
Target Therapy in Malignant Pleural Mesothelioma: Hope or Mirage?

 , , , and
, , , and 
Abstract
:1. Introduction
2. Main Molecular Targets
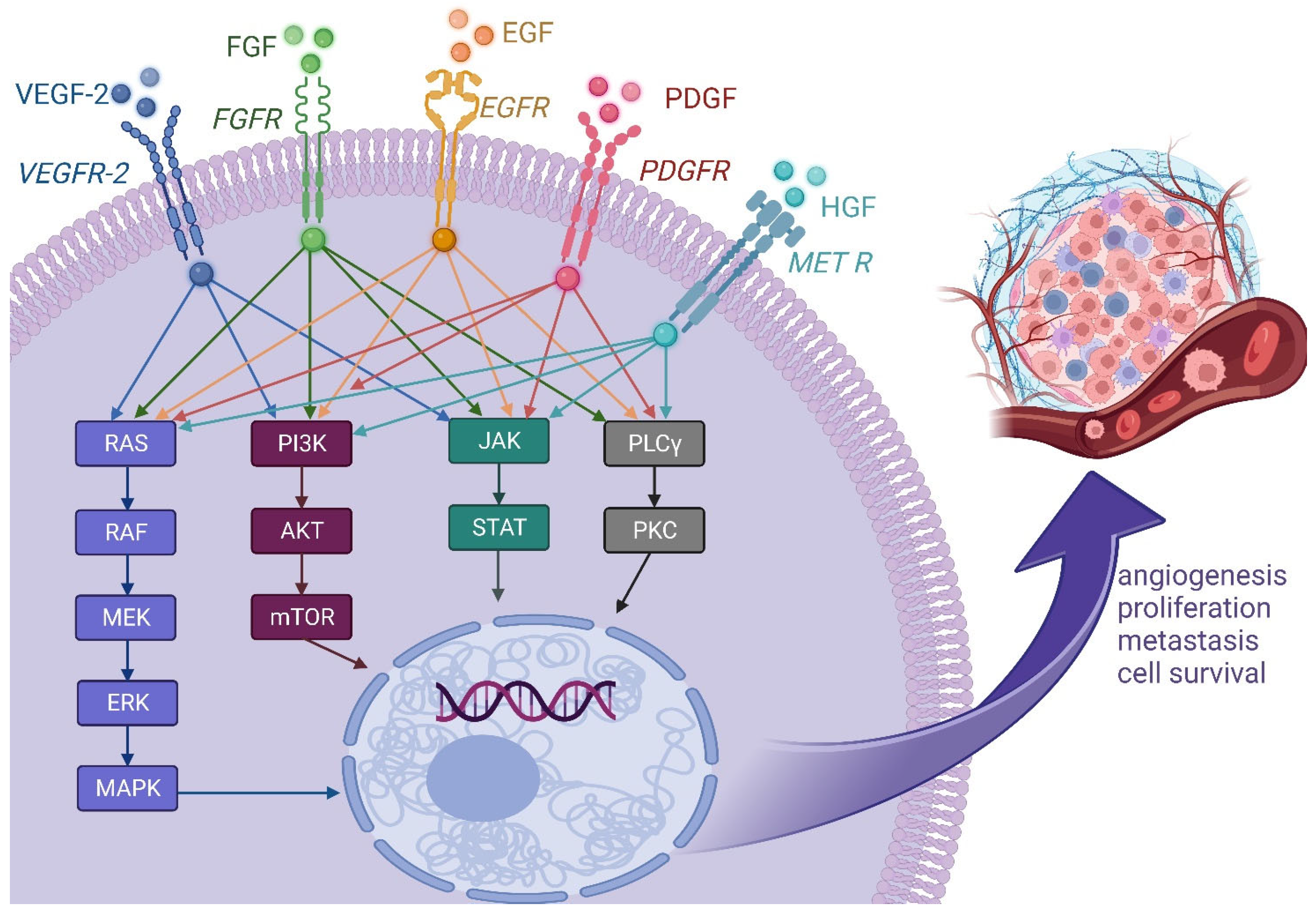
2.1. Growth Factor Pathways
2.1.1. Vascular Endothelial Growth Factor
2.1.2. Platelet-Derived Growth Factor
2.1.3. Epidermal Growth Factor Receptor
2.1.4. Fibroblast Growth Factor
2.1.5. Hepatocyte Growth Factor HGF/MET Axis
2.1.6. Multi-Kinase Inhibitors

{kind=link}
{kind=link}
| Molecular Target | Drug | Mechanism of Action | Trial | Combination | Refs. |
|---|---|---|---|---|---|
| VEGF | Bevacizumab | binds to and neutralizes all human VEGF-A isoforms | double-blind, randomized phase II control: placebo | Gemcitabine Cisplatin | [21] |
| single-arm phase II control: chemotherapy alone | Carboplatin Pemetrexed | [22] | |||
| MAPS study open-label, randomized phase III control: chemotherapy alone | Cisplatin Pemetrexed | [23] | |||
| Ramucirumab | selective against the extracellular domain of VEGFR-2 | RAMES study randomized, double-blind phase II control: placebo | Gemcitabine | [26] | |
| PDGF | Imatinib | selectively inhibits PDGFR | single-arm phase II pemetrexed-pretreated patients control: gemcitabine alone | Gemcitabine | [41] |
| phase I | Cisplatin Pemetrexed | [42] | |||
| EGFR | Gefitinib | competitively inhibits the ATP-binding pocket of EGFR | single arm phase II control: Cancer and Leukemia Group B database | - | [48] |
| Erlotinib | single arm phase II control: standard cisplatin plus pemetrexed chemotherapy database | [49] | |||
| Cetuximab | blocks the binding of activating ligands of EGFR | single-arm phase II | Cisplatin/ Carboplatin Pemetrexed | [52] | |
| FGF | GSK3052230 | sequesters FGFs and blocks their ability to activate FGFR | non-randomized, open-label phase Ib | Cisplatin Pemetrexed | [58] |
| HGF | Tivantinib | MET inhibitor | phase I dose-escalation trial | Carboplatin Pemetrexed | [66] |
| single-arm phase II trial | - | [67] | |||
| Multi-target | Cediranib | Inhibits VEGFR and PDGFR | double-blind phase II control: placebo with platinum-pemetrexed | Platinum Pemetrexed | [74] |
| Nintedanib | Inhibits VEGFR, PDGFR and FGFR | LUME-Meso randomized, double-blind phase II/III control: placebo | Cisplatin Pemetrexed | [76,77] |
2.2. Phosphatidylinositol 3-Kinase/Mammalian Target of Rapamycin/AKT
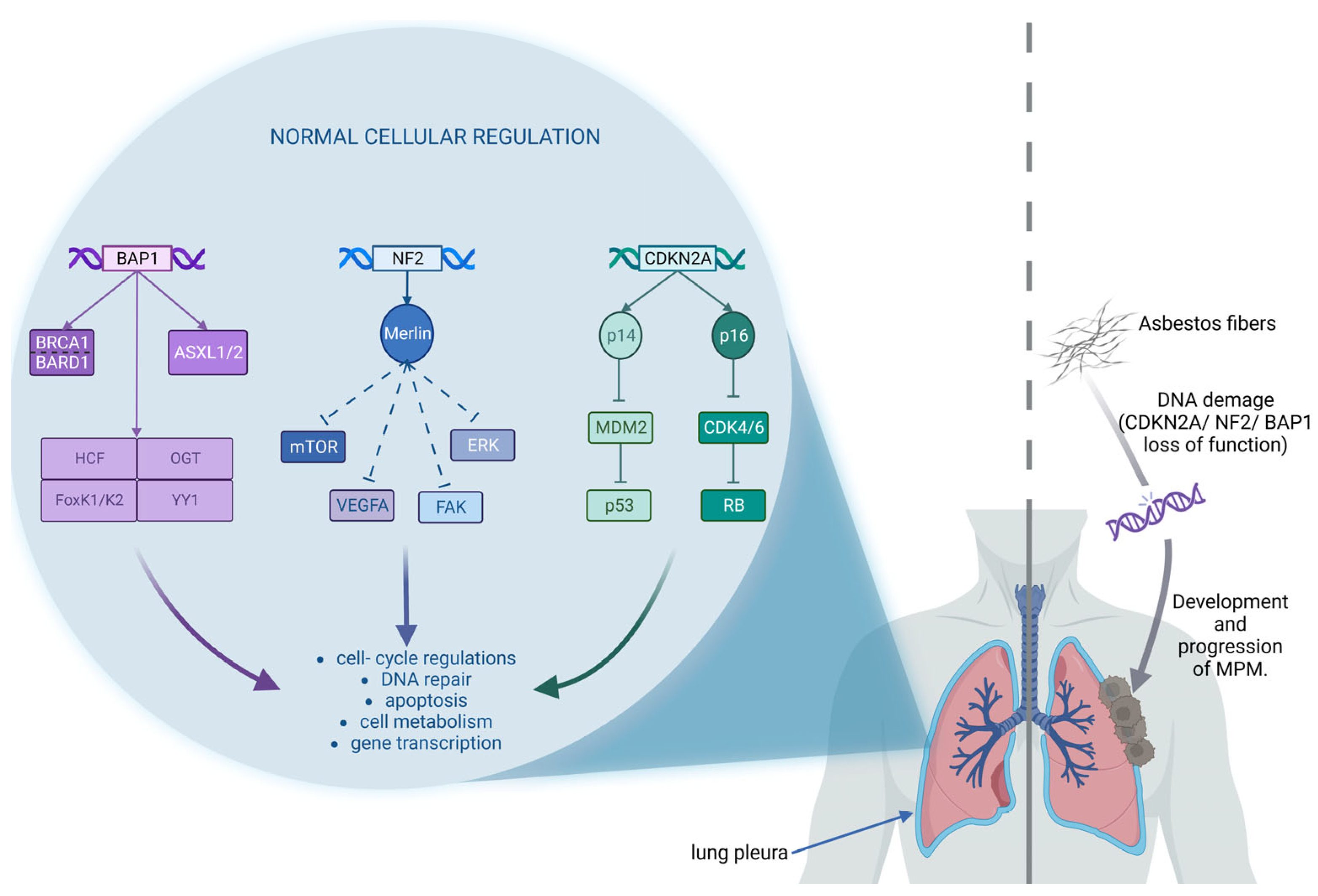
2.3. Genomic Alterations
2.3.1. CDKN2A/ARF and CDK4/6-Cyclin D1 System
2.3.2. NF2 and Focal Adhesion Kinase
2.3.3. BRCA1-Associated Protein 1 (BAP1) and DNA Damage System
2.4. Epigenetic Alterations
2.4.1. Histone Deacetylase Inhibitors
2.4.2. Enhancer of Zeste Homolog 2 (EZH2)
| Target | Drug | Trial | Refs. |
|---|---|---|---|
| CDK4/6 | Abemaciclib | MiST2: single-arm, open-label, phase II | [87] |
| FAK | GSK2256098 | phase I | [90] |
| phase I, in combination with Trametinib (MEK inhibitor) | [91] | ||
| Defactinib | COMMAND: double-blind, randomized, phase II | [92] | |
| PARP | Rucaparib | MiST1: single-arm, phase IIa | [94] |
| Olaparib | single arm, phase II | [96] | |
| Niraparib | two-cohort, phase II | [98] | |
| histone- deacetylase | Vorinostat | VANTAGE-014: double blind, phase III | [101] |
| Belinostat | single arm, phase II | [102] | |
| EZH2 | Tazemetostat | 2-parts; single arm, phase II | [105] |
2.5. Mesothelin
2.6. Arginine
3. Potential New Targets
3.1. AXL
3.2. Lactate Dehydrogenase (LDH)
3.3. Glucose Transporters (GLUT)
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Janes, S.M.; Alrifai, D.; Fennell, D.A. Perspectives on the Treatment of Malignant Pleural Mesothelioma. N. Engl. J. Med. 2021, 385, 1207–1218. [Google Scholar] [CrossRef]
- Fassina, A.; Cappellesso, R.; Guzzardo, V.; Dalla Via, L.; Piccolo, S.; Ventura, L.; Fassan, M. Epithelial–Mesenchymal Transition in Malignant Mesothelioma. Mod. Pathol. 2012, 25, 86–99. [Google Scholar] [CrossRef]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III Study of Pemetrexed in Combination with Cisplatin versus Cisplatin Alone in Patients with Malignant Pleural Mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef]
- Ceresoli, G.L.; Zucali, P.A.; Favaretto, A.G.; Grossi, F.; Bidoli, P.; Del Conte, G.; Ceribelli, A.; Bearz, A.; Morenghi, E.; Cavina, R.; et al. Phase II Study of Pemetrexed Plus Carboplatin in Malignant Pleural Mesothelioma. J. Clin. Oncol. 2006, 24, 1443–1448. [Google Scholar] [CrossRef]
- Iranzo, P.; Callejo, A.; Assaf, J.D.; Molina, G.; Lopez, D.E.; Garcia-Illescas, D.; Pardo, N.; Navarro, A.; Martinez-Marti, A.; Cedres, S.; et al. Overview of Checkpoint Inhibitors Mechanism of Action: Role of Immune-Related Adverse Events and Their Treatment on Progression of Underlying Cancer. Front. Med. 2022, 9, 875974. [Google Scholar] [CrossRef]
- Baas, P.; Scherpereel, A.; Nowak, A.K.; Fujimoto, N.; Peters, S.; Tsao, A.S.; Mansfield, A.S.; Popat, S.; Jahan, T.; Antonia, S.; et al. First-Line Nivolumab plus Ipilimumab in Unresectable Malignant Pleural Mesothelioma (CheckMate 743): A Multicentre, Randomised, Open-Label, Phase 3 Trial. Lancet 2021, 397, 375–386. [Google Scholar] [CrossRef]
- Davis, A.; Ke, H.; Kao, S.; Pavlakis, N. An Update on Emerging Therapeutic Options for Malignant Pleural Mesothelioma. Lung Cancer Targets Ther. 2022, 13, 1–12. [Google Scholar] [CrossRef]
- Borcoman, E.; Le Tourneau, C. Precision Medicine Strategies in Oncology: Mixed Approaches to Matched Therapies. Future Oncol. 2018, 14, 105–109. [Google Scholar] [CrossRef]
- Zucali, P.A.; Ceresoli, G.L.; De Vincenzo, F.; Simonelli, M.; Lorenzi, E.; Gianoncelli, L.; Santoro, A. Advances in the Biology of Malignant Pleural Mesothelioma. Cancer Treat. Rev. 2011, 37, 543–558. [Google Scholar] [CrossRef]
- Fitzpatrick, D.R.; Peroni, D.J.; Bielefeldt-Ohmann, H. The Role of Growth Factors and Cytokines in the Tumorigenesis and Immunobiology of Malignant Mesothelioma. Am. J. Respir. Cell Mol. Biol. 1995, 12, 455–460. [Google Scholar] [CrossRef]
- Ray, M.; Kindler, H.L. Malignant Pleural Mesothelioma: An Update on Biomarkers and Treatment. Chest 2009, 136, 888–896. [Google Scholar] [CrossRef]
- Strizzi, L.; Catalano, A.; Vianale, G.; Orecchia, S.; Casalini, A.; Tassi, G.; Puntoni, R.; Mutti, L.; Procopio, A. Vascular Endothelial Growth Factor Is an Autocrine Growth Factor in Human Malignant Mesothelioma. J. Pathol. 2001, 193, 468–475. [Google Scholar] [CrossRef]
- Dowell, J.E.; Kindler, H.L. Antiangiogenic Therapies for Mesothelioma. Hematol. Oncol. Clin. N. Am. 2005, 19, 1137–1145. [Google Scholar] [CrossRef]
- Holmes, D.I.; Zachary, I. The Vascular Endothelial Growth Factor (VEGF) Family: Angiogenic Factors in Health and Disease. Genome Biol. 2005, 6, 209. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The Biology of VEGF and Its Receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Zachary, I. VEGF Signalling: Integration and Multi-Tasking in Endothelial Cell Biology. Biochem. Soc. Trans. 2003, 31, 1171–1177. [Google Scholar] [CrossRef]
- Cao, Y. Antiangiogenic Cancer Therapy. Semin. Cancer Biol. 2004, 14, 139–145. [Google Scholar] [CrossRef]
- Kumar-Singh, S.; Weyler, J.; Martin, M.J.H.; Vermeulen, P.B.; Van Marck, E. Angiogenic Cytokines in Mesothelioma: A Study of VEGF, FGF-1 and -2, and TGF? Expression. J. Pathol. 1999, 189, 72–78. [Google Scholar] [CrossRef]
- Ansari, M.J.; Bokov, D.; Markov, A.; Jalil, A.T.; Shalaby, M.N.; Suksatan, W.; Chupradit, S.; AL-Ghamdi, H.S.; Shomali, N.; Zamani, A.; et al. Cancer Combination Therapies by Angiogenesis Inhibitors; a Comprehensive Review. Cell Commun. Signal. 2022, 20, 49. [Google Scholar] [CrossRef]
- Favoni, R.E.; Daga, A.; Malatesta, P.; Florio, T. Preclinical Studies Identify Novel Targeted Pharmacological Strategies for Treatment of Human Malignant Pleural Mesothelioma. Br. J. Pharm. 2012, 166, 532–553. [Google Scholar] [CrossRef]
- Kindler, H.L.; Karrison, T.G.; Gandara, D.R.; Lu, C.; Krug, L.M.; Stevenson, J.P.; Jänne, P.A.; Quinn, D.I.; Koczywas, M.N.; Brahmer, J.R.; et al. Multicenter, Double-Blind, Placebo-Controlled, Randomized Phase II Trial of Gemcitabine/Cisplatin Plus Bevacizumab or Placebo in Patients With Malignant Mesothelioma. J. Clin. Oncol. 2012, 30, 2509–2515. [Google Scholar] [CrossRef]
- Ceresoli, G.L.; Zucali, P.A.; Mencoboni, M.; Botta, M.; Grossi, F.; Cortinovis, D.; Zilembo, N.; Ripa, C.; Tiseo, M.; Favaretto, A.G.; et al. Phase II Study of Pemetrexed and Carboplatin plus Bevacizumab as First-Line Therapy in Malignant Pleural Mesothelioma. Br. J. Cancer 2013, 109, 552–558. [Google Scholar] [CrossRef]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for Newly Diagnosed Pleural Mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A Randomised, Controlled, Open-Label, Phase 3 Trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular Endothelial Growth Factor Receptor 2 Mediates Macrophage Infiltration into Orthotopic Pancreatic Tumors in Mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar] [CrossRef]
- Falcon, B.L.; Chintharlapalli, S.; Uhlik, M.T.; Pytowski, B. Antagonist Antibodies to Vascular Endothelial Growth Factor Receptor 2 (VEGFR-2) as Anti-Angiogenic Agents. Pharmacol. Ther. 2016, 164, 204–225. [Google Scholar] [CrossRef]
- Pinto, C.; Zucali, P.A.; Pagano, M.; Grosso, F.; Pasello, G.; Garassino, M.C.; Tiseo, M.; Soto Parra, H.; Grossi, F.; Cappuzzo, F.; et al. Gemcitabine with or without Ramucirumab as Second-Line Treatment for Malignant Pleural Mesothelioma (RAMES): A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Oncol. 2021, 22, 1438–1447. [Google Scholar] [CrossRef]
- Fredriksson, L.; Li, H.; Eriksson, U. The PDGF Family: Four Gene Products Form Five Dimeric Isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef]
- Papadopoulos, N.; Lennartsson, J. The PDGF/PDGFR Pathway as a Drug Target. Mol. Asp. Med. 2018, 62, 75–88. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Lennartsson, J. Structural and Functional Properties of Platelet-Derived Growth Factor and Stem Cell Factor Receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009100. [Google Scholar] [CrossRef]
- Versnel, M.A.; Claesson-Welsh, L.; Hammacher, A.; Bouts, M.J.; van der Kwast, T.H.; Eriksson, A.; Willemsen, R.; Weima, S.M.; Hoogsteden, H.C.; Hagemeijer, A. Human Malignant Mesothelioma Cell Lines Express PDGF Beta-Receptors Whereas Cultured Normal Mesothelial Cells Express Predominantly PDGF Alpha-Receptors. Oncogene 1991, 6, 2005–2011. [Google Scholar]
- Dorai, T.; Kobayashi, H.; Holland, J.F.; Ohnuma, T. Modulation of Platelet-Derived Growth Factor-Beta MRNA Expression and Cell Growth in a Human Mesothelioma Cell Line by a Hammerhead Ribozyme. Mol. Pharm. 1994, 46, 437–444. [Google Scholar]
- Melaiu, O.; Catalano, C.; De Santi, C.; Cipollini, M.; Figlioli, G.; Pellè, L.; Barone, E.; Evangelista, M.; Guazzelli, A.; Boldrini, L.; et al. Inhibition of the Platelet-Derived Growth Factor Receptor Beta (PDGFRB) Using Gene Silencing, Crenolanib Besylate, or Imatinib Mesylate Hampers the Malignant Phenotype of Mesothelioma Cell Lines. Genes Cancer 2017, 8, 438–452. [Google Scholar] [CrossRef]
- Filiberti, R.; Marroni, P.; Neri, M.; Ardizzoni, A.; Betta, P.G.; Cafferata, M.A.; Canessa, P.A.; Puntoni, R.; Ivaldi, G.P.; Paganuzzi, M. Serum PDGF-AB in Pleural Mesothelioma. Tumor Biol. 2005, 26, 221–226. [Google Scholar] [CrossRef]
- Bertino, P.; Porta, C.; Barbone, D.; Germano, S.; Busacca, S.; Pinato, S.; Tassi, G.; Favoni, R.; Gaudino, G.; Mutti, L. Preliminary Data Suggestive of a Novel Translational Approach to Mesothelioma Treatment: Imatinib Mesylate with Gemcitabine or Pemetrexed. Thorax 2007, 62, 690–695. [Google Scholar] [CrossRef]
- Pietras, K.; Rubin, K.; Sjöblom, T.; Buchdunger, E.; Sjöquist, M.; Heldin, C.-H.; Ostman, A. Inhibition of PDGF Receptor Signaling in Tumor Stroma Enhances Antitumor Effect of Chemotherapy. Cancer Res. 2002, 62, 5476–5484. [Google Scholar]
- Villano, J.L.; Husain, A.N.; Stadler, W.M.; Hanson, L.L.; Vogelzang, N.J.; Kindler, H.L. A Phase II Trial of Imatinib Mesylate in Patients (Pts) with Malignant Mesothelioma (MM). J. Clin. Oncol. 2004, 22, 7200. [Google Scholar] [CrossRef]
- Mathy, A.; Baas, P.; Dalesio, O.; Zandwijk, N. van Limited Efficacy of Imatinib Mesylate in Malignant Mesothelioma: A Phase II Trial. Lung Cancer 2005, 50, 83–86. [Google Scholar] [CrossRef]
- Bertino, P.; Piccardi, F.; Porta, C.; Favoni, R.; Cilli, M.; Mutti, L.; Gaudino, G. Imatinib Mesylate Enhances Therapeutic Effects of Gemcitabine in Human Malignant Mesothelioma Xenografts. Clin. Cancer Res. 2008, 14, 541–548. [Google Scholar] [CrossRef]
- George, S.; Desai, J.; Paul Eder, J.; Manola, J.; Ryan, D.P.; Appleman, L.J.; Demetri, G.D. Selective Kinase Inhibition with Daily Imatinib Intensifies Toxicity of Chemotherapy in Patients with Solid Tumours. Eur. J. Cancer 2006, 42, 864–870. [Google Scholar] [CrossRef]
- Ali, Y.; Lin, Y.; Gharibo, M.M.; Gounder, M.K.; Stein, M.N.; Lagattuta, T.F.; Egorin, M.J.; Rubin, E.H.; Poplin, E.A. Phase I and Pharmacokinetic Study of Imatinib Mesylate (Gleevec) and Gemcitabine in Patients with Refractory Solid Tumors. Clin. Cancer Res. 2007, 13, 5876–5882. [Google Scholar] [CrossRef]
- Zucali, P.A.; Perrino, M.; De Vincenzo, F.; Giordano, L.; Cordua, N.; D’Antonio, F.; Santoro, A. A Phase II Study of the Combination of Gemcitabine and Imatinib Mesylate in Pemetrexed-Pretreated Patients with Malignant Pleural Mesothelioma. Lung Cancer 2020, 142, 132–137. [Google Scholar] [CrossRef]
- Tsao, A.S.; Harun, N.; Lee, J.J.; Heymach, J.; Pisters, K.; Hong, W.K.; Fujimoto, J.; Wistuba, I. Phase I Trial of Cisplatin, Pemetrexed, and Imatinib Mesylate in Chemonaive Patients With Unresectable Malignant Pleural Mesothelioma. Clin. Lung Cancer 2014, 15, 197–201. [Google Scholar] [CrossRef]
- Jänne, P.A.; Taffaro, M.L.; Salgia, R.; Johnson, B.E. Inhibition of Epidermal Growth Factor Receptor Signaling in Malignant Pleural Mesothelioma. Cancer Res. 2002, 62, 5242–5247. [Google Scholar] [PubMed]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef]
- Mórocz, I.; Schmitter, D.; Lauber, B.; Stahel, R. Autocrine Stimulation of a Human Lung Mesothelioma Cell Line Is Mediated through the Transforming Growth Factor Alpha/Epidermal Growth Factor Receptor Mitogenic Pathway. Br. J. Cancer 1994, 70, 850–856. [Google Scholar] [CrossRef]
- Giovannetti, E.; Zucali, P.A.; Assaraf, Y.G.; Leon, L.G.; Smid, K.; Alecci, C.; Giancola, F.; Destro, A.; Gianoncelli, L.; Lorenzi, E.; et al. Preclinical Emergence of Vandetanib as a Potent Antitumour Agent in Mesothelioma: Molecular Mechanisms Underlying Its Synergistic Interaction with Pemetrexed and Carboplatin. Br. J. Cancer 2011, 105, 1542–1553. [Google Scholar] [CrossRef]
- Cole, G.W.; Alleva, A.M.; Reddy, R.M.; Maxhimer, J.B.; Zuo, J.; Schrump, D.S.; Nguyen, D.M. The Selective Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor PD153035 Suppresses Expression of Prometastasis Phenotypes in Malignant Pleural Mesothelioma Cells in Vitro. J. Thorac. Cardiovasc. Surg. 2005, 129, 1010–1017. [Google Scholar] [CrossRef]
- Govindan, R.; Kratzke, R.A.; Herndon, J.E.; Niehans, G.A.; Vollmer, R.; Watson, D.; Green, M.R.; Kindler, H.L. Gefitinib in Patients with Malignant Mesothelioma: A Phase II Study by the Cancer and Leukemia Group B. Clin. Cancer Res. 2005, 11, 2300–2304. [Google Scholar] [CrossRef]
- Garland, L.L.; Rankin, C.; Gandara, D.R.; Rivkin, S.E.; Scott, K.M.; Nagle, R.B.; Klein-Szanto, A.J.P.; Testa, J.R.; Altomare, D.A.; Borden, E.C. Phase II Study of Erlotinib in Patients With Malignant Pleural Mesothelioma: A Southwest Oncology Group Study. J. Clin. Oncol. 2007, 25, 2406–2413. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, K.; Arao, T.; Shimoyama, T.; Tamura, T.; Nishio, K. Antibody-Dependent Cellular Cytotoxicity of Cetuximab against Tumor Cells with Wild-Type or Mutant Epidermal Growth Factor Receptor. Cancer Sci. 2007, 98, 1275–1280. [Google Scholar] [CrossRef]
- Kurai, J.; Chikumi, H.; Hashimoto, K.; Takata, M.; Sako, T.; Yamaguchi, K.; Kinoshita, N.; Watanabe, M.; Touge, H.; Makino, H.; et al. Therapeutic Antitumor Efficacy of Anti-Epidermal Growth Factor Receptor Antibody, Cetuximab, against Malignant Pleural Mesothelioma. Int. J. Oncol. 2012, 41, 1610–1618. [Google Scholar] [CrossRef]
- De Paepe, A.; Vermaelen, K.Y.; Cornelissen, R.; Germonpre, P.R.; Janssens, A.; Lambrechts, M.; Bootsma, G.; Van Meerbeeck, J.P.; Surmont, V. Cetuximab plus Platinum-Based Chemotherapy in Patients with Malignant Pleural Mesothelioma: A Single Arm Phase II Trial. J. Clin. Oncol. 2017, 35, e20030. [Google Scholar] [CrossRef]
- Giacomini, A.; Chiodelli, P.; Matarazzo, S.; Rusnati, M.; Presta, M.; Ronca, R. Blocking the FGF/FGFR System as a “Two-Compartment” Antiangiogenic/Antitumor Approach in Cancer Therapy. Pharm. Res. 2016, 107, 172–185. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor Signaling Pathway. WIREs Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef]
- Szymczyk, J.; Sluzalska, K.D.; Materla, I.; Opalinski, L.; Otlewski, J.; Zakrzewska, M. FGF/FGFR-Dependent Molecular Mechanisms Underlying Anti-Cancer Drug Resistance. Cancers 2021, 13, 5796. [Google Scholar] [CrossRef]
- Schelch, K.; Hoda, M.A.; Klikovits, T.; Münzker, J.; Ghanim, B.; Wagner, C.; Garay, T.; Laszlo, V.; Setinek, U.; Dome, B.; et al. Fibroblast Growth Factor Receptor Inhibition Is Active against Mesothelioma and Synergizes with Radio- and Chemotherapy. Am. J. Respir. Crit. Care Med. 2014, 190, 763–772. [Google Scholar] [CrossRef]
- Schelch, K.; Wagner, C.; Hager, S.; Pirker, C.; Siess, K.; Lang, E.; Lin, R.; Kirschner, M.B.; Mohr, T.; Brcic, L.; et al. FGF2 and EGF Induce Epithelial–Mesenchymal Transition in Malignant Pleural Mesothelioma Cells via a MAPKinase/MMP1 Signal. Carcinogenesis 2018, 39, 534–545. [Google Scholar] [CrossRef]
- Van Brummelen, E.M.J.; Levchenko, E.; Dómine, M.; Fennell, D.A.; Kindler, H.L.; Viteri, S.; Gadgeel, S.; López, P.G.; Kostorov, V.; Morgensztern, D.; et al. A Phase Ib Study of GSK3052230, an FGF Ligand Trap in Combination with Pemetrexed and Cisplatin in Patients with Malignant Pleural Mesothelioma. Investig. New Drugs 2020, 38, 457–467. [Google Scholar] [CrossRef]
- Available online: Https://Clinicaltrials.Gov/ (accessed on 2 May 2023).
- Moosavi, F.; Giovannetti, E.; Peters, G.J.; Firuzi, O. Combination of HGF/MET-Targeting Agents and Other Therapeutic Strategies in Cancer. Crit. Rev. Oncol. Hematol. 2021, 160, 103234. [Google Scholar] [CrossRef]
- Gaudino, G.; Yang, H.; Carbone, M. HGF/Met Signaling Is a Key Player in Malignant Mesothelioma Carcinogenesis. Biomedicines 2014, 2, 327–344. [Google Scholar] [CrossRef]
- Tolnay, E.; Kuhnen, C.; Wiethege, T.; König, J.-E.; Voss, B.; Müller, K.-M. Hepatocyte Growth Factor/Scatter Factor and Its Receptor c-Met Are Overexpressed and Associated with an Increased Microvessel Density in Malignant Pleural Mesothelioma. J. Cancer Res. Clin. Oncol. 1998, 124, 291–296. [Google Scholar] [CrossRef]
- Katayama, R.; Aoyama, A.; Yamori, T.; Qi, J.; Oh-hara, T.; Song, Y.; Engelman, J.A.; Fujita, N. Cytotoxic Activity of Tivantinib (ARQ 197) Is Not Due Solely to c-MET Inhibition. Cancer Res. 2013, 73, 3087–3096. [Google Scholar] [CrossRef]
- Suraokar, M.B.; Nunez, M.I.; Diao, L.; Chow, C.W.; Kim, D.; Behrens, C.; Lin, H.; Lee, S.; Raso, G.; Moran, C.; et al. Expression Profiling Stratifies Mesothelioma Tumors and Signifies Deregulation of Spindle Checkpoint Pathway and Microtubule Network with Therapeutic Implications. Ann. Oncol. 2014, 25, 1184–1192. [Google Scholar] [CrossRef]
- Leon, L.; Gemelli, M.; Sciarrillo, R.; Avan, A.; Funel, N.; Giovannetti, E. Synergistic Activity of the C-Met and Tubulin Inhibitor Tivantinib (ARQ197) with Pemetrexed in Mesothelioma Cells. Curr. Drug. Targets 2014, 15, 1331–1340. [Google Scholar] [CrossRef]
- Zucali, P.A.; Simonelli, M.; De Vincenzo, F.; Fatuzzo, G.; Bertossi, M.; Perrino, M.; Miggiano, C.; Giordano, L.; Bonifacio, C.; Mrakic Sposta, F.; et al. Phase I Trial of Tivantinib in Combination with Carboplatin and Pemetrexed as First-Line Treatment in Patients with Advanced Nonsquamous Non Small Cell Lung Cancer or Malignant Pleural Mesothelioma. J. Clin. Oncol. 2015, 33, 2549. [Google Scholar] [CrossRef]
- Maron, S.B.; Karrison, T.; Kanteti, R.; Rao, K.A.; Gandara, D.R.; Koczywas, M.; Salgia, R.; Kindler, H.L. ARQ 197 in Patients with Previously-Treated Malignant Mesothelioma (MM): A Phase II Trial from the University of Chicago Phase II Consortium. J. Clin. Oncol. 2015, 33, 7511. [Google Scholar] [CrossRef]
- Jahan, T.; Gu, L.; Kratzke, R.; Dudek, A.; Otterson, G.A.; Wang, X.; Green, M.; Vokes, E.E.; Kindler, H.L. Vatalanib in Malignant Mesothelioma: A Phase II Trial by the Cancer and Leukemia Group B (CALGB 30107). Lung Cancer 2012, 76, 393–396. [Google Scholar] [CrossRef]
- Dudek, A.Z.; Pang, H.; Kratzke, R.A.; Otterson, G.A.; Hodgson, L.; Vokes, E.E.; Kindler, H.L. Phase II Study of Dasatinib in Patients with Previously Treated Malignant Mesothelioma (Cancer and Leukemia Group B 30601): A Brief Report. J. Thorac. Oncol. 2012, 7, 755–759. [Google Scholar] [CrossRef]
- Nowak, A.K.; Millward, M.J.; Creaney, J.; Francis, R.J.; Dick, I.M.; Hasani, A.; van der Schaaf, A.; Segal, A.; Musk, A.W.; Byrne, M.J. A Phase II Study of Intermittent Sunitinib Malate as Second-Line Therapy in Progressive Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2012, 7, 1449–1456. [Google Scholar] [CrossRef]
- Laurie, S.A.; Gupta, A.; Chu, Q.; Lee, C.W.; Morzycki, W.; Feld, R.; Foo, A.H.; Seely, J.; Goffin, J.R.; Laberge, F.; et al. Brief Report: A Phase II Study of Sunitinib in Malignant Pleural Mesothelioma. The NCIC Clinical Trials Group. J. Thorac. Oncol. 2011, 6, 1950–1954. [Google Scholar] [CrossRef]
- Papa, S.; Popat, S.; Shah, R.; Prevost, A.T.; Lal, R.; McLennan, B.; Cane, P.; Lang-Lazdunski, L.; Viney, Z.; Dunn, J.T.; et al. Phase 2 Study of Sorafenib in Malignant Mesothelioma Previously Treated with Platinum-Containing Chemotherapy. J. Thorac. Oncol. 2013, 8, 783–787. [Google Scholar] [CrossRef]
- Garland, L.L.; Chansky, K.; Wozniak, A.J.; Tsao, A.S.; Gadgeel, S.M.; Verschraegen, C.F.; DaSilva, M.A.; Redman, M.; Gandara, D.R. Phase II Study of Cediranib in Patients with Malignant Pleural Mesothelioma: SWOG S0509. J. Thorac. Oncol. 2011, 6, 1938–1945. [Google Scholar] [CrossRef]
- Tsao, A.S.; Miao, J.; Wistuba, I.I.; Vogelzang, N.J.; Heymach, J.V.; Fossella, F.V.; Lu, C.; Velasco, M.R.; Box-Noriega, B.; Hueftle, J.G.; et al. Phase II Trial of Cediranib in Combination With Cisplatin and Pemetrexed in Chemotherapy-Naïve Patients With Unresectable Malignant Pleural Mesothelioma (SWOG S0905). J. Clin. Oncol. 2019, 37, 2537–2547. [Google Scholar] [CrossRef]
- Laszlo, V.; Valko, Z.; Kovacs, I.; Ozsvar, J.; Hoda, M.A.; Klikovits, T.; Lakatos, D.; Czirok, A.; Garay, T.; Stiglbauer, A.; et al. Nintedanib Is Active in Malignant Pleural Mesothelioma Cell Models and Inhibits Angiogenesis and Tumor Growth In Vivo. Clin. Cancer Res. 2018, 24, 3729–3740. [Google Scholar] [CrossRef]
- Grosso, F.; Steele, N.; Novello, S.; Nowak, A.K.; Popat, S.; Greillier, L.; John, T.; Leighl, N.B.; Reck, M.; Taylor, P.; et al. Nintedanib Plus Pemetrexed/Cisplatin in Patients With Malignant Pleural Mesothelioma: Phase II Results From the Randomized, Placebo-Controlled LUME-Meso Trial. J. Clin. Oncol. 2017, 35, 3591–3600. [Google Scholar] [CrossRef]
- Scagliotti, G.V.; Gaafar, R.; Nowak, A.K.; Nakano, T.; van Meerbeeck, J.; Popat, S.; Vogelzang, N.J.; Grosso, F.; Aboelhassan, R.; Jakopovic, M.; et al. Nintedanib in Combination with Pemetrexed and Cisplatin for Chemotherapy-Naive Patients with Advanced Malignant Pleural Mesothelioma (LUME-Meso): A Double-Blind, Randomised, Placebo-Controlled Phase 3 Trial. Lancet Respir. Med. 2019, 7, 569–580. [Google Scholar] [CrossRef]
- Ocana, A.; Vera-Badillo, F.; Al-Mubarak, M.; Templeton, A.J.; Corrales-Sanchez, V.; Diez-Gonzalez, L.; Cuenca-Lopez, M.D.; Seruga, B.; Pandiella, A.; Amir, E. Activation of the PI3K/MTOR/AKT Pathway and Survival in Solid Tumors: Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e95219. [Google Scholar] [CrossRef]
- Onen, H.; Yilmaz, A.; Alp, E.; Celik, A.; Demiroz, S.; Konac, E.; Kurul, I.; Menevse, E. EF24 and RAD001 Potentiates the Anticancer Effect of Platinum-Based Agents in Human Malignant Pleural Mesothelioma (MSTO-211H) Cells and Protects Nonmalignant Mesothelial (MET-5A) Cells. Hum. Exp. Toxicol. 2015, 34, 117–126. [Google Scholar] [CrossRef]
- Ou, S.-H.I.; Moon, J.; Garland, L.L.; Mack, P.C.; Testa, J.R.; Tsao, A.S.; Wozniak, A.J.; Gandara, D.R. SWOG S0722: Phase II Study of MTOR Inhibitor Everolimus (RAD001) in Advanced Malignant Pleural Mesothelioma (MPM). J. Thorac. Oncol. 2015, 10, 387–391. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, L.; Li, H.; Eilers, G.; Kuang, Y.; Shi, S.; Yan, Z.; Li, X.; Corson, J.M.; Meng, F.; et al. Multipoint Targeting of the PI3K/MTOR Pathway in Mesothelioma. Br. J. Cancer 2014, 110, 2479–2488. [Google Scholar] [CrossRef]
- Zauderer, M.G.; Alley, E.W.; Bendell, J.; Capelletto, E.; Bauer, T.M.; Callies, S.; Szpurka, A.M.; Kang, S.; Willard, M.D.; Wacheck, V.; et al. Phase 1 Cohort Expansion Study of LY3023414, a Dual PI3K/MTOR Inhibitor, in Patients with Advanced Mesothelioma. Investig. New Drugs 2021, 39, 1081–1088. [Google Scholar] [CrossRef]
- Olofsson, K.; Mark, J. Specificity of Asbestos-Induced Chromosomal Aberrations in Short-Term Cultured Human Mesothelial Cells. Cancer Genet. Cytogenet. 1989, 41, 33–39. [Google Scholar] [CrossRef]
- Bronte, G.; Incorvaia, L.; Rizzo, S.; Passiglia, F.; Galvano, A.; Rizzo, F.; Rolfo, C.; Fanale, D.; Listì, A.; Natoli, C.; et al. The Resistance Related to Targeted Therapy in Malignant Pleural Mesothelioma: Why Has Not the Target Been Hit Yet? Crit. Rev. Oncol. Hematol. 2016, 107, 20–32. [Google Scholar] [CrossRef]
- Vachani, A.; Moon, E.; Albelda, S.M. Gene Therapy for Mesothelioma. Curr. Treat. Options Oncol. 2011, 12, 173–180. [Google Scholar] [CrossRef]
- Robertson, K.D.; Jones, P.A. Tissue-Specific Alternative Splicing in the Human INK4a/ARF Cell Cycle Regulatory Locus. Oncogene 1999, 18, 3810–3820. [Google Scholar] [CrossRef]
- Fennell, D.A.; King, A.; Mohammed, S.; Greystoke, A.; Anthony, S.; Poile, C.; Nusrat, N.; Scotland, M.; Bhundia, V.; Branson, A.; et al. Abemaciclib in Patients with P16ink4A-Deficient Mesothelioma (MiST2): A Single-Arm, Open-Label, Phase 2 Trial. Lancet Oncol. 2022, 23, 374–381. [Google Scholar] [CrossRef]
- Ladanyi, M.; Zauderer, M.G.; Krug, L.M.; Ito, T.; McMillan, R.; Bott, M.; Giancotti, F. New Strategies in Pleural Mesothelioma: BAP1 and NF2 as Novel Targets for Therapeutic Development and Risk Assessment. Clin. Cancer Res. 2012, 18, 4485–4490. [Google Scholar] [CrossRef]
- Kanteti, R.; Mirzapoiazova, T.; Riehm, J.J.; Dhanasingh, I.; Mambetsariev, B.; Wang, J.; Kulkarni, P.; Kaushik, G.; Seshacharyulu, P.; Ponnusamy, M.P.; et al. Focal Adhesion Kinase a Potential Therapeutic Target for Pancreatic Cancer and Malignant Pleural Mesothelioma. Cancer Biol. Ther. 2018, 19, 316–327. [Google Scholar] [CrossRef]
- Soria, J.C.; Gan, H.K.; Blagden, S.P.; Plummer, R.; Arkenau, H.T.; Ranson, M.; Evans, T.R.J.; Zalcman, G.; Bahleda, R.; Hollebecque, A.; et al. A Phase I, Pharmacokinetic and Pharmacodynamic Study of GSK2256098, a Focal Adhesion Kinase Inhibitor, in Patients with Advanced Solid Tumors. Ann. Oncol. 2016, 27, 2268–2274. [Google Scholar] [CrossRef]
- Mak, G.; Soria, J.-C.; Blagden, S.P.; Plummer, R.; Fleming, R.A.; Nebot, N.; Zhang, J.; Mazumdar, J.; Rogan, D.; Gazzah, A.; et al. A Phase Ib Dose-Finding, Pharmacokinetic Study of the Focal Adhesion Kinase Inhibitor GSK2256098 and Trametinib in Patients with Advanced Solid Tumours. Br. J. Cancer 2019, 120, 975–981. [Google Scholar] [CrossRef]
- Fennell, D.A.; Baas, P.; Taylor, P.; Nowak, A.K.; Gilligan, D.; Nakano, T.; Pachter, J.A.; Weaver, D.T.; Scherpereel, A.; Pavlakis, N.; et al. Maintenance Defactinib Versus Placebo After First-Line Chemotherapy in Patients With Merlin-Stratified Pleural Mesothelioma: COMMAND—A Double-Blind, Randomized, Phase II Study. J. Clin. Oncol. 2019, 37, 790–798. [Google Scholar] [CrossRef]
- Han, A.; Purwin, T.J.; Aplin, A.E. Roles of the BAP1 Tumor Suppressor in Cell Metabolism. Cancer Res. 2021, 81, 2807–2814. [Google Scholar] [CrossRef]
- Fennell, D.A.; King, A.; Mohammed, S.; Branson, A.; Brookes, C.; Darlison, L.; Dawson, A.G.; Gaba, A.; Hutka, M.; Morgan, B.; et al. Rucaparib in Patients with BAP1-Deficient or BRCA1-Deficient Mesothelioma (MiST1): An Open-Label, Single-Arm, Phase 2a Clinical Trial. Lancet Respir. Med. 2021, 9, 593–600. [Google Scholar] [CrossRef]
- Srinivasan, G.; Sidhu, G.S.; Williamson, E.A.; Jaiswal, A.S.; Najmunnisa, N.; Wilcoxen, K.; Jones, D.; George, T.J.; Hromas, R. Synthetic Lethality in Malignant Pleural Mesothelioma with PARP1 Inhibition. Cancer Chemother. Pharm. 2017, 80, 861–867. [Google Scholar] [CrossRef]
- Ghafoor, A.; Mian, I.; Wagner, C.; Mallory, Y.; Agra, M.G.; Morrow, B.; Wei, J.S.; Khan, J.; Thomas, A.; Sengupta, M.; et al. Phase 2 Study of Olaparib in Malignant Mesothelioma and Correlation of Efficacy With Germline or Somatic Mutations in BAP1 Gene. JTO Clin. Res. Rep. 2021, 2, 100231. [Google Scholar] [CrossRef]
- George, T.J.; Lee, J.-H.; Hosein, P.J.; DeRemer, D.L.; Chatzkel, J.A.; Ramnaraign, B.H.; Rogers, S.C.; Markham, M.J.; Daily, K.C.; Ezenwajiaku, N.; et al. Results of a Phase II Trial of the PARP Inhibitor, Niraparib, in BAP1 and Other DNA Damage Response Pathway Deficient Neoplasms. J. Clin. Oncol. 2022, 40, 3122. [Google Scholar] [CrossRef]
- Passiglia, F.; Bironzo, P.; Righi, L.; Listì, A.; Arizio, F.; Novello, S.; Volante, M.; Scagliotti, G.V. A Prospective Phase II Single-Arm Study of Niraparib Plus Dostarlimab in Patients With Advanced Non–Small-Cell Lung Cancer and/or Malignant Pleural Mesothelioma, Positive for PD-L1 Expression and Germline or Somatic Mutations in the DNA Repair Genes: Rationale and Study Design. Clin. Lung Cancer 2021, 22, e63–e66. [Google Scholar] [CrossRef]
- Villar-Garea, A.; Esteller, M. Histone Deacetylase Inhibitors: Understanding a New Wave of Anticancer Agents. Int. J. Cancer 2004, 112, 171–178. [Google Scholar] [CrossRef]
- Paik, P.K.; Krug, L.M. Histone Deacetylase Inhibitors in Malignant Pleural Mesothelioma: Preclinical Rationale and Clinical Trials. J. Thorac. Oncol. 2010, 5, 275–279. [Google Scholar] [CrossRef]
- Krug, L.M.; Kindler, H.L.; Calvert, H.; Manegold, C.; Tsao, A.S.; Fennell, D.; Öhman, R.; Plummer, R.; Eberhardt, W.E.E.; Fukuoka, K.; et al. Vorinostat in Patients with Advanced Malignant Pleural Mesothelioma Who Have Progressed on Previous Chemotherapy (VANTAGE-014): A Phase 3, Double-Blind, Randomised, Placebo-Controlled Trial. Lancet Oncol. 2015, 16, 447–456. [Google Scholar] [CrossRef]
- Ramalingam, S.S.; Belani, C.P.; Ruel, C.; Frankel, P.; Gitlitz, B.; Koczywas, M.; Espinoza-Delgado, I.; Gandara, D. Phase II Study of Belinostat (PXD101), a Histone Deacetylase Inhibitor, for Second Line Therapy of Advanced Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2009, 4, 97–101. [Google Scholar] [CrossRef]
- Yang, Y.; Li, G. Post-Translational Modifications of PRC2: Signals Directing Its Activity. Epigenet. Chromatin 2020, 13, 47. [Google Scholar] [CrossRef]
- LaFave, L.M.; Béguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 Function Leads to EZH2-Dependent Transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef]
- Zauderer, M.G.; Szlosarek, P.W.; Le Moulec, S.; Popat, S.; Taylor, P.; Planchard, D.; Scherpereel, A.; Koczywas, M.; Forster, M.; Cameron, R.B.; et al. EZH2 Inhibitor Tazemetostat in Patients with Relapsed or Refractory, BAP1-Inactivated Malignant Pleural Mesothelioma: A Multicentre, Open-Label, Phase 2 Study. Lancet Oncol. 2022, 23, 758–767. [Google Scholar] [CrossRef]
- Hassan, R.; Kreitman, R.J.; Pastan, I.; Willingham, M.C. Localization of Mesothelin in Epithelial Ovarian Cancer. Appl. Immunohistochem. Mol. Morphol. 2005, 13, 243–247. [Google Scholar] [CrossRef]
- Argani, P.; Iacobuzio-Donahue, C.; Ryu, B.; Rosty, C.; Goggins, M.; Wilentz, R.E.; Murugesan, S.R.; Leach, S.D.; Jaffee, E.; Yeo, C.J.; et al. Mesothelin Is Overexpressed in the Vast Majority of Ductal Adenocarcinomas of the Pancreas: Identification of a New Pancreatic Cancer Marker by Serial Analysis of Gene Expression (SAGE). Clin. Cancer Res. 2001, 7, 3862–3868. [Google Scholar]
- Servais, E.L.; Colovos, C.; Rodriguez, L.; Bograd, A.J.; Nitadori, J.; Sima, C.; Rusch, V.W.; Sadelain, M.; Adusumilli, P.S. Mesothelin Overexpression Promotes Mesothelioma Cell Invasion and MMP-9 Secretion in an Orthotopic Mouse Model and in Epithelioid Pleural Mesothelioma Patients. Clin. Cancer Res. 2012, 18, 2478–2489. [Google Scholar] [CrossRef]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I Study of SS1P, a Recombinant Anti-Mesothelin Immunotoxin Given as a Bolus I.V. Infusion to Patients with Mesothelin-Expressing Mesothelioma, Ovarian, and Pancreatic Cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Hassan, R.; FitzGerald, D.J.; Pastan, I. Phase I Trial of Continuous Infusion Anti-Mesothelin Recombinant Immunotoxin SS1P. Clin. Cancer Res. 2009, 15, 5274–5279. [Google Scholar] [CrossRef]
- Hassan, R.; Sharon, E.; Thomas, A.; Zhang, J.; Ling, A.; Miettinen, M.; Kreitman, R.J.; Steinberg, S.M.; Hollevoet, K.; Pastan, I. Phase 1 Study of the Antimesothelin Immunotoxin SS1P in Combination with Pemetrexed and Cisplatin for Front-Line Therapy of Pleural Mesothelioma and Correlation of Tumor Response with Serum Mesothelin, Megakaryocyte Potentiating Factor, and Cancer Antigen. Cancer 2014, 120, 3311–3319. [Google Scholar] [CrossRef]
- Mossoba, M.E.; Onda, M.; Taylor, J.; Massey, P.R.; Treadwell, S.; Sharon, E.; Hassan, R.; Pastan, I.; Fowler, D.H. Pentostatin Plus Cyclophosphamide Safely and Effectively Prevents Immunotoxin Immunogenicity in Murine Hosts. Clin. Cancer Res. 2011, 17, 3697–3705. [Google Scholar] [CrossRef]
- Hassan, R.; Miller, A.C.; Sharon, E.; Thomas, A.; Reynolds, J.C.; Ling, A.; Kreitman, R.J.; Miettinen, M.M.; Steinberg, S.M.; Fowler, D.H.; et al. Major Cancer Regressions in Mesothelioma After Treatment with an Anti-Mesothelin Immunotoxin and Immune Suppression. Sci. Transl. Med. 2013, 5, 208ra147. [Google Scholar] [CrossRef]
- Hassan, R.; Alewine, C.; Mian, I.; Spreafico, A.; Siu, L.L.; Gomez-Roca, C.; Delord, J.; Italiano, A.; Lassen, U.; Soria, J.; et al. Phase 1 Study of the Immunotoxin LMB-100 in Patients with Mesothelioma and Other Solid Tumors Expressing Mesothelin. Cancer 2020, 126, 4936–4947. [Google Scholar] [CrossRef]
- Zhang, J.; Khanna, S.; Jiang, Q.; Alewine, C.; Miettinen, M.; Pastan, I.; Hassan, R. Efficacy of Anti-Mesothelin Immunotoxin RG7787 plus Nab-Paclitaxel against Mesothelioma Patient–Derived Xenografts and Mesothelin as a Biomarker of Tumor Response. Clin. Cancer Res. 2017, 23, 1564–1574. [Google Scholar] [CrossRef]
- Leshem, Y.; O’Brien, J.; Liu, X.; Bera, T.K.; Terabe, M.; Berzofsky, J.A.; Bossenmaier, B.; Niederfellner, G.; Tai, C.-H.; Reiter, Y.; et al. Combining Local Immunotoxins Targeting Mesothelin with CTLA-4 Blockade Synergistically Eradicates Murine Cancer by Promoting Anticancer Immunity. Cancer Immunol. Res. 2017, 5, 685–694. [Google Scholar] [CrossRef]
- Hassan, R.; Cohen, S.J.; Phillips, M.; Pastan, I.; Sharon, E.; Kelly, R.J.; Schweizer, C.; Weil, S.; Laheru, D. Phase I Clinical Trial of the Chimeric Anti-Mesothelin Monoclonal Antibody MORAb-009 in Patients with Mesothelin-Expressing Cancers. Clin. Cancer Res. 2010, 16, 6132–6138. [Google Scholar] [CrossRef]
- Hassan, R.; Kindler, H.L.; Jahan, T.; Bazhenova, L.; Reck, M.; Thomas, A.; Pastan, I.; Parno, J.; O’Shannessy, D.J.; Fatato, P.; et al. Phase II Clinical Trial of Amatuximab, a Chimeric Antimesothelin Antibody with Pemetrexed and Cisplatin in Advanced Unresectable Pleural Mesothelioma. Clin. Cancer Res. 2014, 20, 5927–5936. [Google Scholar] [CrossRef]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab Ravtansine: A Novel Mesothelin-Targeting Antibody–Drug Conjugate Cures Tumors with Heterogeneous Target Expression Favored by Bystander Effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef]
- Hassan, R.; Blumenschein, G.R.; Moore, K.N.; Santin, A.D.; Kindler, H.L.; Nemunaitis, J.J.; Seward, S.M.; Thomas, A.; Kim, S.K.; Rajagopalan, P.; et al. First-in-Human, Multicenter, Phase I Dose-Escalation and Expansion Study of Anti-Mesothelin Antibody–Drug Conjugate Anetumab Ravtansine in Advanced or Metastatic Solid Tumors. J. Clin. Oncol. 2020, 38, 1824–1835. [Google Scholar] [CrossRef]
- Hassan, R.; Wang, D.; Wrangle, J.; Thomas, A.; Byars, A.; Asschert, L.; Atienza, R.; Rajagopalan, P.; Walter, A.; Zhang, J.; et al. Abstract A095: Phase Ib Study of Anetumab Ravtansine in Combination with Pemetrexed and Cisplatin in Patients with Mesothelin-Expressing Epithelial Mesothelioma or Nonsquamous Non-Small Cell Lung Cancer. Mol. Cancer Ther. 2018, 17, A095. [Google Scholar] [CrossRef]
- Kindler, H.L.; Novello, S.; Bearz, A.; Ceresoli, G.L.; Aerts, J.G.J.V.; Spicer, J.; Taylor, P.; Nackaerts, K.; Greystoke, A.; Jennens, R.; et al. Anetumab Ravtansine versus Vinorelbine in Patients with Relapsed, Mesothelin-Positive Malignant Pleural Mesothelioma (ARCS-M): A Randomised, Open-Label Phase 2 Trial. Lancet Oncol. 2022, 23, 540–552. [Google Scholar] [CrossRef]
- Yeo, D.; Castelletti, L.; van Zandwijk, N.; Rasko, J.E.J. Hitting the Bull’s-Eye: Mesothelin’s Role as a Biomarker and Therapeutic Target for Malignant Pleural Mesothelioma. Cancers 2021, 13, 3932. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Klabatsa, A.; Pallaska, A.; Sheaff, M.; Smith, P.; Crook, T.; Grimshaw, M.J.; Steele, J.P.; Rudd, R.M.; Balkwill, F.R.; et al. In Vivo Loss of Expression of Argininosuccinate Synthetase in Malignant Pleural Mesothelioma Is a Biomarker for Susceptibility to Arginine Depletion. Clin. Cancer Res. 2006, 12, 7126–7131. [Google Scholar] [CrossRef]
- Phillips, M.M.; Sheaff, M.T.; Szlosarek, P.W. Targeting Arginine-Dependent Cancers with Arginine-Degrading Enzymes: Opportunities and Challenges. Cancer Res. Treat. 2013, 45, 251–262. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation With Pegylated Arginine Deiminase in Patients With Argininosuccinate Synthetase 1–Deficient Malignant Pleural Mesothelioma. JAMA Oncol. 2017, 3, 58. [Google Scholar] [CrossRef]
- Beddowes, E.; Spicer, J.; Chan, P.Y.; Khadeir, R.; Corbacho, J.G.; Repana, D.; Steele, J.P.; Schmid, P.; Szyszko, T.; Cook, G.; et al. Phase 1 Dose-Escalation Study of Pegylated Arginine Deiminase, Cisplatin, and Pemetrexed in Patients With Argininosuccinate Synthetase 1–Deficient Thoracic Cancers. J. Clin. Oncol. 2017, 35, 1778–1785. [Google Scholar] [CrossRef]
- Szlosarek, P.W.; Phillips, M.M.; Pavlyk, I.; Steele, J.; Shamash, J.; Spicer, J.; Kumar, S.; Pacey, S.; Feng, X.; Johnston, A.; et al. Expansion Phase 1 Study of Pegargiminase Plus Pemetrexed and Cisplatin in Patients With Argininosuccinate Synthetase 1–Deficient Mesothelioma: Safety, Efficacy, and Resistance Mechanisms. JTO Clin. Res. Rep. 2020, 1, 100093. [Google Scholar] [CrossRef]
- Lemke, G.; Rothlin, C.V. Immunobiology of the TAM Receptors. Nat. Rev. Immunol. 2008, 8, 327–336. [Google Scholar] [CrossRef]
- Schmidt, T.; Ben-Batalla, I.; Schultze, A.; Loges, S. Macrophage–Tumor Crosstalk: Role of TAMR Tyrosine Kinase Receptors and of Their Ligands. Cell. Mol. Life Sci. 2012, 69, 1391–1414. [Google Scholar] [CrossRef]
- Li, Y.; Ye, X.; Tan, C.; Hongo, J.-A.; Zha, J.; Liu, J.; Kallop, D.; Ludlam, M.J.C.; Pei, L. Axl as a Potential Therapeutic Target in Cancer: Role of Axl in Tumor Growth, Metastasis and Angiogenesis. Oncogene 2009, 28, 3442–3455. [Google Scholar] [CrossRef]
- Pass, H.I.; Liu, Z.; Wali, A.; Bueno, R.; Land, S.; Lott, D.; Siddiq, F.; Lonardo, F.; Carbone, M.; Draghici, S. Gene Expression Profiles Predict Survival and Progression of Pleural Mesothelioma. Clin. Cancer Res. 2004, 10, 849–859. [Google Scholar] [CrossRef]
- Song, W.; Wang, H.; Lu, M.; Ni, X.; Bahri, N.; Zhu, S.; Chen, L.; Wu, Y.; Qiu, J.; Fletcher, J.A.; et al. AXL Inactivation Inhibits Mesothelioma Growth and Migration via Regulation of P53 Expression. Cancers 2020, 12, 2757. [Google Scholar] [CrossRef]
- Oien, D.B.; Garay, T.; Eckstein, S.; Chien, J. Cisplatin and Pemetrexed Activate AXL and AXL Inhibitor BGB324 Enhances Mesothelioma Cell Death from Chemotherapy. Front. Pharm. 2018, 8, 970. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Potter, M.; Newport, E.; Morten, K.J. The Warburg Effect: 80 Years On. Biochem. Soc. Trans. 2016, 44, 1499–1505. [Google Scholar] [CrossRef]
- Zhuo, Y.; Lin, L.; Wei, S.; Zhang, M. Pretreatment Elevated Serum Lactate Dehydrogenase as a Significant Prognostic Factor in Malignant Mesothelioma. Medicine 2016, 95, e5706. [Google Scholar] [CrossRef]
- Comandatore, A.; Franczak, M.; Smolenski, R.T.; Morelli, L.; Peters, G.J.; Giovannetti, E. Lactate Dehydrogenase and Its Clinical Significance in Pancreatic and Thoracic Cancers. Semin. Cancer Biol. 2022, 86, 93–100. [Google Scholar] [CrossRef]
- Farabegoli, F.; Vettraino, M.; Manerba, M.; Fiume, L.; Roberti, M.; Di Stefano, G. Galloflavin, a New Lactate Dehydrogenase Inhibitor, Induces the Death of Human Breast Cancer Cells with Different Glycolytic Attitude by Affecting Distinct Signaling Pathways. Eur. J. Pharm. Sci. 2012, 47, 729–738. [Google Scholar] [CrossRef]
- Boudreau, A.; Purkey, H.E.; Hitz, A.; Robarge, K.; Peterson, D.; Labadie, S.; Kwong, M.; Hong, R.; Gao, M.; Del Nagro, C.; et al. Metabolic Plasticity Underpins Innate and Acquired Resistance to LDHA Inhibition. Nat. Chem. Biol. 2016, 12, 779–786. [Google Scholar] [CrossRef]
- Yang, Y.; Su, D.; Zhao, L.; Zhang, D.; Xu, J.; Wan, J.; Fan, S.; Chen, M. Different Effects of LDH-A Inhibition by Oxamate in Non-Small Cell Lung Cancer Cells. Oncotarget 2014, 5, 11886–11896. [Google Scholar] [CrossRef]
- Giovannetti, E.; Leon, L.G.; Gómez, V.E.; Zucali, P.A.; Minutolo, F.; Peters, G.J. A Specific Inhibitor of Lactate Dehydrogenase Overcame the Resistance toward Gemcitabine in Hypoxic Mesothelioma Cells, and Modulated the Expression of the Human Equilibrative Transporter-1. Nucl. Nucl. Nucleic Acids 2016, 35, 643–651. [Google Scholar] [CrossRef]
- Li Petri, G.; El Hassouni, B.; Sciarrillo, R.; Funel, N.; Mantini, G.; Zeeuw van der Laan, E.A.; Cascioferro, S.; Avan, A.; Zucali, P.A.; Zaffaroni, N.; et al. Impact of Hypoxia on Chemoresistance of Mesothelioma Mediated by the Proton-Coupled Folate Transporter, and Preclinical Activity of New Anti-LDH-A Compounds. Br. J. Cancer 2020, 123, 644–656. [Google Scholar] [CrossRef]
- Franczak, M.A.; Krol, O.; Harasim, G.; Jedrzejewska, A.; Zaffaroni, N.; Granchi, C.; Minutolo, F.; Avan, A.; Giovannetti, E.; Smolenski, R.T.; et al. Metabolic Effects of New Glucose Transporter (GLUT-1) and Lactate Dehydrogenase-A (LDH-A) Inhibitors against Chemoresistant Malignant Mesothelioma. Int. J. Mol. Sci. 2023, 24, 7771. [Google Scholar] [CrossRef]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) Family of Membrane Transporters. Mol. Asp. Med. 2013, 34, 121–138. [Google Scholar] [CrossRef]
- Hussein, Y.R.; Bandyopadhyay, S.; Semaan, A.; Ahmed, Q.; Albashiti, B.; Jazaerly, T.; Nahleh, Z.; Ali-Fehmi, R. Glut-1 Expression Correlates with Basal-like Breast Cancer. Transl. Oncol. 2011, 4, 321–327. [Google Scholar] [CrossRef]
- Zhang, T.-B.; Zhao, Y.; Tong, Z.-X.; Guan, Y.-F. Inhibition of Glucose-Transporter 1 (GLUT-1) Expression Reversed Warburg Effect in Gastric Cancer Cell MKN45. Int. J. Clin. Exp. Med. 2015, 8, 2423–2428. [Google Scholar]
- Zhang, B.; Xie, Z.; Li, B. The Clinicopathologic Impacts and Prognostic Significance of GLUT1 Expression in Patients with Lung Cancer: A Meta-Analysis. Gene 2019, 689, 76–83. [Google Scholar] [CrossRef]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The Prognostic Value of GLUT1 in Cancers: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef]
- Zhong, S.-C.; Ao, X.-J.; Yu, S.-H. Diagnostic Value of GLUT-1 in Distinguishing Malignant Mesothelioma from Reactive Mesothelial Cells: A Meta-Analysis. Biomarkers 2020, 25, 157–163. [Google Scholar] [CrossRef]
- Zhao, F.; Ming, J.; Zhou, Y.; Fan, L. Inhibition of Glut1 by WZB117 Sensitizes Radioresistant Breast Cancer Cells to Irradiation. Cancer Chemother. Pharm. 2016, 77, 963–972. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Idowu, M.; Oh, U.; Wang, X.-Y.; Temkin, S.; Fang, X. Ovarian Cancer Relies on Glucose Transporter 1 to Fuel Glycolysis and Growth: Anti-Tumor Activity of BAY-876. Cancers 2018, 11, 33. [Google Scholar] [CrossRef]
- Matsumoto, T.; Jimi, S.; Migita, K.; Takamatsu, Y.; Hara, S. Inhibition of Glucose Transporter 1 Induces Apoptosis and Sensitizes Multiple Myeloma Cells to Conventional Chemotherapeutic Agents. Leuk. Res. 2016, 41, 103–110. [Google Scholar] [CrossRef]
- Stella, G.M.; Bortolotto, C. Managing Malignant Pleural Mesothelioma in the Age of Personalized Medicine: Where Are We and What Is Still Missing? Cancers 2022, 14, 5540. [Google Scholar] [CrossRef]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive Genomic Analysis of Malignant Pleural Mesothelioma Identifies Recurrent Mutations, Gene Fusions and Splicing Alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef]
- Cortese, J.F.; Gowda, A.L.; Wali, A.; Eliason, J.F.; Pass, H.I.; Everson, R.B. Common EGFR Mutations Conferring Sensitivity to Gefitinib in Lung Adenocarcinoma Are Not Prevalent in Human Malignant Mesothelioma. Int. J. Cancer 2006, 118, 521–522. [Google Scholar] [CrossRef]
- Okuda, K.; Sasaki, H.; Kawano, O.; Yukiue, H.; Yokoyama, T.; Yano, M.; Fujii, Y. Epidermal Growth Factor Receptor Gene Mutation, Amplification and Protein Expression in Malignant Pleural Mesothelioma. J. Cancer Res. Clin. Oncol. 2008, 134, 1105–1111. [Google Scholar] [CrossRef]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. Mutations and Benefit from Cetuximab in Advanced Colorectal Cancer. N. Engl. J. Med. 2008, 359, 1757–1765. [Google Scholar] [CrossRef]
- Alfieri, R.; Giovannetti, E.; Bonelli, M.; Cavazzoni, A. New Treatment Opportunities in Phosphatase and Tensin Homolog (PTEN)-Deficient Tumors: Focus on PTEN/Focal Adhesion Kinase Pathway. Front. Oncol. 2017, 7, 170. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borea, F.; Franczak, M.A.; Garcia, M.; Perrino, M.; Cordua, N.; Smolenski, R.T.; Peters, G.J.; Dziadziuszko, R.; Santoro, A.; Zucali, P.A.; et al. Target Therapy in Malignant Pleural Mesothelioma: Hope or Mirage? Int. J. Mol. Sci. 2023, 24, 9165. https://doi.org/10.3390/ijms24119165
Borea F, Franczak MA, Garcia M, Perrino M, Cordua N, Smolenski RT, Peters GJ, Dziadziuszko R, Santoro A, Zucali PA, et al. Target Therapy in Malignant Pleural Mesothelioma: Hope or Mirage? International Journal of Molecular Sciences. 2023; 24(11):9165. https://doi.org/10.3390/ijms24119165
Chicago/Turabian StyleBorea, Federica, Marika A. Franczak, Maria Garcia, Matteo Perrino, Nadia Cordua, Ryszard T. Smolenski, Godefridus J. Peters, Rafal Dziadziuszko, Armando Santoro, Paolo A. Zucali, and et al. 2023. "Target Therapy in Malignant Pleural Mesothelioma: Hope or Mirage?" International Journal of Molecular Sciences 24, no. 11: 9165. https://doi.org/10.3390/ijms24119165
APA StyleBorea, F., Franczak, M. A., Garcia, M., Perrino, M., Cordua, N., Smolenski, R. T., Peters, G. J., Dziadziuszko, R., Santoro, A., Zucali, P. A., & Giovannetti, E. (2023). Target Therapy in Malignant Pleural Mesothelioma: Hope or Mirage? International Journal of Molecular Sciences, 24(11), 9165. https://doi.org/10.3390/ijms24119165