
KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy

 ,
, 
Abstract
:1. Introduction
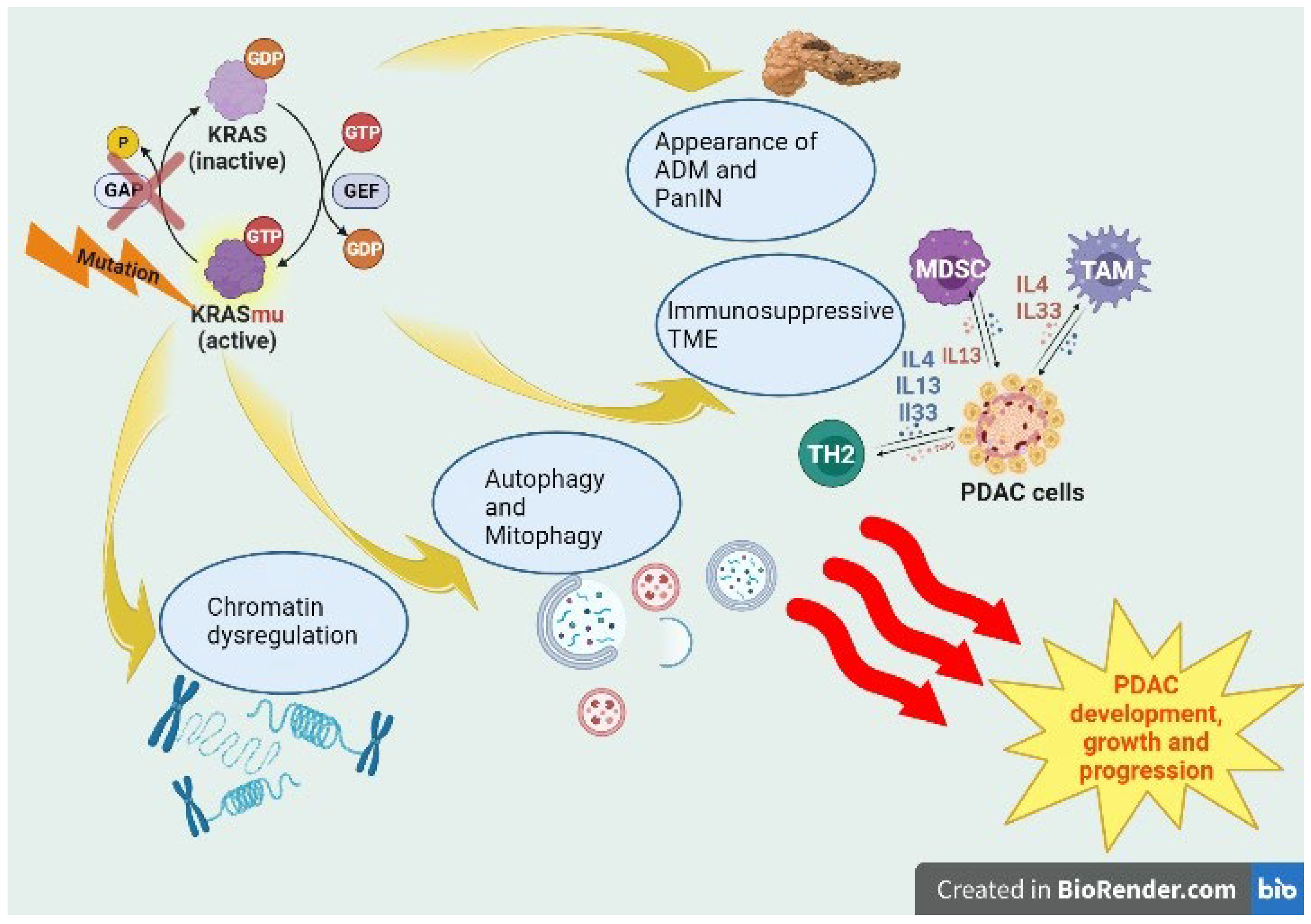
2. KRAS-Dependent Tumorigenesis in PDAC
2.1. KRASmu, Inflammation and Precursor Lesions
2.2. KRASmu and Metabolism
2.3. KRASmu and TME
2.4. KRASmu and Chromatin
3. KRAS Signaling Inhibitors in PDAC
3.1. KRASG12C Inhibition
3.2. KRASG12D Inhibition
3.3. Other Inhibitors
4. Mechanisms of Escaping in Resistance to KRAS Inhibitors
5. Alternative Targets in KRAS Signaling Pathways and Future Perspectives
5.1. Alternative Targets in KRAS Signaling Pathway
5.2. Future Perspectives
6. KRAS Dependency in PDAC
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ha, C.S.R.; Müller-Nurasyid, M.; Petrera, A.; Hauck, S.M.; Marini, F.; Bartsch, D.K.; Slater, E.P.; Strauch, K. Proteomics Biomarker Discovery for Individualized Prevention of Familial Pancreatic Cancer Using Statistical Learning. PLoS ONE 2023, 18, e0280399. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA. Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Park, W.; Chawla, A.; O’Reilly, E.M. Pancreatic Cancer: A Review. Jama 2021, 326, 851–862. [Google Scholar] [CrossRef]
- Sung, H.; Siegel, R.L.; Rosenberg, P.S.; Jemal, A. Emerging Cancer Trends among Young Adults in the USA: Analysis of a Population-Based Cancer Registry. Lancet Public Health 2019, 4, e137–e147. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus Gemcitabine for Metastatic Pancreatic Cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Wang-Gillam, A.; Li, C.-P.; Bodoky, G.; Dean, A.; Shan, Y.-S.; Jameson, G.; Macarulla, T.; Lee, K.-H.; Cunningham, D.; Blanc, J.F.; et al. Nanoliposomal Irinotecan with Fluorouracil and Folinic Acid in Metastatic Pancreatic Cancer after Previous Gemcitabine-Based Therapy (NAPOLI-1): A Global, Randomised, Open-Label, Phase 3 Trial. Lancet Lond. Engl. 2016, 387, 545–557. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.-M.; Gingras, M.-C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.C.; Quinn, M.C.; et al. Genomic Analyses Identify Molecular Subtypes of Pancreatic Cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Li, F.; Zhang, M.; Xia, X.; Wu, J.; Gao, X.; Zhou, H.; Zhang, Z.; Huang, N.; Yang, X.; et al. A Novel Protein RASON Encoded by a LncRNA Controls Oncogenic RAS Signaling in KRAS Mutant Cancers. Cell Res. 2023, 33, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of Oncogenic KRAS in the Diagnosis, Prognosis and Treatment of Pancreatic Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Costamagna, A.; Natalini, D.; Camacho Leal, M.D.P.; Simoni, M.; Gozzelino, L.; Cappello, P.; Novelli, F.; Ambrogio, C.; Defilippi, P.; Turco, E.; et al. Docking Protein P130Cas Regulates Acinar to Ductal Metaplasia During Pancreatic Adenocarcinoma Development and Pancreatitis. Gastroenterology 2022, 162, 1242–1255.e11. [Google Scholar] [CrossRef]
- Awad, M.M.; Liu, S.; Rybkin, I.I.; Arbour, K.C.; Dilly, J.; Zhu, V.W.; Johnson, M.L.; Heist, R.S.; Patil, T.; Riely, G.J.; et al. Acquired Resistance to KRASG12C Inhibition in Cancer. N. Engl. J. Med. 2021, 384, 2382–2393. [Google Scholar] [CrossRef] [PubMed]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-Dependent Transformation of Adult Pancreatic Cells by Oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-Induced Inflammation Contributes to Pancreatic Cancer by Inhibiting Oncogene-Induced Senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Del Poggetto, E.; Ho, I.-L.; Balestrieri, C.; Yen, E.-Y.; Zhang, S.; Citron, F.; Shah, R.; Corti, D.; Diaferia, G.R.; Li, C.-Y.; et al. Epithelial Memory of Inflammation Limits Tissue Damage While Promoting Pancreatic Tumorigenesis. Science 2021, 373, eabj0486. [Google Scholar] [CrossRef]
- Storz, P.; Crawford, H.C. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology 2020, 158, 2072–2081. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Wang, L.; Xie, D.; Wei, D. Pancreatic Acinar-to-Ductal Metaplasia and Pancreatic Cancer. Methods Mol. Biol. Clifton NJ 2019, 1882, 299–308. [Google Scholar] [CrossRef]
- Strobel, O.; Dor, Y.; Alsina, J.; Stirman, A.; Lauwers, G.; Trainor, A.; Castillo, C.F.-D.; Warshaw, A.L.; Thayer, S.P. In Vivo Lineage Tracing Defines the Role of Acinar-to-Ductal Transdifferentiation in Inflammatory Ductal Metaplasia. Gastroenterology 2007, 133, 1999–2009. [Google Scholar] [CrossRef]
- Shi, G.; DiRenzo, D.; Qu, C.; Barney, D.; Miley, D.; Konieczny, S.F. Maintenance of Acinar Cell Organization Is Critical to Preventing Kras-Induced Acinar-Ductal Metaplasia. Oncogene 2013, 32, 1950–1958. [Google Scholar] [CrossRef]
- Ge, W.; Goga, A.; He, Y.; Silva, P.N.; Hirt, C.K.; Herrmanns, K.; Guccini, I.; Godbersen, S.; Schwank, G.; Stoffel, M. MiR-802 Suppresses Acinar-to-Ductal Reprogramming During Early Pancreatitis and Pancreatic Carcinogenesis. Gastroenterology 2022, 162, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Schneeweis, C.; Wirth, M.; Müller, S.; Geismann, C.; Neuß, T.; Steiger, K.; Krämer, O.H.; Schmid, R.M.; Rad, R.; et al. Important Role of Nfkb2 in the KrasG12D-Driven Carcinogenesis in the Pancreas. Pancreatology 2021, 21, 912–919. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Sun, Z.; Zhang, M.; Qu, X.; Yang, S.; Wang, L.; Jing, Y.; Li, L.; Deng, W.; Liu, F.; et al. Deficient Rnf43 Potentiates Hyperactive Kras-Mediated Pancreatic Preneoplasia Initiation and Malignant Transformation. Anim. Models Exp. Med. 2022, 5, 61–71. [Google Scholar] [CrossRef]
- Yan, H.H.; Jung, K.H.; Lee, J.E.; Son, M.K.; Fang, Z.; Park, J.H.; Kim, S.J.; Kim, J.Y.; Lim, J.H.; Hong, S.-S. ANGPTL4 Accelerates KRASG12D-Induced Acinar to Ductal Metaplasia and Pancreatic Carcinogenesis. Cancer Lett. 2021, 519, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Curbelo, D.; Ho, Y.-J.; Burdziak, C.; Maag, J.L.V.; Morris, J.P.; Chandwani, R.; Chen, H.-A.; Tsanov, K.M.; Barriga, F.M.; Luan, W.; et al. A Gene-Environment-Induced Epigenetic Program Initiates Tumorigenesis. Nature 2021, 590, 642–648. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. P53 Status Determines the Role of Autophagy in Pancreatic Tumour Development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Carbone, C.; Piro, G.; Agostini, A.; Delfino, P.; De Sanctis, F.; Nasca, V.; Spallotta, F.; Sette, C.; Martini, M.; Ugel, S.; et al. Intratumoral Injection of TLR9 Agonist Promotes an Immunopermissive Microenvironment Transition and Causes Cooperative Antitumor Activity in Combination with Anti-PD1 in Pancreatic Cancer. J. Immunother. Cancer 2021, 9, e002876. [Google Scholar] [CrossRef]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene Ablation-Resistant Pancreatic Cancer Cells Depend on Mitochondrial Function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef]
- Jin, J.; Byun, J.-K.; Choi, Y.-K.; Park, K.-G. Targeting Glutamine Metabolism as a Therapeutic Strategy for Cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.-T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic Deletion of Malic Enzyme 2 Confers Collateral Lethality in Pancreatic Cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef]
- Liu, Y.; Deguchi, Y.; Wei, D.; Liu, F.; Moussalli, M.J.; Deguchi, E.; Li, D.; Wang, H.; Valentin, L.A.; Colby, J.K.; et al. Rapid Acceleration of KRAS-Mutant Pancreatic Carcinogenesis via Remodeling of Tumor Immune Microenvironment by PPARδ. Nat. Commun. 2022, 13, 2665. [Google Scholar] [CrossRef]
- Berta, M.A.; Baker, C.M.; Cottle, D.L.; Watt, F.M. Dose and Context Dependent Effects of Myc on Epidermal Stem Cell Proliferation and Differentiation. EMBO Mol. Med. 2010, 2, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Bannoura, S.F.; Khan, H.Y.; Azmi, A.S. KRAS G12D Targeted Therapies for Pancreatic Cancer: Has the Fortress Been Conquered? Front. Oncol. 2022, 12, 1013902. [Google Scholar] [CrossRef] [PubMed]
- Sangrador, I.; Molero, X.; Campbell, F.; Franch-Expósito, S.; Rovira-Rigau, M.; Samper, E.; Domínguez-Fraile, M.; Fillat, C.; Castells, A.; Vaquero, E.C. Zeb1 in Stromal Myofibroblasts Promotes Kras-Driven Development of Pancreatic Cancer. Cancer Res. 2018, 78, 2624–2637. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Carugo, A.; Tepper, J.; Robinson, F.S.; Li, L.; Svelto, M.; Nezi, L.; Corti, D.; Minelli, R.; Pettazzoni, P.; et al. Synthetic Vulnerabilities of Mesenchymal Subpopulations in Pancreatic Cancer. Nature 2017, 542, 362–366. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Liu, Z.; Wang, J. Targeting KRAS in PDAC: A New Way to Cure It? Cancers 2022, 14, 4982. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef]
- Bannoura, S.F.; Uddin, M.H.; Nagasaka, M.; Fazili, F.; Al-Hallak, M.N.; Philip, P.A.; El-Rayes, B.; Azmi, A.S. Targeting KRAS in Pancreatic Cancer: New Drugs on the Horizon. Cancer Metastasis Rev. 2021, 40, 819–835. [Google Scholar] [CrossRef]
- Sakamoto, K.; Masutani, T.; Hirokawa, T. Generation of KS-58 as the First K-Ras(G12D)-Inhibitory Peptide Presenting Anti-Cancer Activity in Vivo. Sci. Rep. 2020, 10, 21671. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, Y.J. Adoptive Cell Therapy Targeting Neoantigens: A Frontier for Cancer Research. Front. Immunol. 2020, 11, 176. [Google Scholar] [CrossRef]
- Wang, Q.J.; Yu, Z.; Griffith, K.; Hanada, K.; Restifo, N.P.; Yang, J.C. Identification of T-Cell Receptors Targeting KRAS-Mutated Human Tumors. Cancer Immunol. Res. 2016, 4, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Heitman, J. The Cyclophilins. Genome Biol. 2005, 6, 226. [Google Scholar] [CrossRef]
- Ahearn, I.M.; Haigis, K.; Bar-Sagi, D.; Philips, M.R. Regulating the Regulator: Post-Translational Modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef]
- Wang, W.-H.; Yuan, T.; Qian, M.-J.; Yan, F.-J.; Yang, L.; He, Q.-J.; Yang, B.; Lu, J.-J.; Zhu, H. Post-Translational Modification of KRAS: Potential Targets for Cancer Therapy. Acta Pharmacol. Sin. 2021, 42, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The Clinical KRAS(G12C) Inhibitor AMG 510 Drives Anti-Tumour Immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Lito, P.; Solomon, M.; Li, L.-S.; Hansen, R.; Rosen, N. Allele-Specific Inhibitors Inactivate Mutant KRAS G12C by a Trapping Mechanism. Science 2016, 351, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Soares, H.P.; Ming, M.; Mellon, M.; Young, S.H.; Han, L.; Sinnet-Smith, J.; Rozengurt, E. Dual PI3K/MTOR Inhibitors Induce Rapid Overactivation of the MEK/ERK Pathway in Human Pancreatic Cancer Cells through Suppression of MTORC2. Mol. Cancer Ther. 2015, 14, 1014–1023. [Google Scholar] [CrossRef]
- Oncogenic KRAS Engages an RSK1/NF1 Pathway to Inhibit Wild-Type RAS Signaling in Pancreatic Cancer—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/34021083/ (accessed on 4 March 2023).
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 Inhibition Diminishes KRASG12C Cycling and Promotes Tumor Microenvironment Remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef]
- Zhao, H.; Yang, L.; Baddour, J.; Achreja, A.; Bernard, V.; Moss, T.; Marini, J.C.; Tudawe, T.; Seviour, E.G.; San Lucas, F.A.; et al. Tumor Microenvironment Derived Exosomes Pleiotropically Modulate Cancer Cell Metabolism. eLife 2016, 5, e10250. [Google Scholar] [CrossRef]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef]
- Hou, P.; Ma, X.; Zhang, Q.; Wu, C.-J.; Liao, W.; Li, J.; Wang, H.; Zhao, J.; Zhou, X.; Guan, C.; et al. USP21 Deubiquitinase Promotes Pancreas Cancer Cell Stemness via Wnt Pathway Activation. Genes Dev. 2019, 33, 1361–1366. [Google Scholar] [CrossRef]
- Crawford, H.C. Anticipating Resistance to KRAS Inhibition: A Novel Role for USP21 in Macropinocytosis Regulation. Genes Dev. 2021, 35, 1325–1326. [Google Scholar] [CrossRef] [PubMed]
- Hennig, A.; Markwart, R.; Esparza-Franco, M.A.; Ladds, G.; Rubio, I. Ras Activation Revisited: Role of GEF and GAP Systems. Biol. Chem. 2015, 396, 831–848. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Nishimura, R.; Kashishian, A.; Batzer, A.G.; Kim, W.J.; Cooper, J.A.; Schlessinger, J. A New Function for a Phosphotyrosine Phosphatase: Linking GRB2-Sos to a Receptor Tyrosine Kinase. Mol. Cell. Biol. 1994, 14, 509–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-N.P.; LaMarche, M.J.; Chan, H.M.; Fekkes, P.; Garcia-Fortanet, J.; Acker, M.G.; Antonakos, B.; Chen, C.H.-T.; Chen, Z.; Cooke, V.G.; et al. Allosteric Inhibition of SHP2 Phosphatase Inhibits Cancers Driven by Receptor Tyrosine Kinases. Nature 2016, 535, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Kerr, D.L.; Haderk, F.; Bivona, T.G. Allosteric SHP2 Inhibitors in Cancer: Targeting the Intersection of RAS, Resistance, and the Immune Microenvironment. Curr. Opin. Chem. Biol. 2021, 62, 1–12. [Google Scholar] [CrossRef]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS Nucleotide Cycling Underlies the SHP2 Phosphatase Dependence of Mutant BRAF-, NF1- and RAS-Driven Cancers. Nat. Cell Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Liu, C.; Lu, H.; Wang, H.; Loo, A.; Zhang, X.; Yang, G.; Kowal, C.; Delach, S.; Wang, Y.; Goldoni, S.; et al. Combinations with Allosteric SHP2 Inhibitor TNO155 to Block Receptor Tyrosine Kinase Signaling. Clin. Cancer Res. 2021, 27, 342–354. [Google Scholar] [CrossRef]
- Chamberlain, P.P.; Hamann, L.G. Development of Targeted Protein Degradation Therapeutics. Nat. Chem. Biol. 2019, 15, 937–944. [Google Scholar] [CrossRef]
- Chiarantini, L.; Cerasi, A.; Fraternale, A.; Millo, E.; Benatti, U.; Sparnacci, K.; Laus, M.; Ballestri, M.; Tondelli, L. Comparison of Novel Delivery Systems for Antisense Peptide Nucleic Acids. J. Control. Release 2005, 109, 24–36. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, Z.; Li, D.; Wang, E.; Wang, J. Rational Drug Design: The Search for Ras Protein Hydrolysis Intermediate Conformation Inhibitors with Both Affinity and Specificity. Curr. Pharm. Des. 2013, 19, 2246–2258. [Google Scholar] [CrossRef]
- Li, D.; Liu, Z.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A Small-Molecule Induces Apoptosis and Suppresses Metastasis in Pancreatic Cancer Cells. Eur. J. Pharm. Sci. 2013, 48, 658–667. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, D.; Zheng, X.; Wang, E.; Wang, J. Selective Induction of Apoptosis: Promising Therapy in Pancreatic Cancer. Curr. Pharm. Des. 2013, 19, 2259–2268. [Google Scholar] [CrossRef]
- Khan, H.Y.; Nagasaka, M.; Li, Y.; Aboukameel, A.; Uddin, M.H.; Sexton, R.; Bannoura, S.; Mzannar, Y.; Al-Hallak, M.N.; Kim, S.; et al. Inhibitor of the Nuclear Transport Protein XPO1 Enhances the Anticancer Efficacy of KRAS G12C Inhibitors in Preclinical Models of KRAS G12C-Mutant Cancers. Cancer Res. Commun. 2022, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef] [PubMed]
- M.D. Anderson Cancer Center. Phase I Study of Mesenchymal Stromal Cells-Derived Exosomes with KrasG12D SiRNA for Metastatic Pancreas Cancer Patients Harboring KrasG12D Mutation. 2023. Available online: https://clinicaltrials.gov (accessed on 5 March 2023).
- XP-524 Is a Dual-BET/EP300 Inhibitor That Represses Oncogenic KRAS and Potentiates Immune Checkpoint Inhibition in Pancreatic Cancer—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/35064087/ (accessed on 4 March 2023).
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Müller, F.; Lim, J.K.M.; Bebber, C.M.; Seidel, E.; Tishina, S.; Dahlhaus, A.; Stroh, J.; Beck, J.; Yapici, F.I.; Nakayama, K.; et al. Elevated FSP1 Protects KRAS-Mutated Cells from Ferroptosis during Tumor Initiation. Cell Death Differ. 2023, 30, 442–456. [Google Scholar] [CrossRef]
- Yan, H.; Yu, C.-C.; Fine, S.A.; Youssof, A.L.; Yang, Y.-R.; Yan, J.; Karg, D.C.; Cheung, E.C.; Friedman, R.A.; Ying, H.; et al. Loss of the Wild-Type KRAS Allele Promotes Pancreatic Cancer Progression through Functional Activation of YAP1. Oncogene 2021, 40, 6759–6771. [Google Scholar] [CrossRef]
- Cordenonsi, M.; Zanconato, F.; Azzolin, L.; Forcato, M.; Rosato, A.; Frasson, C.; Inui, M.; Montagner, M.; Parenti, A.R.; Poletti, A.; et al. The Hippo Transducer TAZ Confers Cancer Stem Cell-Related Traits on Breast Cancer Cells. Cell 2011, 147, 759–772. [Google Scholar] [CrossRef]
- Salcedo Allende, M.T.; Zeron-Medina, J.; Hernandez, J.; Macarulla, T.; Balsells, J.; Merino, X.; Allende, H.; Tabernero, J.; Ramon, Y.; Cajal, S. Overexpression of Yes Associated Protein 1, an Independent Prognostic Marker in Patients with Pancreatic Ductal Adenocarcinoma, Correlated with Liver Metastasis and Poor Prognosis. Pancreas 2017, 46, 913–920. [Google Scholar] [CrossRef]
- Hou, P.; Ma, X.; Yang, Z.; Zhang, Q.; Wu, C.-J.; Li, J.; Tan, L.; Yao, W.; Yan, L.; Zhou, X.; et al. USP21 Deubiquitinase Elevates Macropinocytosis to Enable Oncogenic KRAS Bypass in Pancreatic Cancer. Genes Dev. 2021, 35, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Kapoor, A.; Zhang, Q.; Li, J.; Wu, C.-J.; Li, J.; Lan, Z.; Tang, M.; Ma, X.; Ackroyd, J.J.; et al. Tumor Microenvironment Remodeling Enables Bypass of Oncogenic KRAS Dependency in Pancreatic Cancer. Cancer Discov. 2020, 10, 1058–1077. [Google Scholar] [CrossRef] [PubMed]
- Pettazzoni, P.; Viale, A.; Shah, P.; Carugo, A.; Ying, H.; Wang, H.; Genovese, G.; Seth, S.; Minelli, R.; Green, T.; et al. Genetic Events That Limit the Efficacy of MEK and RTK Inhibitor Therapies in a Mouse Model of KRAS-Driven Pancreatic Cancer. Cancer Res. 2015, 75, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a031435. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| ClinicalTrials.Gov Identifier | Title | Phase | Drugs | Targets |
|---|---|---|---|---|
| NCT03785249 | Phase 1/2 Study of MRTX849 in Patients with Cancer Having a KRAS G12C Mutation KRYSTAL-1 | I/II | MRTX849 (Adagrasib) | KRASG12C |
| NCT03948763 | A Study of mRNA-5671/V941 as Monotherapy and in Combination with Pembrolizumab (V941-001) | I | mRNA-5671/V941 Pembrolizumab | KRASmu PD-1 |
| NCT03592888 | DC Vaccine in Pancreatic Cancer | I | mDC3/8 | KRASmu |
| NCT04117087 | Pooled Mutant KRAS-Targeted Long Peptide Vaccine Combined with Nivolumab and Ipilimumab for Patients with Resected MMR-p Colorectal and Pancreatic Cancer | I | KRAS peptide vaccine Nivolumab Ipilimumab | KRASmu PD-1 CTLA-4 |
| NCT03745326 | Administering Peripheral Blood Lymphocytes Transduced with a Murine T-Cell Receptor Recognizing the G12D Variant of Mutated RAS in HLA-A*11:01 Patients | I/II | Cyclophosphamide Fludarabine Aldesleukin Anti-KRAS G12D mTCR PBL | KRASG12D |
| NCT03190941 | Administering Peripheral Blood Lymphocytes Transduced with a Murine T-Cell Receptor Recognizing the G12V Variant of Mutated RAS in HLA-A*11:01 Patients | I/II | Cyclophosphamide Fludarabine Aldesleukin Anti-KRAS G12V mTCR PBL | KRASG12V |
| NCT04330664 | Adagrasib in Combination with TNO155 in Patients with Cancer (KRYSTAL 2) | I/II | MRTX849 (Adagrasib) TNO155 | KRASG12C SHP2 |
| NCT04185883 | Sotorasib Activity in Subjects with Advanced Solid Tumors with KRAS p.G12C Mutation (CodeBreak 101) | I/II | Sotorasib AMG 404 Trametinib RMC-4630 Afatinib Pembrolizumab Panitumumab Carboplatin, pemetrexed, docetaxel, paclitaxel Atezolizumab Everolimus Palbociclib MVASI® (bevacizumab-awwb) TNO155 FOLFIRI FOLFOX BI 1701963 | KRASG12C PD-1 MAP2K1 SHP2 EGFR PD-L1 mTOR CDK4/6 VEGF SOS1 |
| ClinicalTrials.Gov Identifier | Title | Phase | Drugs | Targets |
|---|---|---|---|---|
| NCT04111458 | A Study to Test Different Doses of BI 1701963 Alone and Combined with Trametinib in Patients with Different Types of Advanced Cancer (Solid Tumors with KRAS Mutation) | I | BI 1701963 Trametinib | SOS1 MAP2K1 |
| NCT03634982 | Dose Escalation of RMC-4630 Monotherapy in Relapsed/Refractory Solid Tumors | I | RMC-4630 | SHP2 |
| NCT04916236 | Combination Therapy of RMC-4630 and LY3214996 in Metastatic KRAS Mutant Cancers (SHERPA) | I | RMC-4630 LY3214996 | SHP2 ERK |
| NCT04000529 | Phase Ib Study of TNO155 in Combination with Spartalizumab or Ribociclib in Selected Malignancies | I | TNO155 Spartalizumab Ribociclib | SHP2 CDK4/6 PD-1 |
| NCT03114319 | Dose Finding Study of TNO155 in Adult Patients with Advanced Solid Tumors | I | TNO155 EGF816 (nazartinib) | SHP2 EGFR |
| NCT04670679 | A Dose Escalation/Expansion Study of ERAS-601 in Patients with Advanced or Metastatic Solid Tumors (FLAGSHP-1) | I | ERAS-601 Cetuximab Pembrolizumab | SHP2 EGFR PD-1 |
| NCT04121286 | A Study of JAB-3312 in Adult Patients with Advanced Solid Tumors in China | I | JAB-3312 | SHP2 |
| NCT04528836 | First-in-Human Study of the SHP2 Inhibitor BBP-398 in Patients with Advanced Solid Tumors | I | BBP-398 (Formerly known as IACS-15509) | SHP2 |
| NCT04252339 | RLY-1971 in Subjects with Advanced or Metastatic Solid Tumors | I | RLY-1971 | SHP2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurreri, E.; Genovese, G.; Perelli, L.; Agostini, A.; Piro, G.; Carbone, C.; Tortora, G. KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy. Int. J. Mol. Sci. 2023, 24, 9313. https://doi.org/10.3390/ijms24119313
Gurreri E, Genovese G, Perelli L, Agostini A, Piro G, Carbone C, Tortora G. KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy. International Journal of Molecular Sciences. 2023; 24(11):9313. https://doi.org/10.3390/ijms24119313
Chicago/Turabian StyleGurreri, Enrico, Giannicola Genovese, Luigi Perelli, Antonio Agostini, Geny Piro, Carmine Carbone, and Giampaolo Tortora. 2023. "KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy" International Journal of Molecular Sciences 24, no. 11: 9313. https://doi.org/10.3390/ijms24119313
APA StyleGurreri, E., Genovese, G., Perelli, L., Agostini, A., Piro, G., Carbone, C., & Tortora, G. (2023). KRAS-Dependency in Pancreatic Ductal Adenocarcinoma: Mechanisms of Escaping in Resistance to KRAS Inhibitors and Perspectives of Therapy. International Journal of Molecular Sciences, 24(11), 9313. https://doi.org/10.3390/ijms24119313