
Molecular and Functional Relevance of NaV1.8-Induced Atrial Arrhythmogenic Triggers in a Human SCN10A Knock-Out Stem Cell Model
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. CRISPR/Cas9 Based Homozygous Knock-Out of SCN10A in Human Atrial iPSC-Cardiomyocytes
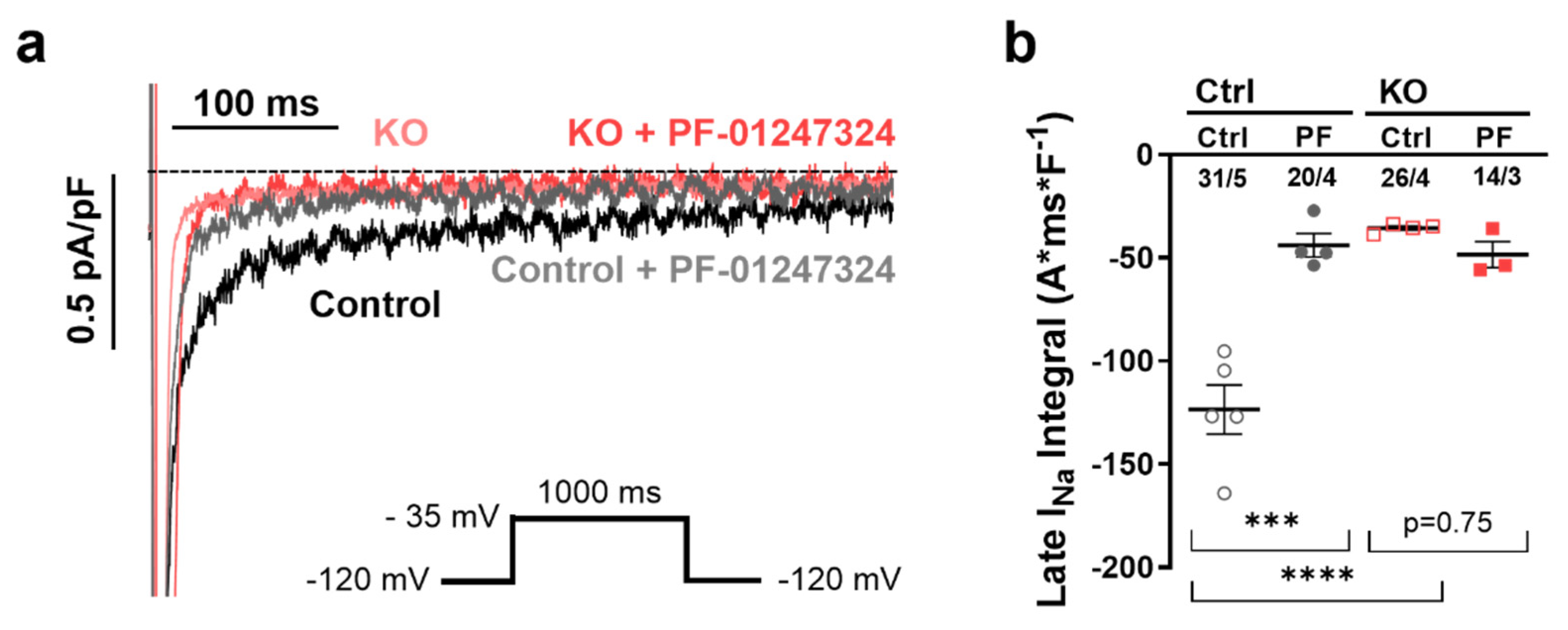
2.2. Influence of NaV1.8 on INaL in Human Atrial iPSC-Cardiomyocytes
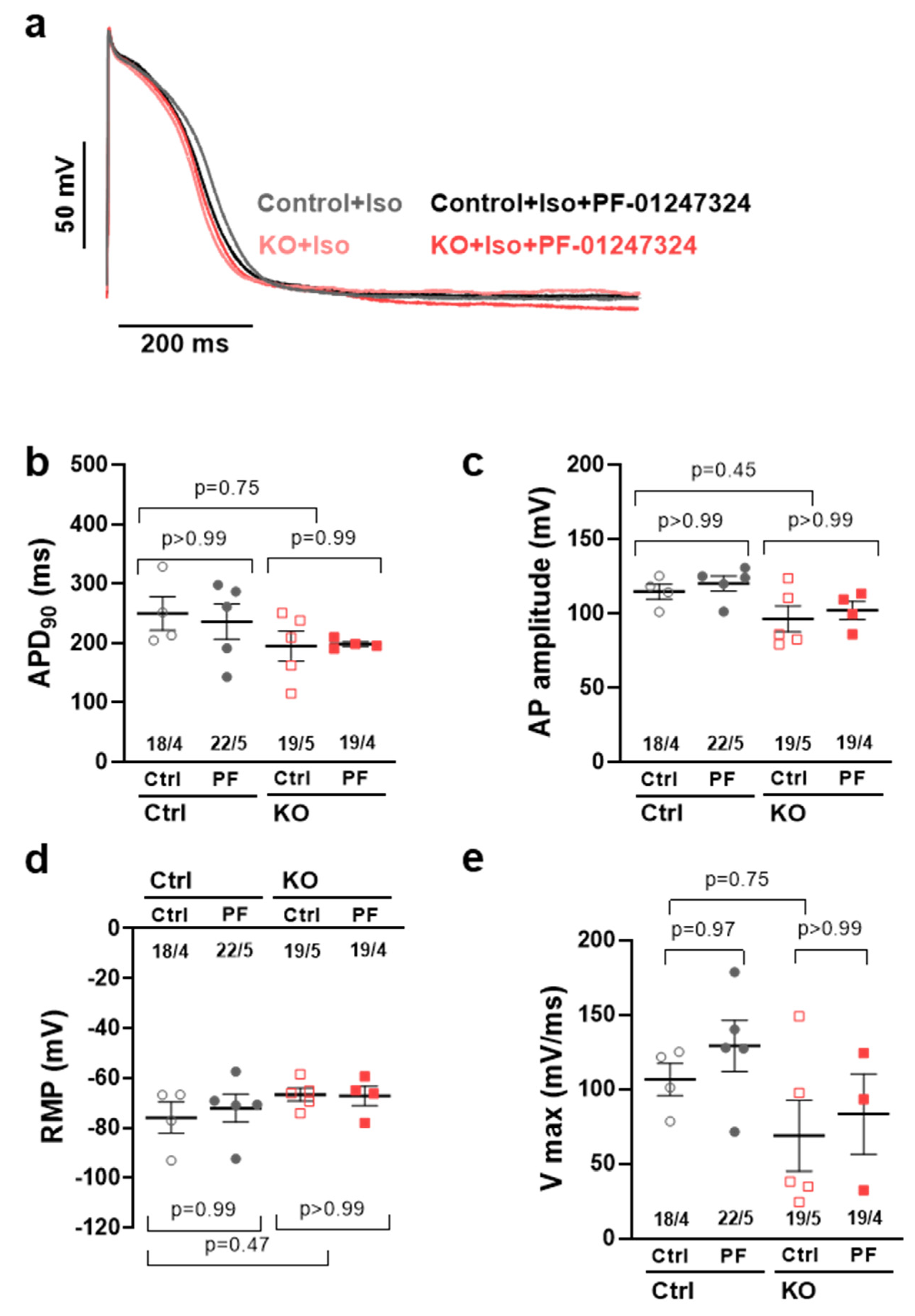
2.3. Effects of NaV1.8 on the Atrial Action Potential
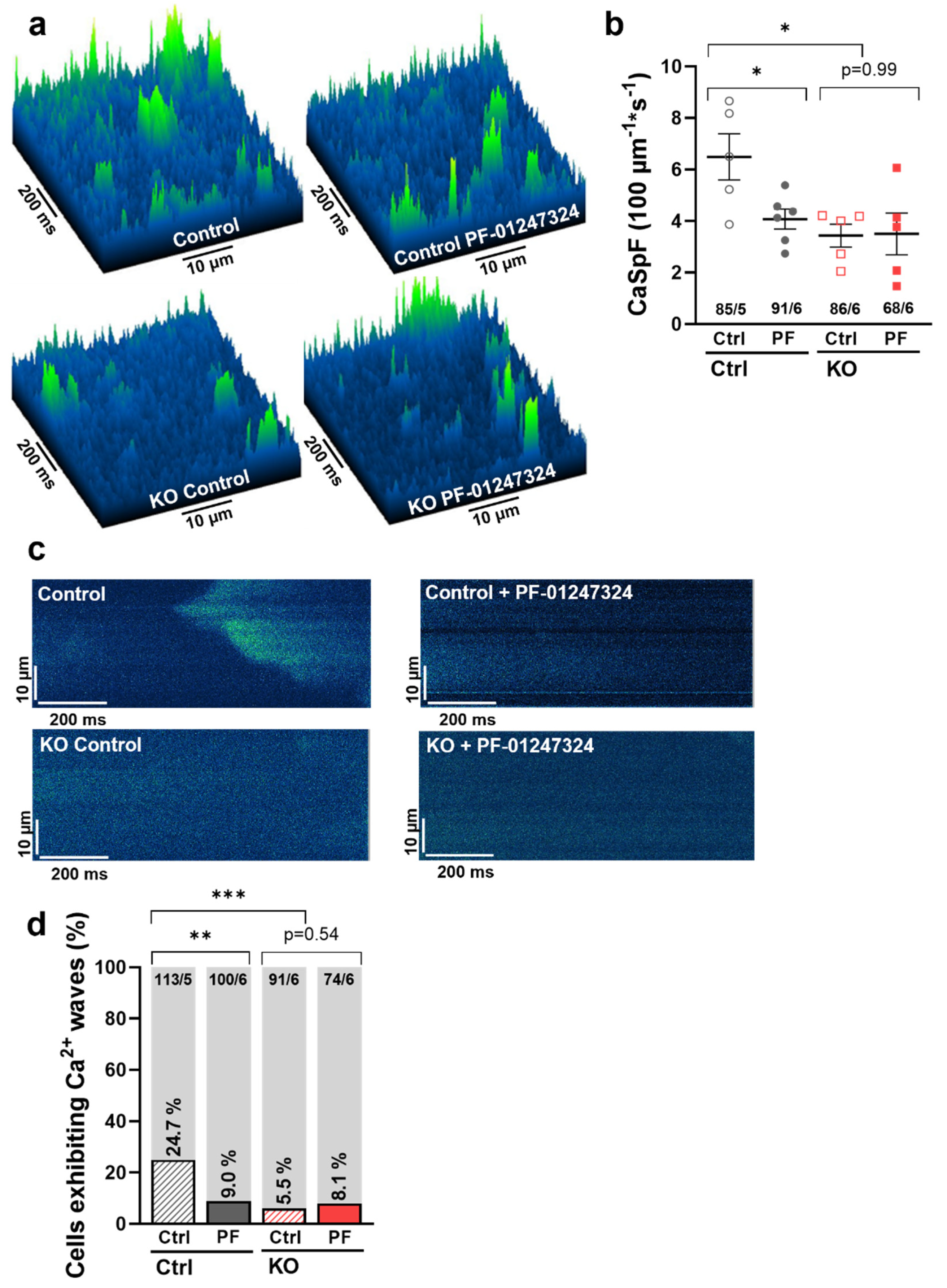
2.4. Effects of NaV1.8 on Atrial Sarcoplasmic Reticulum Ca2+ Leak and Arrhythmogenesis
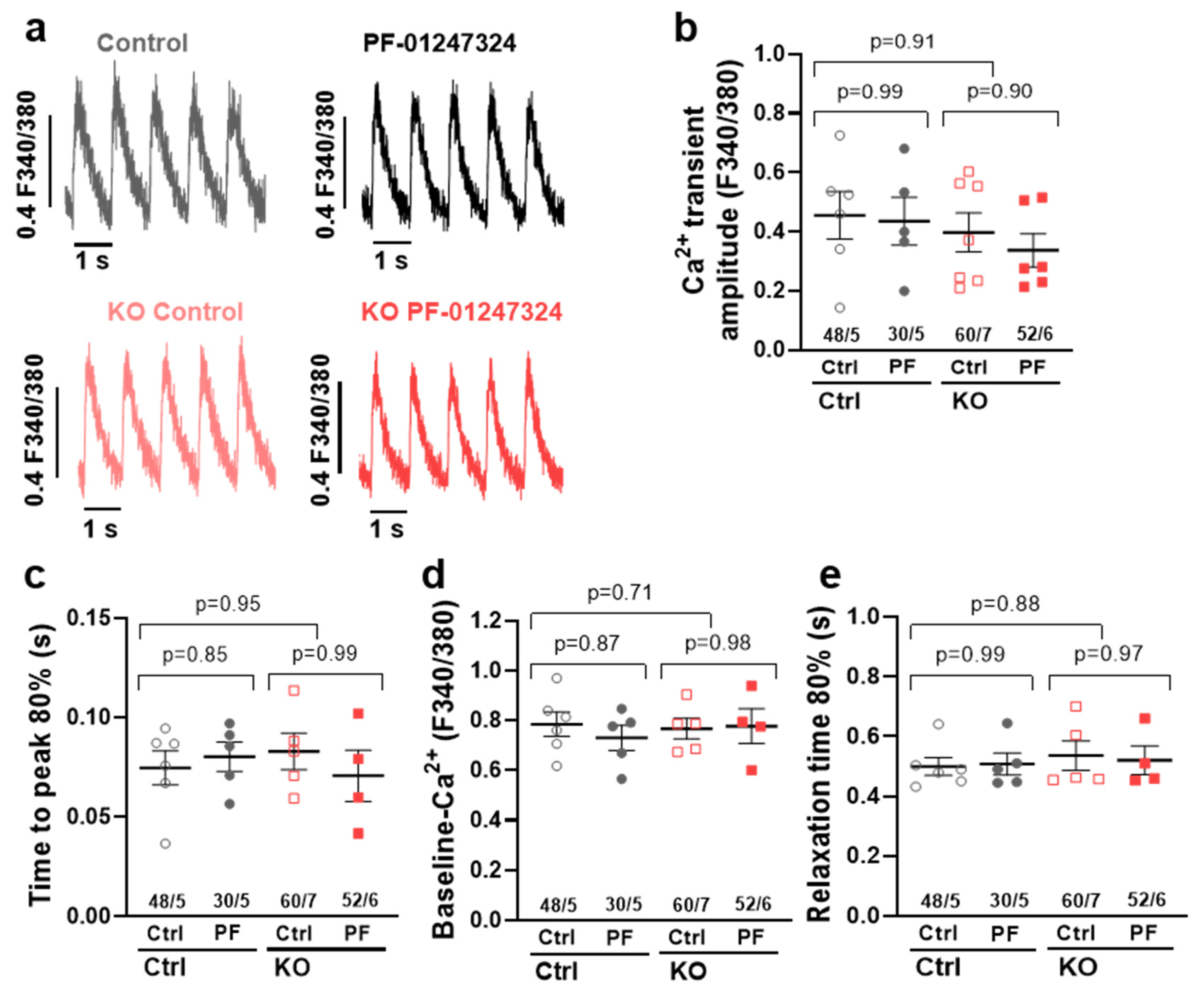
2.5. Influence of SCN10A KO on Intracellular Ca2+ Transients
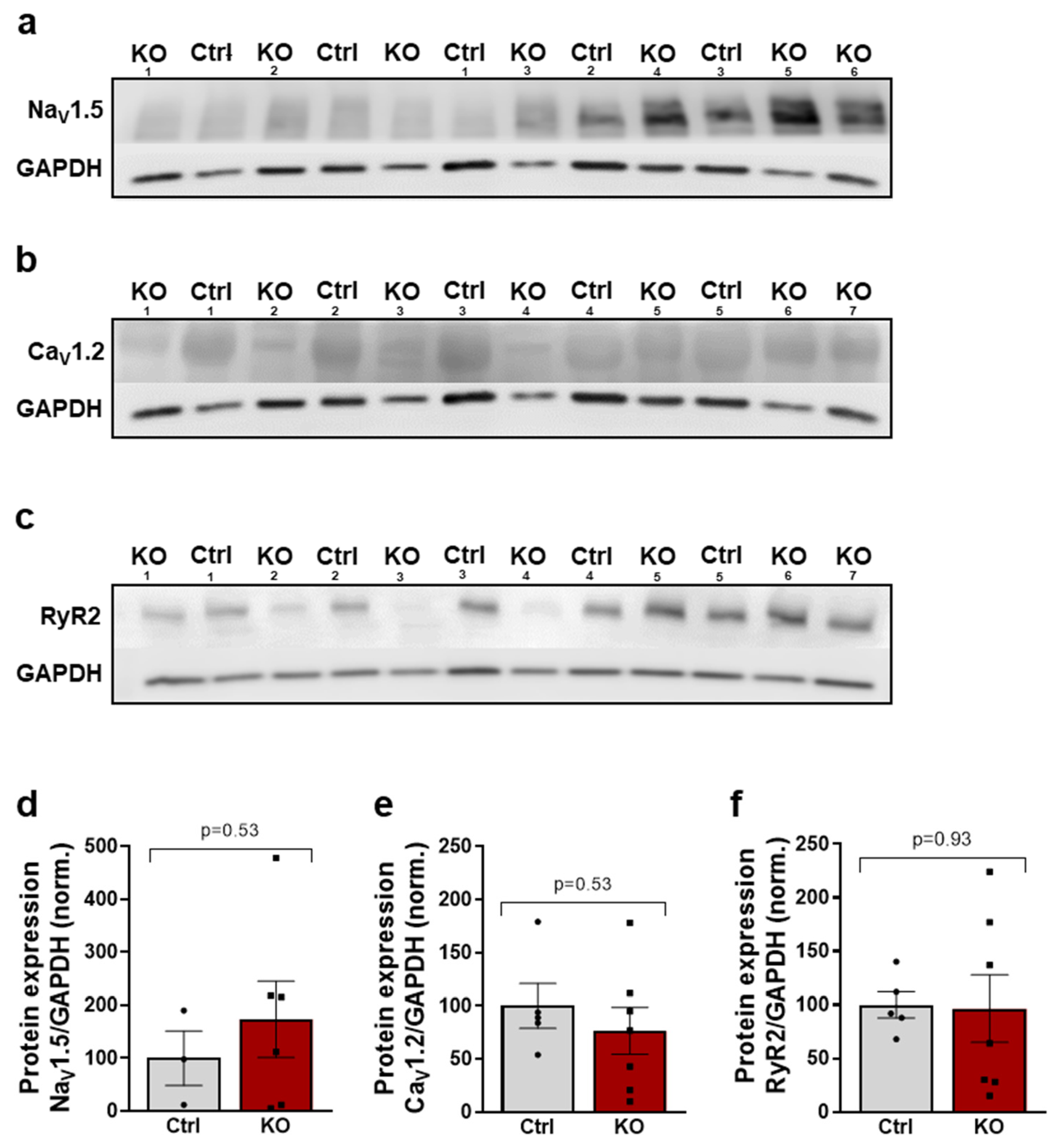
2.6. The Expression of Key Proteins of Excitation–Contraction Coupling Is Not Altered by a SCN10A KO
3. Discussion
3.1. NaV1.8 and Atrial INaL
3.2. NaV1.8 and Atrial Action Potential Duration
3.3. NaV1.8 and Atrial Ca2+ Handling
3.4. Clinical Relevance
4. Materials and Methods
4.1. Generation of Homozygous Knockout iPSCs Using CRISPR/Cas9 and Directed Differentiation into Atrial iPSC-Cardiomyocytes
4.2. Pharmacological Intervention
4.3. Patch-Clamp Experiments
4.4. Confocal Ca2+ Imaging
4.5. Epifluorescence Microscopy for Ca2+ Transient Measurements
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Valdivia, C.R.; Chu, W.W.; Pu, J.; Foell, J.D.; Haworth, R.A.; Wolff, M.R.; Kamp, T.J.; Makielski, J.C. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J. Mol. Cell. Cardiol. 2005, 38, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Maltsev, V.A.; Silverman, N.; Sabbah, H.N.; Undrovinas, A.I. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: Implications for repolarization variability. Eur. J. Heart Fail. 2007, 9, 219–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Shryock, J.C.; Belardinelli, L. An increase of late sodium current induces delayed afterdepolarizations and sustained triggered activity in atrial myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H2031–H2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sossalla, S.; Wagner, S.; Rasenack, E.C.; Ruff, H.; Weber, S.L.; Schondube, F.A.; Tirilomis, T.; Tenderich, G.; Hasenfuss, G.; Belardinelli, L.; et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts—role of late sodium current and intracellular ion accumulation. J. Mol. Cell. Cardiol. 2008, 45, 32–43. [Google Scholar] [CrossRef]
- Maltsev, V.A.; Sabbah, H.N.; Higgins, R.S.; Silverman, N.; Lesch, M.; Undrovinas, A.I. Novel, ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation 1998, 98, 2545–2552. [Google Scholar] [CrossRef]
- Undrovinas, A.I.; Maltsev, V.A.; Sabbah, H.N. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: Role of sustained inward current. Cell Mol. Life Sci. 1999, 55, 494–505. [Google Scholar] [CrossRef]
- Despa, S.; Islam, M.A.; Weber, C.R.; Pogwizd, S.M.; Bers, D.M. Intracellular Na(+) concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation 2002, 105, 2543–2548. [Google Scholar] [CrossRef] [Green Version]
- Toischer, K.; Hartmann, N.; Wagner, S.; Fischer, T.H.; Herting, J.; Danner, B.C.; Sag, C.M.; Hund, T.J.; Mohler, P.J.; Belardinelli, L.; et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J. Mol. Cell. Cardiol. 2013, 61, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Djouhri, L.; Fang, X.; Okuse, K.; Wood, J.N.; Berry, C.M.; Lawson, S.N. The TTX-resistant sodium channel Nav1.8 (SNS/PN3): Expression and correlation with membrane properties in rat nociceptive primary afferent neurons. J. Physiol. 2003, 550, 739–752. [Google Scholar] [CrossRef]
- Dib-Hajj, S.D.; Binshtok, A.M.; Cummins, T.R.; Jarvis, M.F.; Samad, T.; Zimmermann, K. Voltage-gated sodium channels in pain states: Role in pathophysiology and targets for treatment. Brain Res. Rev. 2009, 60, 65–83. [Google Scholar] [CrossRef]
- Akopian, A.N.; Sivilotti, L.; Wood, J.N. A tetrodotoxin-resistant voltage-gated sodium channel expressed by sensory neurons. Nature 1996, 379, 257–262. [Google Scholar] [CrossRef]
- Liu, M.; Wood, J.N. The roles of sodium channels in nociception: Implications for mechanisms of neuropathic pain. Pain Med. 2011, 12 (Suppl. S3), S93–S99. [Google Scholar] [CrossRef] [Green Version]
- Sotoodehnia, N.; Isaacs, A.; de Bakker, P.I.; Dorr, M.; Newton-Cheh, C.; Nolte, I.M.; van der Harst, P.; Muller, M.; Eijgelsheim, M.; Alonso, A.; et al. Common variants in 22 loci are associated with QRS duration and cardiac ventricular conduction. Nat. Genet. 2010, 42, 1068–1076. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.C.; Zhao, J.; Terracciano, C.M.; Bezzina, C.R.; Zhang, W.; Kaba, R.; Navaratnarajah, M.; Lotlikar, A.; Sehmi, J.S.; Kooner, M.K.; et al. Genetic variation in SCN10A influences cardiac conduction. Nat. Genet. 2010, 42, 149–152. [Google Scholar] [CrossRef]
- van den Boogaard, M.; Wong, L.Y.; Tessadori, F.; Bakker, M.L.; Dreizehnter, L.K.; Wakker, V.; Bezzina, C.R.; ’t Hoen, P.A.; Bakkers, J.; Barnett, P.; et al. Genetic variation in T-box binding element functionally affects SCN5A/SCN10A enhancer. J. Clin. Investig. 2012, 122, 2519–2530. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.D.; Denny, J.C.; Zuvich, R.L.; Crawford, D.C.; Schildcrout, J.S.; Bastarache, L.; Ramirez, A.H.; Mosley, J.D.; Pulley, J.M.; Basford, M.A.; et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation 2013, 127, 1377–1385. [Google Scholar] [CrossRef] [Green Version]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martinez, H.; Pfeiffer, R.; Dezi, F.; Pfeiffer, J.; Buch, T.; Betzenhauser, M.J.; Belardinelli, L.; Kahlig, K.M.; Rajamani, S.; et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J. Am. Coll. Cardiol. 2014, 64, 66–79. [Google Scholar] [CrossRef] [Green Version]
- Pabel, S.; Ahmad, S.; Tirilomis, P.; Stehle, T.; Mustroph, J.; Knierim, M.; Dybkova, N.; Bengel, P.; Holzamer, A.; Hilker, M.; et al. Inhibition of NaV1.8 prevents atrial arrhythmogenesis in human and mice. Basic Res. Cardiol. 2020, 115, 20. [Google Scholar] [CrossRef]
- Dybkova, N.; Ahmad, S.; Pabel, S.; Tirilomis, P.; Hartmann, N.; Fischer, T.H.; Bengel, P.; Tirilomis, T.; Ljubojevic, S.; Renner, A.; et al. Differential regulation of sodium channels as a novel proarrhythmic mechanism in the human failing heart. Cardiovasc. Res. 2018, 114, 1728–1737. [Google Scholar] [CrossRef]
- Bengel, P.; Ahmad, S.; Tirilomis, P.; Trum, M.; Dybkova, N.; Wagner, S.; Maier, L.S.; Hasenfuss, G.; Sossalla, S. Contribution of the neuronal sodium channel NaV1.8 to sodium- and calcium-dependent cellular proarrhythmia. J. Mol. Cell. Cardiol. 2020, 144, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Bengel, P.; Dybkova, N.; Tirilomis, P.; Ahmad, S.; Hartmann, N.; Mohamed, B.A.; Krekeler, M.C.; Maurer, W.; Pabel, S.; Trum, M.; et al. Detrimental proarrhythmogenic interaction of Ca(2+)/calmodulin-dependent protein kinase II and NaV1.8 in heart failure. Nat. Commun. 2021, 12, 6586. [Google Scholar] [CrossRef] [PubMed]
- Maurer, W.; Hartmann, N.; Argyriou, L.; Sossalla, S.; Streckfuss-Bomeke, K. Generation of homozygous Na(v)1.8 knock-out iPSC lines by CRISPR Cas9 genome editing to investigate a potential new antiarrhythmic strategy. Stem Cell Res. 2022, 60, 102677. [Google Scholar] [CrossRef] [PubMed]
- Casini, S.; Marchal, G.A.; Kawasaki, M.; Nariswari, F.A.; Portero, V.; van den Berg, N.W.E.; Guan, K.; Driessen, A.H.G.; Veldkamp, M.W.; Mengarelli, I.; et al. Absence of Functional Nav1.8 Channels in Non-diseased Atrial and Ventricular Cardiomyocytes. Cardiovasc. Drugs Ther. 2019, 33, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomstrom-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.A.; Dilaveris, P.E.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Reiffel, J.A.; Camm, A.J.; Belardinelli, L.; Zeng, D.; Karwatowska-Prokopczuk, E.; Olmsted, A.; Zareba, W.; Rosero, S.; Kowey, P. The HARMONY Trial: Combined Ranolazine and Dronedarone in the Management of Paroxysmal Atrial Fibrillation: Mechanistic and Therapeutic Synergism. Circ. Arrhythmia Electrophysiol. 2015, 8, 1048–1056. [Google Scholar] [CrossRef]
- Sossalla, S.; Kallmeyer, B.; Wagner, S.; Mazur, M.; Maurer, U.; Toischer, K.; Schmitto, J.D.; Seipelt, R.; Schondube, F.A.; Hasenfuss, G.; et al. Altered Na(+) currents in atrial fibrillation effects of ranolazine on arrhythmias and contractility in human atrial myocardium. J. Am. Coll. Cardiol. 2010, 55, 2330–2342. [Google Scholar] [CrossRef] [Green Version]
- Fischer, T.H.; Herting, J.; Mason, F.E.; Hartmann, N.; Watanabe, S.; Nikolaev, V.O.; Sprenger, J.U.; Fan, P.; Yao, L.; Popov, A.F.; et al. Late INa increases diastolic SR-Ca2+-leak in atrial myocardium by activating PKA and CaMKII. Cardiovasc. Res. 2015, 107, 184–196. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, N.; Mason, F.E.; Braun, I.; Pabel, S.; Voigt, N.; Schotola, H.; Fischer, T.H.; Dobrev, D.; Danner, B.C.; Renner, A.; et al. The combined effects of ranolazine and dronedarone on human atrial and ventricular electrophysiology. J. Mol. Cell. Cardiol. 2016, 94, 95–106. [Google Scholar] [CrossRef]
- White, C.M.; Nguyen, E. Novel Use of Ranolazine as an Antiarrhythmic Agent in Atrial Fibrillation. Ann. Pharmacother. 2017, 51, 245–252. [Google Scholar] [CrossRef]
- Ahmad, S.; Tirilomis, P.; Pabel, S.; Dybkova, N.; Hartmann, N.; Molina, C.E.; Tirilomis, T.; Kutschka, I.; Frey, N.; Maier, L.S.; et al. The functional consequences of sodium channel Na(V) 1.8 in human left ventricular hypertrophy. ESC Heart Fail. 2019, 6, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Jabbari, J.; Olesen, M.S.; Yuan, L.; Nielsen, J.B.; Liang, B.; Macri, V.; Christophersen, I.E.; Nielsen, N.; Sajadieh, A.; Ellinor, P.T.; et al. Common and rare variants in SCN10A modulate the risk of atrial fibrillation. Circ. Cardiovasc. Genet. 2015, 8, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Maier, L.S.; Sossalla, S. The late Na current as a therapeutic target: Where are we? J. Mol. Cell. Cardiol. 2013, 61, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Pfeufer, A.; van Noord, C.; Marciante, K.D.; Arking, D.E.; Larson, M.G.; Smith, A.V.; Tarasov, K.V.; Muller, M.; Sotoodehnia, N.; Sinner, M.F.; et al. Genome-wide association study of PR interval. Nat. Genet. 2010, 42, 153–159. [Google Scholar] [CrossRef]
- Dybkova, N.; Wagner, S.; Backs, J.; Hund, T.J.; Mohler, P.J.; Sowa, T.; Nikolaev, V.O.; Maier, L.S. Tubulin polymerization disrupts cardiac beta-adrenergic regulation of late INa. Cardiovasc. Res. 2014, 103, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Yang, T.; Atack, T.C.; Stroud, D.M.; Zhang, W.; Hall, L.; Roden, D.M. Blocking Scn10a channels in heart reduces late sodium current and is antiarrhythmic. Circ. Res. 2012, 111, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Savio-Galimberti, E.; Weeke, P.; Muhammad, R.; Blair, M.; Ansari, S.; Short, L.; Atack, T.C.; Kor, K.; Vanoye, C.G.; Olesen, M.S.; et al. SCN10A/Nav1.8 modulation of peak and late sodium currents in patients with early onset atrial fibrillation. Cardiovasc. Res. 2014, 104, 355–363. [Google Scholar] [CrossRef]
- Kleinsorge, M.; Cyganek, L. Subtype-Directed Differentiation of Human iPSCs into Atrial and Ventricular Cardiomyocytes. STAR Protoc. 2020, 1, 100026. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hartmann, N.; Knierim, M.; Maurer, W.; Dybkova, N.; Hasenfuß, G.; Sossalla, S.; Streckfuss-Bömeke, K. Molecular and Functional Relevance of NaV1.8-Induced Atrial Arrhythmogenic Triggers in a Human SCN10A Knock-Out Stem Cell Model. Int. J. Mol. Sci. 2023, 24, 10189. https://doi.org/10.3390/ijms241210189
Hartmann N, Knierim M, Maurer W, Dybkova N, Hasenfuß G, Sossalla S, Streckfuss-Bömeke K. Molecular and Functional Relevance of NaV1.8-Induced Atrial Arrhythmogenic Triggers in a Human SCN10A Knock-Out Stem Cell Model. International Journal of Molecular Sciences. 2023; 24(12):10189. https://doi.org/10.3390/ijms241210189
Chicago/Turabian StyleHartmann, Nico, Maria Knierim, Wiebke Maurer, Nataliya Dybkova, Gerd Hasenfuß, Samuel Sossalla, and Katrin Streckfuss-Bömeke. 2023. "Molecular and Functional Relevance of NaV1.8-Induced Atrial Arrhythmogenic Triggers in a Human SCN10A Knock-Out Stem Cell Model" International Journal of Molecular Sciences 24, no. 12: 10189. https://doi.org/10.3390/ijms241210189