

Conformational Disorder Analysis of the Conditionally Disordered Protein CP12 from Arabidopsis thaliana in Its Different Redox States

 , ,
, ,  , , and
, , and 
Abstract
:
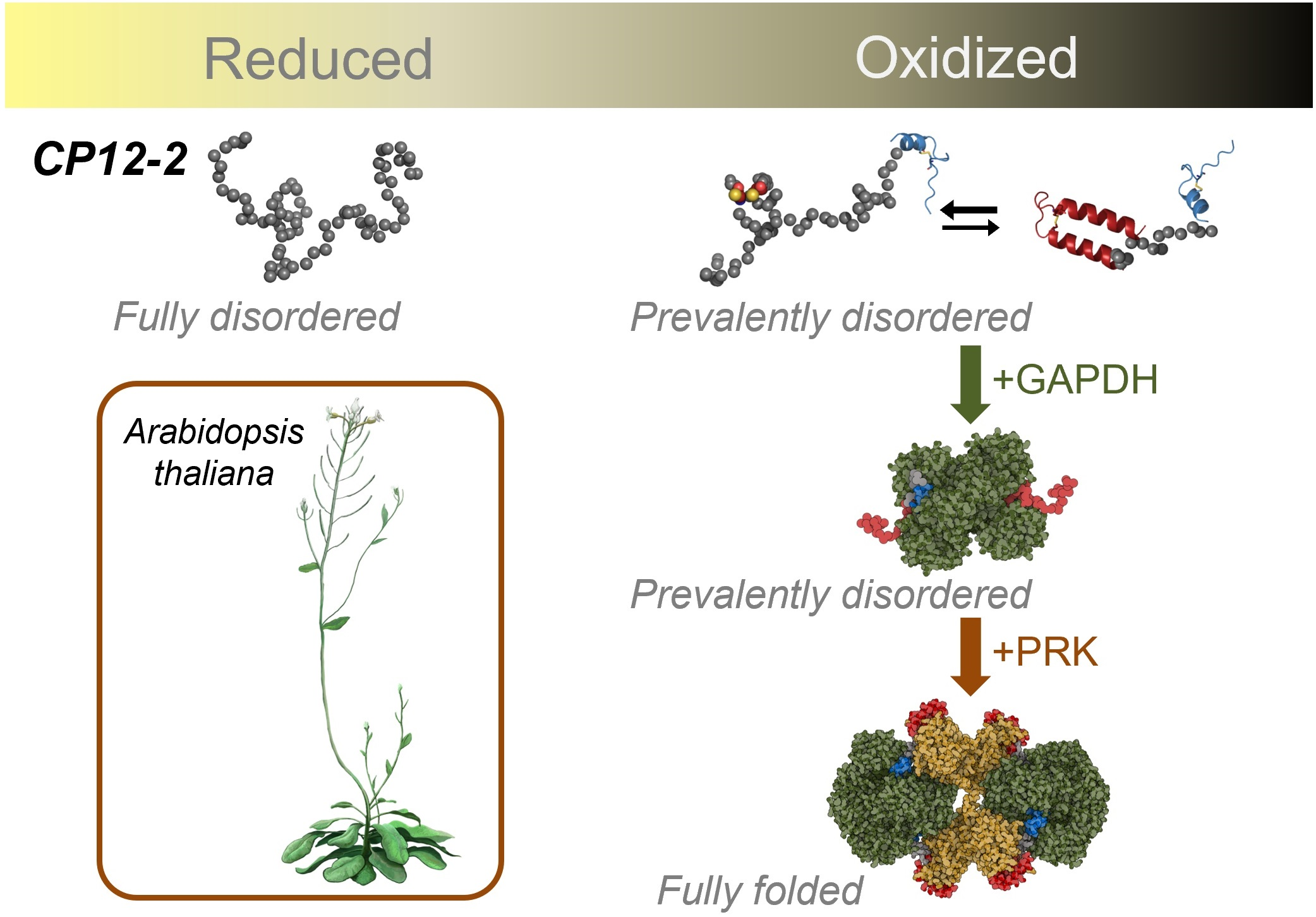{kind=link}
{kind=link}
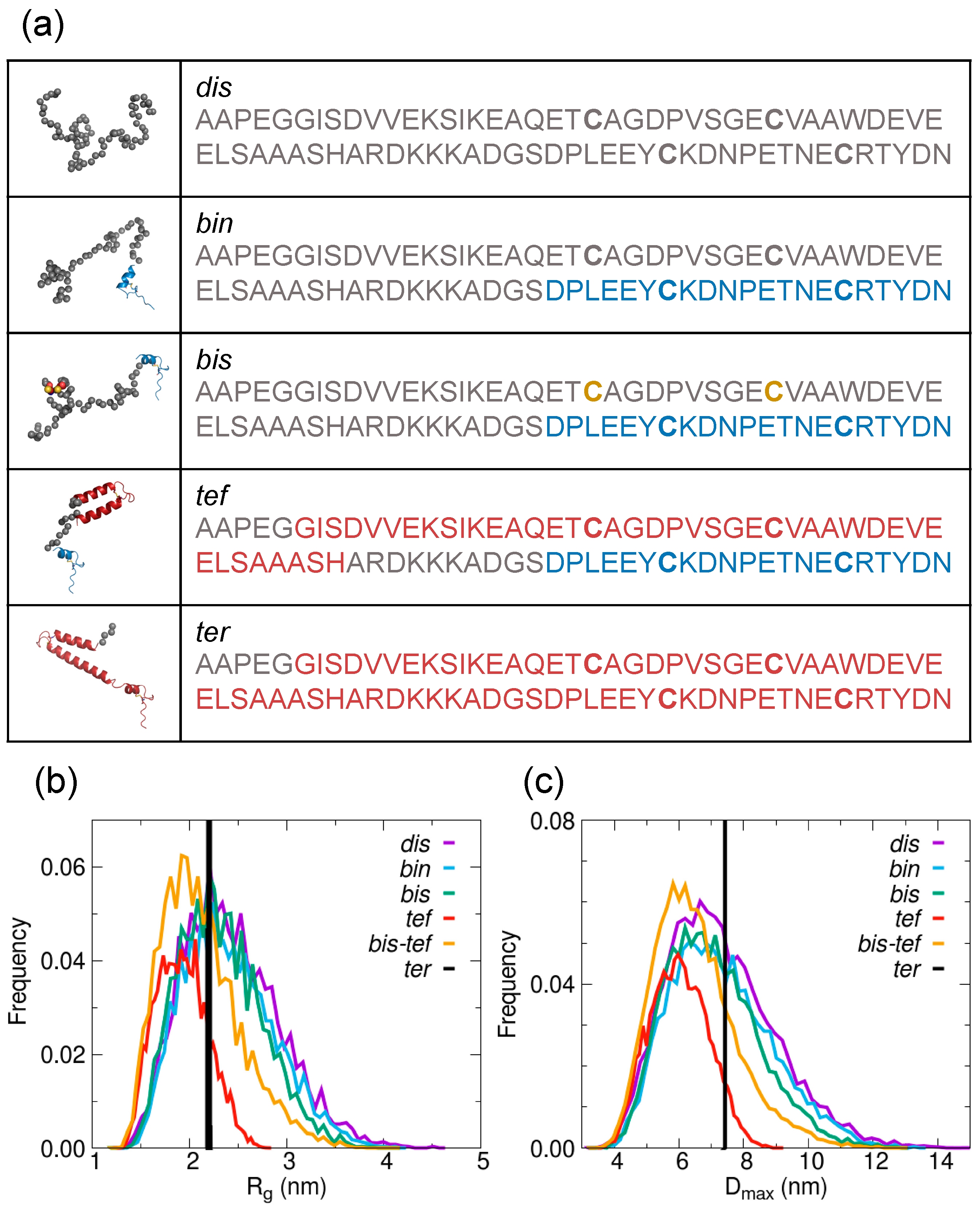
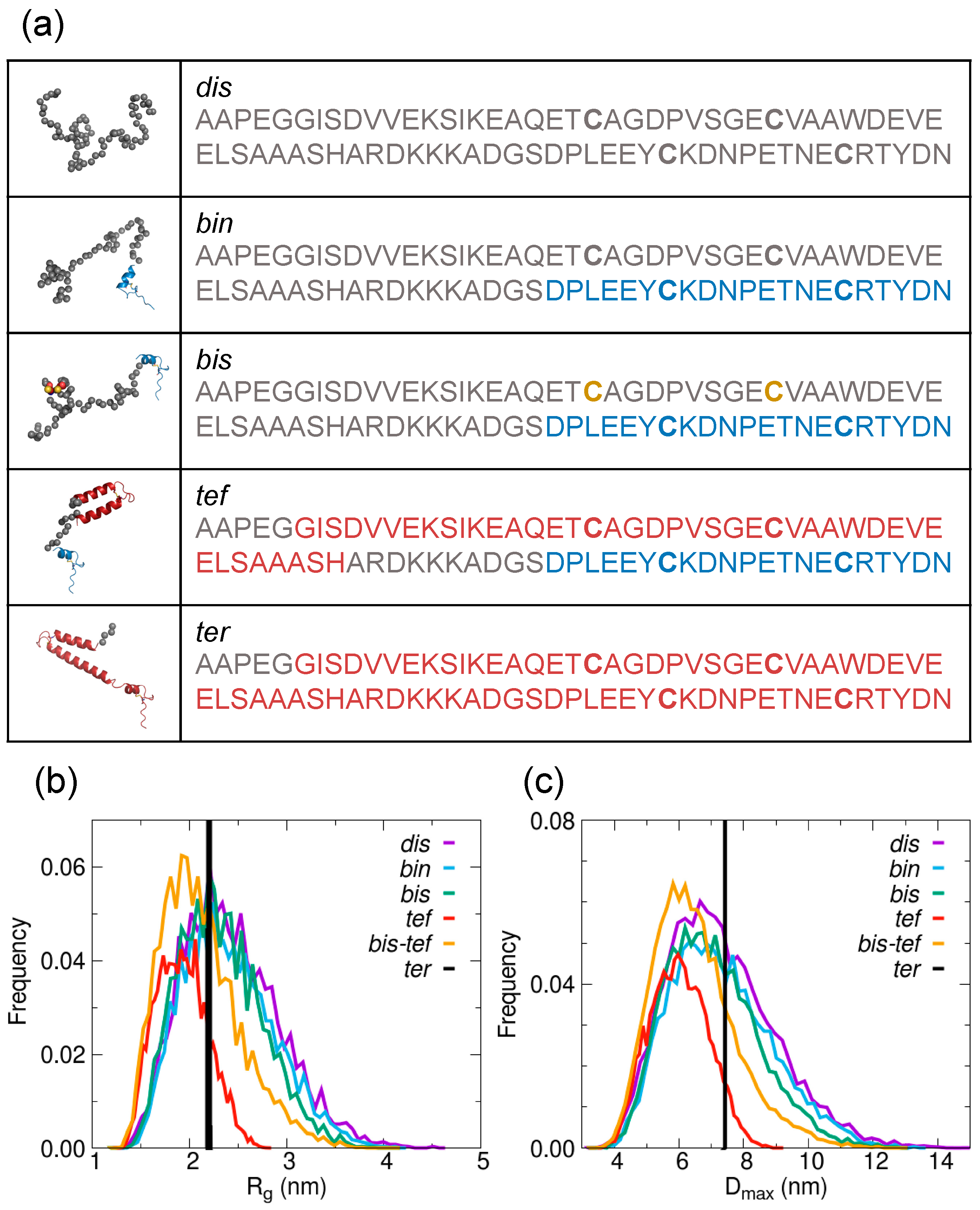{kind=link}
{kind=link}
1. Introduction
2. Results
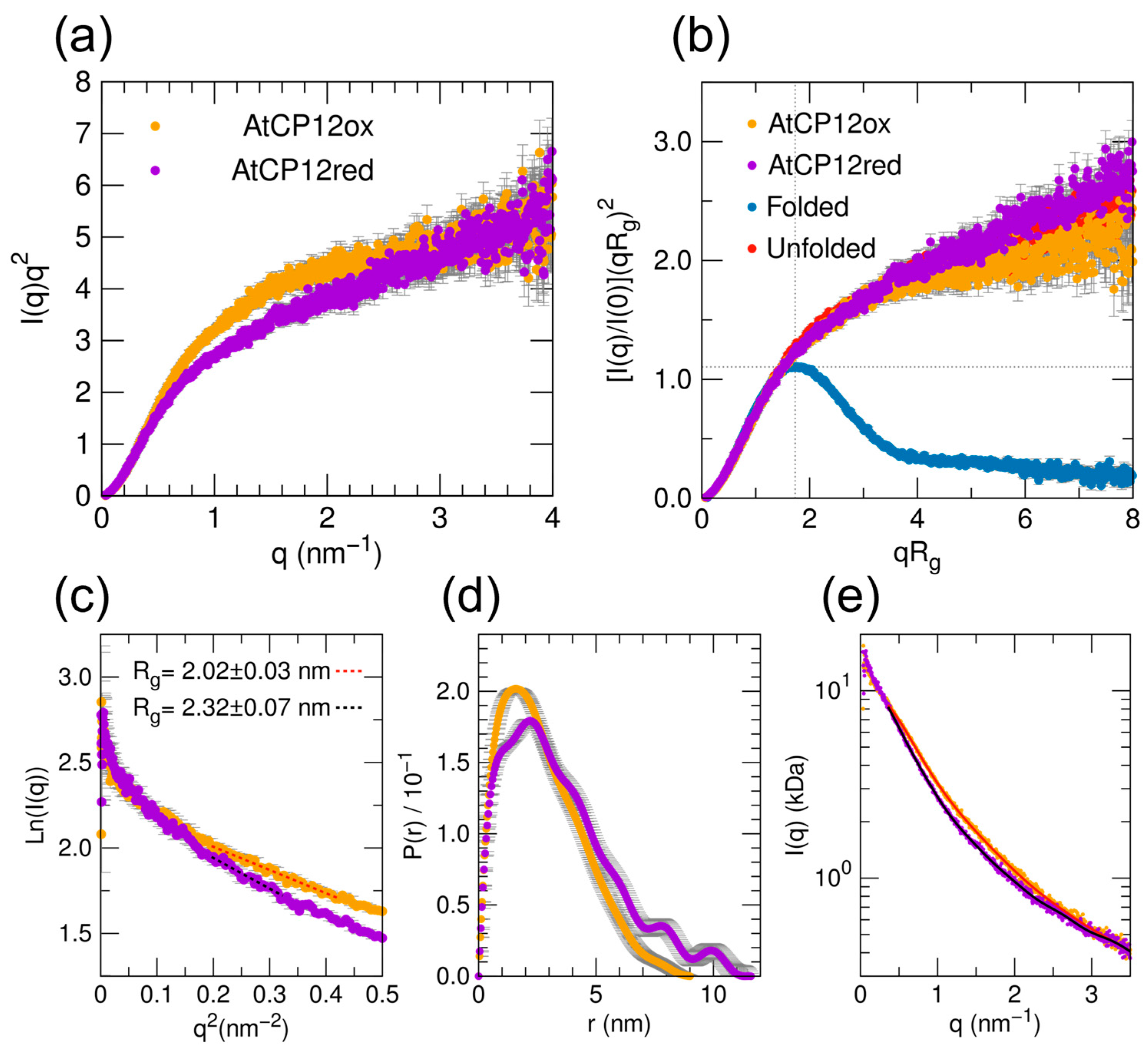
2.1. AtCP12 in Solution Is Disordered and More Expanded in Reduced Form Compared to Its Oxidized Form
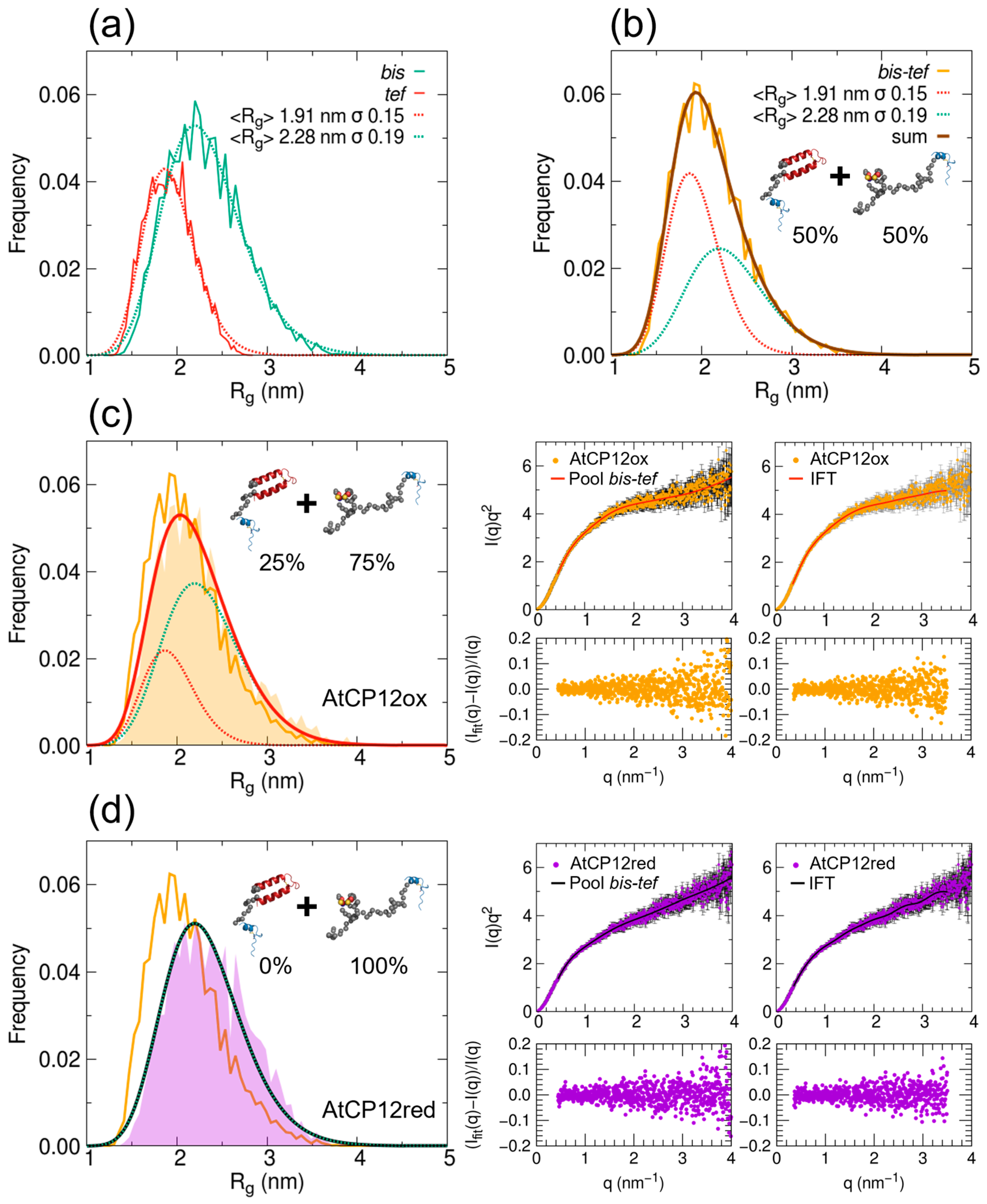
2.2. Estimate of the Contribution from Different Structural Ensembles in the Solution States of AtCP12
3. Discussion
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Samples Preparation
4.3. SAXS Data Collection
4.4. SAXS Data Analysis
4.5. Ensemble Fitting with Multiple Pools
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pohlmeyer, K.; Paap, B.K.; Soll, J.; Wedel, N. CP12: A small nuclear-encoded chloroplast protein provides novel insights into higher-plant GAPDH evolution. Plant Mol. Biol. 1996, 32, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Wedel, N.; Soll, J. Evolutionary conserved light regulation of Calvin cycle activity by NADPH-mediated reversible phosphoribulokinase/CP12/glyceraldehyde-3-phosphate dehydrogenase complex dissociation. Proc. Natl. Acad. Sci. USA 1998, 95, 9699–9704. [Google Scholar] [CrossRef] [PubMed]
- Groben, R.; Kaloudas, D.; Raines, C.A.; Offmann, B.; Maberly, S.C.; Gontero, B. Comparative sequence analysis of CP12, a small protein involved in the formation of a Calvin cycle complex in photosynthetic organisms. Photosynth. Res. 2010, 103, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Marri, L.; Pesaresi, A.; Valerio, C.; Lamba, D.; Pupillo, P.; Trost, P.; Sparla, F. In vitro characterization of Arabidopsis CP12 isoforms reveals common biochemical and molecular properties. J. Plant Physiol. 2010, 167, 939–950. [Google Scholar] [CrossRef]
- Stanley, D.N.; Raines, C.A.; Kerfeld, C.A. Comparative analysis of 126 cyanobacterial genomes reveals evidence of functional diversity among homologs of the redox-regulated CP12 protein. Plant Physiol. 2013, 161, 824–835. [Google Scholar] [CrossRef]
- Reichmann, D.; Jakob, U. The roles of conditional disorder in redox proteins. Curr. Opin. Struct. Biol. 2013, 23, 436–442. [Google Scholar] [CrossRef]
- Launay, H.; Barré, P.; Puppo, C.; Zhang, Y.; Maneville, S.; Gontero, B.; Receveur-Bréchot, V. Cryptic Disorder Out of Disorder: Encounter between conditionally disordered CP12 and glyceraldehyde-3-phosphate dehydrogenase. J. Mol. Biol. 2018, 430, 1218–1234. [Google Scholar] [CrossRef]
- Launay, H.; Shao, H.; Bornet, O.; Cantrelle, F.X.; Lebrun, R.; Receveur-Brechot, V.; Gontero, B. Flexibility of oxidized and reduced states of the chloroplast regulatory protein CP12 in isolation and in cell extracts. Biomolecules 2021, 11, 701. [Google Scholar] [CrossRef]
- Gérard, C.; Carrière, F.; Receveur-Bréchot, V.; Launay, H.; Gontero, B. A trajectory of discovery: Metabolic regulation by the conditionally disordered Chloroplast Protein, CP12. Biomolecules 2022, 12, 1047. [Google Scholar] [CrossRef]
- Singh, P.; Kaloudas, D.; Raines, C.A. Expression analysis of the Arabidopsis CP12 gene family suggests novel roles for these proteins in roots and floral tissues. J. Exp. Bot. 2008, 59, 3975–3985. [Google Scholar] [CrossRef]
- Wedel, N.; Soll, J.; Paap, B.K. CP12 provides a new mode of light regulation of Calvin cycle activity in higher plants. Proc. Natl. Acad. Sci. USA 1997, 94, 10479–10484. [Google Scholar] [CrossRef]
- Scheibe, R.; Wedel, N.; Vetter, S.; Emmerlich, V.; Sauermann, S.M. Co-existence of two regulatory NADP-glyceraldehyde 3-P dehydrogenase complexes in higher plant chloroplasts. Eur. J. Biochem. 2002, 269, 5617–5624. [Google Scholar]
- Howard, T.P.; Lloyd, J.C.; Raines, C.A. Inter-species variation in the oligomeric states of the higher plant Calvin cycle enzymes glyceraldehyde-3-phosphate dehydrogenase and phosphoribulokinase. J. Exp. Bot. 2011, 62, 3799–3805. [Google Scholar] [CrossRef]
- Boggetto, N.; Gontero, B.; Maberly, S.C. Regulation of phosphoribulokinase and glyceraldehyde 3-phosphate dehydrogenase in a freshwater diatom, Asterionella formosa. J. Phycol. 2007, 43, 1227–1235. [Google Scholar] [CrossRef]
- Oesterhelt, C.; Klocke, S.; Holtgrefe, S.; Linke, V.; Weber, A.P.M.; Scheibe, R. Redox regulation of chloroplast enzymes in Galdieria sulphuraria in view of eukaryotic evolution. Plant Cell Physiol. 2007, 48, 1359–1373. [Google Scholar] [CrossRef]
- Tamoi, M.; Miyazaki, T.; Fukamizo, T.; Shigeoka, S. The Calvin cycle in cyanobacteria is regulated by CP12 via the NAD(H)/NADP(H) ratio under light/dark conditions. Plant J. 2005, 42, 504–513. [Google Scholar] [CrossRef]
- McFarlane, C.R.; Shah, N.R.; Kabasakal, B.V.; Echeverria, B.; Cotton, C.A.R.; Bubeck, D.; Murray, J.W. Structural basis of light-induced redox regulation in the Calvin-Benson cycle in cyanobacteria. Proc. Natl. Acad. Sci. USA 2019, 116, 20984–20990. [Google Scholar] [CrossRef]
- Lucius, S.; Theune, M.; Arrivault, S.; Hildebrandt, S.; Mullineaux, C.W.; Gutekunst, K.; Hagemann, M. CP12 fine-tunes the Calvin-Benson cycle and carbohydrate metabolism in cyanobacteria. Front. Plant Sci. 2022, 13, 1028794. [Google Scholar] [CrossRef]
- Marri, L.; Trost, P.; Pupillo, P.; Sparla, F. Reconstitution and Properties of the Recombinant Glyceraldehyde-3-Phosphate Dehydrogenase/CP12/Phosphoribulokinase Supramolecular Complex of Arabidopsis. Plant Physiol. 2005, 139, 1433–1443. [Google Scholar] [CrossRef]
- Howard, T.P.; Metodiev, M.; Lloyd, J.C.; Raines, C.A. Thioredoxin-mediated reversible dissociation of a stromal multiprotein complex in response to changes in light availability. Proc. Natl. Acad. Sci. USA 2008, 105, 4056–4061. [Google Scholar] [CrossRef]
- Clement, R.; Lignon, S.; Mansuelle, P.; Jensen, E.; Pophillat, M.; Lebrun, R.; Denis, Y.; Puppo, C.; Maberly, S.C.; Gontero, B. Responses of the marine diatom Thalassiosira pseudonana to changes in CO2 concentration: A proteomic approach. Sci. Rep. 2017, 7, 42333. [Google Scholar] [CrossRef] [PubMed]
- López-Calcagno, P.E.; Abuzaid, A.O.; Lawson, T.; Raines, C.A. Arabidopsis CP12 mutants have reduced levels of phosphoribulokinase and impaired function of the Calvin–Benson cycle. J. Exp. Bot. 2017, 68, 2285–2298. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Qiu, H.; Zhou, M.; Lin, Y.; Guo, Z.; Lu, S. Chloroplast Protein 12 expression alters growth and chilling tolerance in tropical forage Stylosanthes guianensis (Aublet) Sw. Front. Plant Sci. 2018, 9, 1319. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Huang, W.; Avilan, L.; Receveur-Bréchot, V.; Puppo, C.; Puppo, R.; Lebrun, R.; Gontero, B.; Launay, H. A new type of flexible CP12 protein in the marine diatom Thalassiosira pseudonana. Cell Commun. Signal. 2021, 19, 38. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Xie, Y.; Pan, X.; Zhang, H.; Cao, P.; Su, X.; Chang, W.; Li, M. Photosynthetic phosphoribulokinase structures: Enzymatic mechanisms and the redox regulation of the Calvin-Benson-Bassham cycle. Plant Cell 2020, 32, 1556–1573. [Google Scholar] [CrossRef]
- Graciet, E.; Gans, P.; Wedel, N.; Lebreton, S.; Camadro, J.M.; Gontero, B. The small protein CP12: A protein linker for supramolecular complex assembly. Biochemistry 2003, 42, 8163–8170. [Google Scholar] [CrossRef]
- Moparthi, S.B.; Thieulin-Pardo, G.; de Torres, J.; Ghenuche, P.; Gontero, B.; Wenger, J. FRET analysis of CP12 structural interplay by GAPDH and PRK. Biochem. Biophys. Res. Commun. 2015, 458, 488–493. [Google Scholar] [CrossRef]
- Launay, H.; Barré, P.; Puppo, C.; Manneville, S.; Gontero, B.; Receveur-Bréchot, V. Absence of residual structure in the intrinsically disordered regulatory protein CP12 in its reduced state. Biochem. Biophys. Res. Commun. 2016, 477, 20–26. [Google Scholar] [CrossRef]
- Bernadó, P.; Svergun, D.I. Structural analysis of intrinsically disordered proteins by small-angle X-ray scattering. Mol. Biosyst. 2012, 8, 151–167. [Google Scholar] [CrossRef]
- Marri, L.; Trost, P.; Trivelli, X.; Gonnelli, L.; Pupillo, P.; Sparla, F. Spontaneous assembly of photosynthetic supramolecular complexes as mediated by the intrinsically unstructured protein CP12. J. Biol. Chem. 2008, 283, 1831–1838. [Google Scholar] [CrossRef]
- Marri, L.; Zaffagnini, M.; Collin, V.; Issakidis-Bourguet, E.; Lemaire, S.D.; Pupillo, P.; Sparla, F.; Miginiac-Maslow, M.; Trost, P. Prompt and easy activation by specific thioredoxins of calvin cycle enzymes of Arabidopsis thaliana associated in the GAPDH/CP12/PRK supramolecular complex. Mol. Plant. 2009, 2, 259–269. [Google Scholar] [CrossRef]
- González-Foutel, N.S.; Glavina, J.; Borcherds, W.M.; Safranchik, M.; Barrera-Vilarmau, S.; Sagar, A.; Estaña, A.; Barozet, A.; Garrone, N.A.; Fernandez-Ballester, G.; et al. Conformational Buffering Underlies Functional Selection in Intrinsically Disordered Protein Regions. Nat. Struct. Mol. Biol. 2022, 29, 781–790. [Google Scholar] [CrossRef]
- Receveur-Brechot, V.; Durand, D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 2012, 13, 55–75. [Google Scholar] [CrossRef]
- Durand, D.; Vives, C.; Cannella, D.; Perez, J.; Pebay-Peyroula, E.; Vachette, P.; Fieschi, F. NADPH oxidase activator P67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J. Struct. Biol. 2010, 169, 45–53. [Google Scholar] [CrossRef]
- Bernadó, P.; Blackledge, M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys. J. 2009, 97, 2839–2845. [Google Scholar] [CrossRef]
- Bernadó, P.; Mylonas, E.; Petoukhov, M.V.; Blackledge, M.; Svergun, D.I. Structural characterization of flexible proteins using small-angle X-ray scattering. J. Am. Chem. Soc. 2007, 129, 5656–5664. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015, 2, 207–217. [Google Scholar] [CrossRef]
- Fermani, S.; Trivelli, X.; Sparla, F.; Thumiger, A.; Calvaresi, M.; Marri, L.; Falini, G.; Zerbetto, F.; Trost, P. Conformational selection and folding-upon-binding of intrinsically disordered protein CP12 regulate photosynthetic enzymes assembly. J. Biol. Chem. 2012, 287, 21372–21383. [Google Scholar] [CrossRef]
- Gardebien, F.; Thangudu, R.R.; Gontero, B.; Offmann, B. Construction of a 3D model of CP12, a protein linker. J. Mol. Graph. Model. 2006, 25, 186–195. [Google Scholar] [CrossRef]
- Cammers-Goodwin, A.; Allen, T.J.; Oslick, S.L.; McClure, K.F.; Lee, J.H.; Kemp, D.S. Mechanism of stabilization of helical conformations of polypeptides by water containing trifluoroethanol. J. Am. Chem. Soc. 1996, 118, 3082–3091. [Google Scholar] [CrossRef]
- Del Giudice, A.; Pavel, N.V.; Galantini, L.; Falini, G.; Trost, P.; Fermani, S.; Sparla, F. Unravelling the shape and structural assembly of the photosynthetic GAPDH-CP12-PRK complex from Arabidopsis thaliana by small-angle X-ray scattering analysis. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2372–2385. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the Expasy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Pernot, P.; Round, A.; Barrett, R.; De Maria Antolinos, A.; Gobbo, A.; Gordon, E.; Huet, J.; Kieffer, J.; Lentini, M.; Mattenet, M.; et al. Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron. Radiat. 2013, 20, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Brennich, M.E.; Kieffer, J.; Bonamis, G.; De Maria Antolinos, A.; Hutin, S.; Pernot, P.A. Round, Online data analysis at the ESRF bioSAXS beamline, BM29. J. Appl. Crystallogr. 2016, 49, 203–212. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef]
- Hajizadeh, N.R.; Franke, D.; Jeffries, C.M.; Svergun, D.I. Consensus Bayesian assessment of protein molecular mass from solution X-ray scattering data. Sci. Rep. 2018, 8, 7204. [Google Scholar] [CrossRef]
- Sagar, A.; Jeffries, C.M.; Petoukhov, M.V.; Svergun, D.I.; Bernadó, P. Comment on the Optimal Parameters to Derive Intrinsically Disordered Protein Conformational Ensembles from Small-Angle X-ray Scattering Data Using the Ensemble Optimization Method. J Chem. Theory Comp. 2021, 17, 2014–2021. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Giudice, A.; Gurrieri, L.; Galantini, L.; Fanti, S.; Trost, P.; Sparla, F.; Fermani, S. Conformational Disorder Analysis of the Conditionally Disordered Protein CP12 from Arabidopsis thaliana in Its Different Redox States. Int. J. Mol. Sci. 2023, 24, 9308. https://doi.org/10.3390/ijms24119308
Del Giudice A, Gurrieri L, Galantini L, Fanti S, Trost P, Sparla F, Fermani S. Conformational Disorder Analysis of the Conditionally Disordered Protein CP12 from Arabidopsis thaliana in Its Different Redox States. International Journal of Molecular Sciences. 2023; 24(11):9308. https://doi.org/10.3390/ijms24119308
Chicago/Turabian StyleDel Giudice, Alessandra, Libero Gurrieri, Luciano Galantini, Silvia Fanti, Paolo Trost, Francesca Sparla, and Simona Fermani. 2023. "Conformational Disorder Analysis of the Conditionally Disordered Protein CP12 from Arabidopsis thaliana in Its Different Redox States" International Journal of Molecular Sciences 24, no. 11: 9308. https://doi.org/10.3390/ijms24119308
APA StyleDel Giudice, A., Gurrieri, L., Galantini, L., Fanti, S., Trost, P., Sparla, F., & Fermani, S. (2023). Conformational Disorder Analysis of the Conditionally Disordered Protein CP12 from Arabidopsis thaliana in Its Different Redox States. International Journal of Molecular Sciences, 24(11), 9308. https://doi.org/10.3390/ijms24119308