google Scholar] [CrossRef]
- Hoffmann, R.; Zeiss, G.D.; Van Dine, G.W. The Electronic Structure of Methylenes. J. Am. Chem. Soc. 1968, 90, 1485–1499. [Google Scholar] [CrossRef]
- Gleiter, R.; Hoffmann, R. On Stabilizing a Singlet Methylene. J. Am. Chem. Soc. 1968, 90, 5457–5460. [Google Scholar] [CrossRef]
- Baird, N.C.; Taylor, K.F. Multiplicity of the Ground State and Magnitude of the T1–S0 Gap in Substituted Carbenes. J. Am. Chem. Soc. 1978, 100, 1333–1338. [Google Scholar] [CrossRef]
- Harrison, J.F.; Liedtke, R.C.; Liebman, J.F. The Multiplicity of Substituted Acyclic Carbenes and Related Molecules. J. Am. Chem. Soc. 1979, 101, 7162–7168. [Google Scholar] [CrossRef]
- Mueller, P.H.; Rondan, N.G.; Houk, K.N.; Harrison, J.F.; Hooper, D.; Willen, B.H.; Liebman, J.F. Carbene Singlet–Triplet Gaps. Linear Correlations with Substituent π Donation. J. Am. Chem. Soc. 1981, 103, 5049–5052. [Google Scholar] [CrossRef]
- Boehme, C.; Frenking, G. Electronic Structure of Stable Carbenes, Silylenes, and Germylenes. J. Am. Chem. Soc. 1996, 118, 2039–2046. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J. A LFER analysis of the singlet-triplet gap in a series of sixty-six carbenes. Chem. Phys. Lett. 2018, 691, 33–36. [Google Scholar] [CrossRef]
- Schoeller, W.W. Electrophilicity and nucleophilicity in singlet carbenes. II. Electrophilic selectivity. Tetrahedron Lett. 1980, 21, 1509–1510. [Google Scholar] [CrossRef]
- Goumri-Magnet, S.; Polishchuck, O.; Gornitzka, H.; Marsden, C.J.; Baceiredo, A.; Bertrand, G. The Electrophilic Behavior of Stable Phosphanylcarbenes Towards Phosphorus Lone Pairs. Angew. Chem. Int. Ed. 1999, 38, 3727–3729. [Google Scholar] [CrossRef]
- Moss, R.A.; Wang, L.; Cang, H.; Krogh-Jespersen, K. Extremely reactive carbenes: Electrophiles and nucleophiles. J. Phys. Org. Chem. 2017, 30, e3555. [Google Scholar] [CrossRef]
- Jabłoński, M. The first theoretical proof of the existence of a hydride-carbene bond. Chem. Phys. Lett. 2018, 710, 78–83. [Google Scholar] [CrossRef]
- Jabłoński, M. In search for a hydride-carbene bond. J. Phys. Org. Chem. 2019, 32, e3949. [Google Scholar] [CrossRef]
- Yourdkhani, S.; Jabłoński, M. Physical nature of silane⋯carbene dimers revealed by state-of-the-art ab initio calculations. J. Comput. Chem. 2019, 40, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen Bonding: Fundamentals and Applications; Mingos, D.M.P., Ed.; Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2008. [Google Scholar]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically-driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7757. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, W.; Jin, W.J. σ-Hole Bond vs. π-Hole Bond: A Comparison Based on Halogen Bond. Chem. Rev. 2016, 116, 5072–5104. [Google Scholar] [CrossRef] [PubMed]
- Varadwaj, P.R.; Varadwaj, A.; Marques, H.M. Halogen Bonding: A Halogen-Centered Noncovalent Interaction Yet to Be Understood. Inorganics 2019, 7, 40. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface Electrostatic Potentials of Halogenated Methanes as Indicators of Directional Intermolecular Interactions. Int. J. Quant. Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. An Overview of σ-Hole Bonding, an Important and Widely-Occurring Noncovalent Interaction. In Practical Aspect of Computational Chemistry; Leszczynski, J., Shukla, M.K., Eds.; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef]
- Mallada, B.; Gallardo, A.; Lamanec, M.; de la Torre, B.; Špirko, V.; Hobza, P.; Jelinek, P. Real-space imaging of anisotropic charge of σ-hole by means of Kelvin probe force microscopy. Science 2021, 374, 863–867. [Google Scholar] [CrossRef]
- Sanyal, S.; Esterhuysen, C. Nature of halogen bond adducts of carbones with XCF3 (X = Cl, Br, I) species. Polyhedron 2021, 200, 115107. [Google Scholar] [CrossRef]
- Frosch, J.; Koneczny, M.; Bannenberg, T.; Tamm, M. Halogen Complexes of Anionic N-Heterocyclic Carbenes. Chem. Eur. J. 2021, 27, 4349–4363. [Google Scholar] [CrossRef] [PubMed]
- Savin, K.A. Writing Reaction Mechanisms in Organic Chemistry; Elsevier Inc.: Amsterdam, The Netherlands, 2015. [Google Scholar] [CrossRef]
- Pratihar, S.; Roy, S. Nucleophilicity and Site Selectivity of Commonly Used Arenes and Heteroarenes. J. Org. Chem. 2010, 75, 4957–4963, Erratum in J. Org. Chem. 2011, 76, 4219–4219. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules: A Quantum Theory; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring Nature and Predicting Strength of Hydrogen Bonds: A Correlation Analysis Between Atoms-in-Molecules Descriptors, Binding Energies, and Energy Components of Symmetry-Adapted Perturbation Theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Cryst. 2016, B72, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.L.; Jones, C.; Junk, P.C. Studies of the reactivity of N-heterocyclic carbenes with halogen and halide sources. New J. Chem. 2002, 26, 1296–1303. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Peverati, R.; Truhlar, D.G. Quest for a universal density functional: The accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc. A 2014, 372, 20120476. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Kozuch, S.; Martin, J.M.L. Halogen Bonds: Benchmarks and Theoretical Analysis. J. Chem. Theory Comput. 2013, 9, 1918–1931. [Google Scholar] [CrossRef] [PubMed]
- Domagała, M.; Jabłoński, M.; Dubis, A.T.; Zabel, M.; Pfitzner, A.; Palusiak, M. Testing of Exchange-Correlation Functionals of DFT for a Reliable Description of the Electron Density Distribution in Organic Molecules. Int. J. Mol. Sci. 2022, 23, 14719. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Curtiss, L.A.; McGrath, M.P.; Blandeau, J.-P.; Davis, N.E.; Binning, R.C., Jr.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; Schleyer, P.v.R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3–21+G Basis Set for First-Row Elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Schäfer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Schäfer, A.; Huber, C.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar] [CrossRef]
- Weigend, F.; Furche, F.; Ahlrichs, R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 2003, 119, 12753–12762. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 2006, 8, 1057–1065. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2019. [Google Scholar]
- Contreras, R.; Andres, J.; Safont, V.S.; Campodonico, P.; Santos, J.G. A Theoretical Study on the Relationship between Nucleophilicity and Ionization Potentials in Solution Phase. J. Phys. Chem. A 2003, 107, 5588–5593. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Perez, P. Understanding the Reactivity of Captodative Ethylenes in Polar Cycloaddition Reactions. A Theoretical Study. J. Org. Chem. 2008, 73, 4615–4624. [Google Scholar] [CrossRef] [PubMed]
- Domingo, L.R.; Pérez, P. The nucleophilicity N index in organic chemistry. Org. Biomol. Chem. 2011, 9, 7168–7175. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Maiti, B. Reactivity Dynamics in Atom–Field Interactions: A Quantum Fluid Density Functional Study. J. Phys. Chem. A 2001, 105, 169–183. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpaly, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Gazquez, J.L.; Cedillo, A.; Vela, A.J. Electrodonating and Electroaccepting Powers. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Electronegativity—A perspective. J. Mol. Model. 2018, 24, 214. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAll; Version 15.05.18; TK Gristmill Software: Overland Park, KS, USA, 2015; Available online: aim.tkgristmill.com (accessed on 17 September 2022).
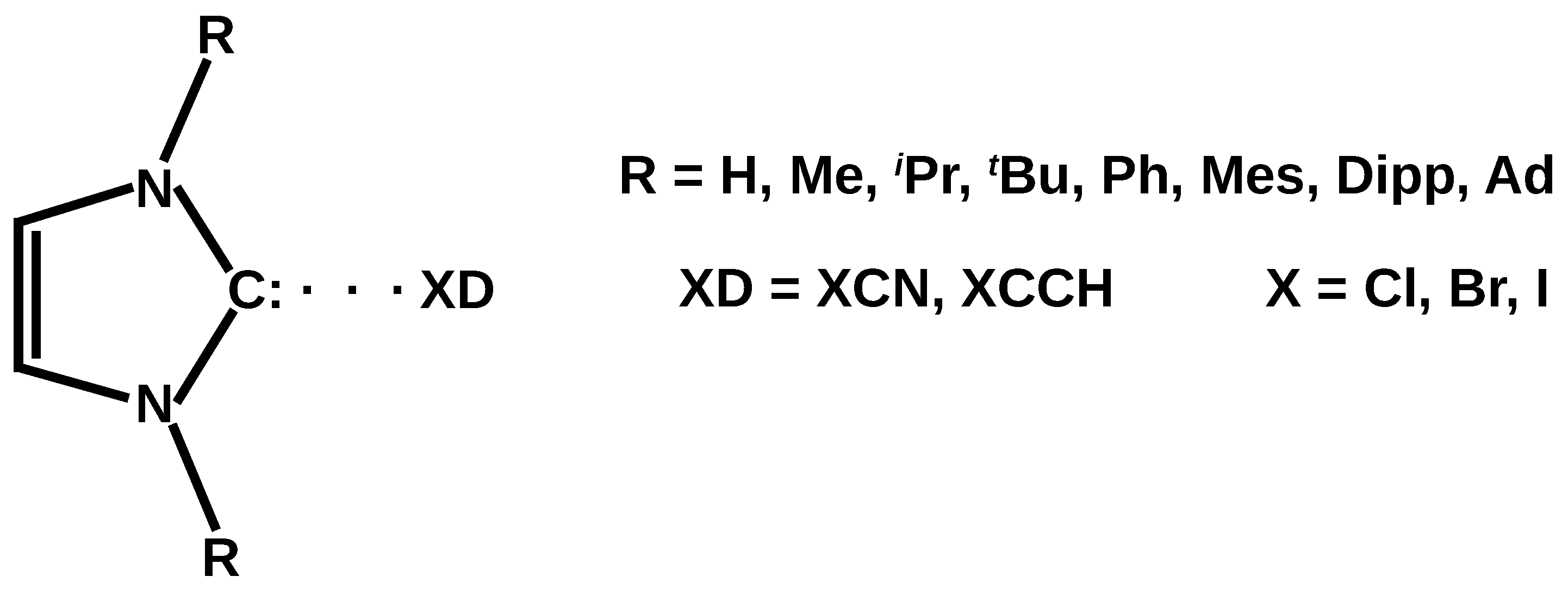


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | ||||||||
|---|---|---|---|---|---|---|---|---|
| Method | I | IMe | IPr | IBu | IPh | IMes | IDipp | IAd |
| I | 3.27 | 3.39 | 3.42 | 3.63 | 3.28 | 3.37 | 3.84 | 3.24 |
| II | 1.22 | 1.30 | 1.31 | 1.36 | 1.28 | 1.30 | 1.25 | 1.32 |
| III | 2.54 | 2.67 | 2.74 | 2.84 | 2.67 | 2.75 | 2.89 | 2.72 |
| IV | 2.39 | 2.53 | 2.58 | 2.68 | 2.52 | 2.58 | 2.68 | 2.53 |
| X-Donor | Parameters | Carbene | |||||||
|---|---|---|---|---|---|---|---|---|---|
| I | IMe | IPr | IBu | IPh | IMes | IDipp | IAd | ||
| ClCN | 5.87 | 6.63 | 6.99 | 6.11 | 6.99 | 9.26 | 8.59 | 6.78 | |
| 2.979 | 2.959 | 2.955 | 3.144 | 2.959 | 3.168 | 3.019 | 3.138 | ||
| 180.0 | 180.0 | 180.0 | 180.0 | 179.4 | 152.9 | 171.7 | 180.0 | ||
| 0.016 | 0.017 | 0.017 | 0.012 | 0.016 | 0.011 | 0.014 | 0.012 | ||
| 0.050 | 0.052 | 0.052 | 0.037 | 0.052 | 0.035 | 0.045 | 0.038 | ||
| 0.0015 | 0.0014 | 0.0014 | 0.0012 | 0.0014 | 0.0013 | 0.0014 | 0.0013 | ||
| BrCN | 8.58 | 9.59 | 10.00 | 7.23 | 9.37 | 11.18 | 10.95 | 7.84 | |
| 2.877 | 2.843 | 2.829 | 3.148 | 2.887 | 2.830 | 2.856 | 3.143 | ||
| 180.0 | 180.0 | 180.0 | 180.0 | 179.2 | 176.2 | 177.0 | 180.0 | ||
| 0.022 | 0.024 | 0.025 | 0.014 | 0.022 | 0.025 | 0.023 | 0.014 | ||
| 0.061 | 0.063 | 0.064 | 0.039 | 0.060 | 0.065 | 0.062 | 0.039 | ||
| 0.0009 | 0.0005 | 0.0004 | 0.0011 | 0.0008 | 0.0006 | 0.0007 | 0.0012 | ||
| ICN | 14.31 | 16.18 | 17.12 | 9.71 | 14.81 | 18.71 | 17.56 | 10.54 | |
| 2.640 | 2.589 | 2.552 | 3.162 | 2.703 | 2.541 | 2.601 | 3.152 | ||
| 180.0 | 180.0 | 178.2 | 180.0 | 177.8 | 179.4 | 180.0 | 180.0 | ||
| 0.044 | 0.049 | 0.052 | 0.017 | 0.039 | 0.053 | 0.047 | 0.017 | ||
| 0.074 | 0.073 | 0.073 | 0.041 | 0.071 | 0.075 | 0.073 | 0.041 | ||
| −0.0064 | −0.0089 | −0.0108 | 0.0006 | −0.0044 | −0.0109 | −0.0083 | 0.0006 | ||
| ClCCH | 3.43 | 4.02 | 4.29 | 3.93 | 4.33 | 5.97 | 5.90 | 4.47 | |
| 3.042 | 3.062 | 3.060 | 3.279 | 3.069 | 3.359 | 3.235 | 3.284 | ||
| 165.9 | 180.0 | 180.0 | 180.0 | 179.3 | 154.6 | 164.3 | 180.0 | ||
| 0.013 | 0.014 | 0.014 | 0.010 | 0.013 | 0.008 | 0.010 | 0.009 | ||
| 0.043 | 0.044 | 0.044 | 0.030 | 0.043 | 0.024 | 0.030 | 0.029 | ||
| 0.0014 | 0.0014 | 0.0014 | 0.0012 | 0.0014 | 0.0011 | 0.0012 | 0.0012 | ||
| BrCCH | 5.30 | 6.16 | 6.40 | 4.70 | 6.09 | 7.34 | 7.39 | 5.32 | |
| 3.023 | 3.004 | 2.996 | 3.319 | 3.039 | 2.999 | 3.006 | 3.324 | ||
| 177.4 | 180.0 | 180.0 | 180.0 | 179.9 | 174.8 | 177.2 | 180.0 | ||
| 0.017 | 0.018 | 0.018 | 0.010 | 0.017 | 0.017 | 0.017 | 0.010 | ||
| 0.049 | 0.051 | 0.051 | 0.029 | 0.048 | 0.051 | 0.051 | 0.029 | ||
| 0.0013 | 0.0012 | 0.0012 | 0.0011 | 0.0012 | 0.0013 | 0.0013 | 0.0011 | ||
| ICCH | 9.15 | 10.47 | 11.01 | 6.62 | 9.77 | 12.05 | 11.66 | 7.35 | |
| 2.902 | 2.861 | 2.827 | 3.372 | 2.951 | 2.822 | 2.884 | 3.371 | ||
| 178.7 | 180.0 | 177.7 | 180.0 | 178.3 | 177.9 | 179.5 | 180.0 | ||
| 0.026 | 0.029 | 0.031 | 0.012 | 0.024 | 0.030 | 0.027 | 0.012 | ||
| 0.063 | 0.065 | 0.066 | 0.030 | 0.059 | 0.068 | 0.064 | 0.030 | ||
| −0.0004 | −0.0012 | −0.0017 | 0.0009 | 0.0000 | −0.0016 | −0.0007 | 0.0009 | ||
| Carbene | |||||||
|---|---|---|---|---|---|---|---|
| I | IMe | IPr | IBu | IPh | IMes | IDipp | IAd |
| 0.964 | 0.984 | 0.993 | 0.255 | 0.937 | 0.828 | 0.932 | 0.274 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabłoński, M. Halogen Bond to Experimentally Significant N-Heterocyclic Carbenes (I, IMe2, IiPr2, ItBu2, IPh2, IMes2, IDipp2, IAd2; I = Imidazol-2-ylidene). Int. J. Mol. Sci. 2023, 24, 9057. https://doi.org/10.3390/ijms24109057
Jabłoński M. Halogen Bond to Experimentally Significant N-Heterocyclic Carbenes (I, IMe2, IiPr2, ItBu2, IPh2, IMes2, IDipp2, IAd2; I = Imidazol-2-ylidene). International Journal of Molecular Sciences. 2023; 24(10):9057. https://doi.org/10.3390/ijms24109057
Chicago/Turabian StyleJabłoński, Mirosław. 2023. "Halogen Bond to Experimentally Significant N-Heterocyclic Carbenes (I, IMe2, IiPr2, ItBu2, IPh2, IMes2, IDipp2, IAd2; I = Imidazol-2-ylidene)" International Journal of Molecular Sciences 24, no. 10: 9057. https://doi.org/10.3390/ijms24109057