

Epidermal Loss of RORα Enhances Skin Inflammation in a MC903-Induced Mouse Model of Atopic Dermatitis
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
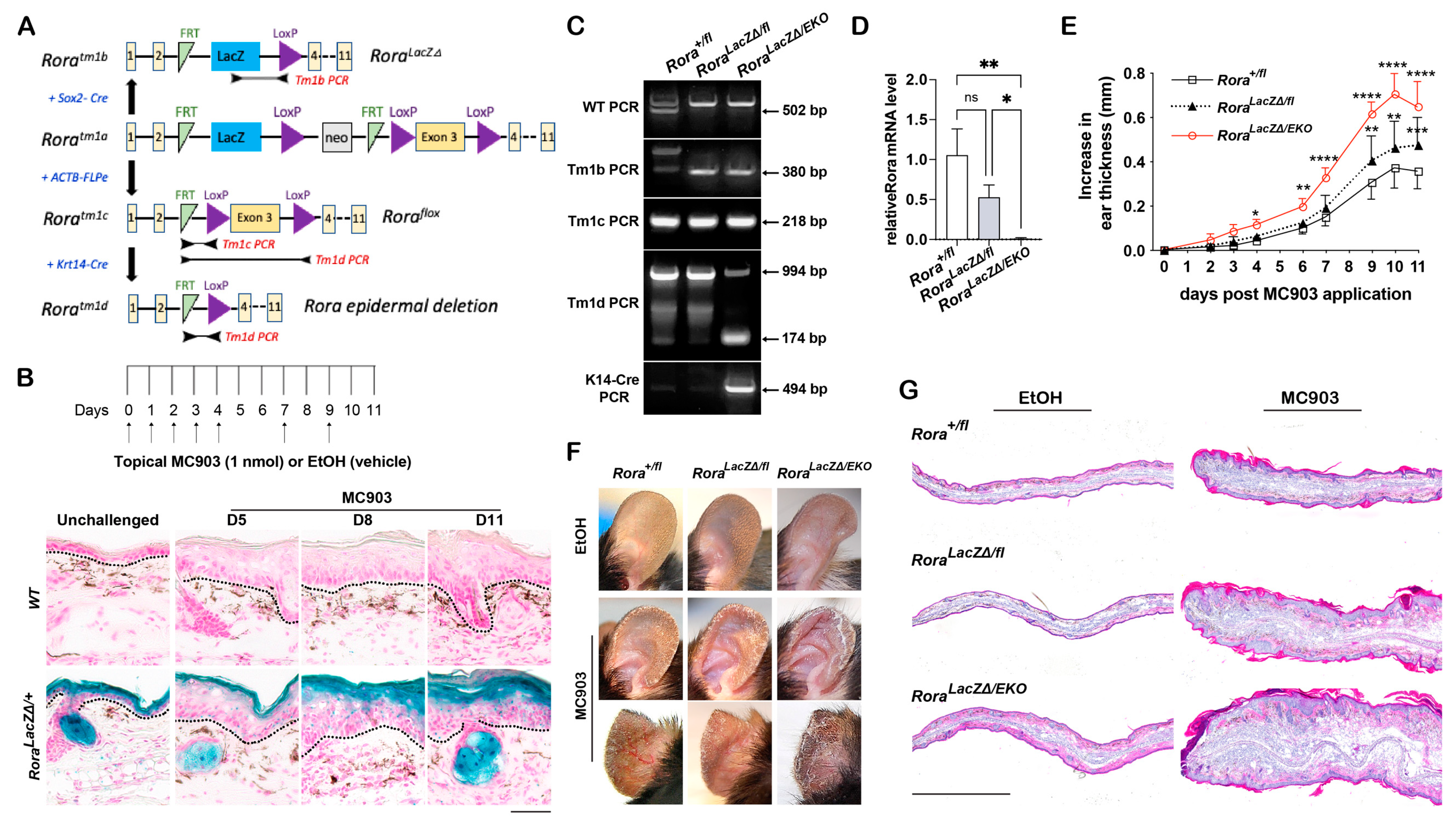
2.1. Epidermal Rora Expression in Mouse Skin in Response to MC903
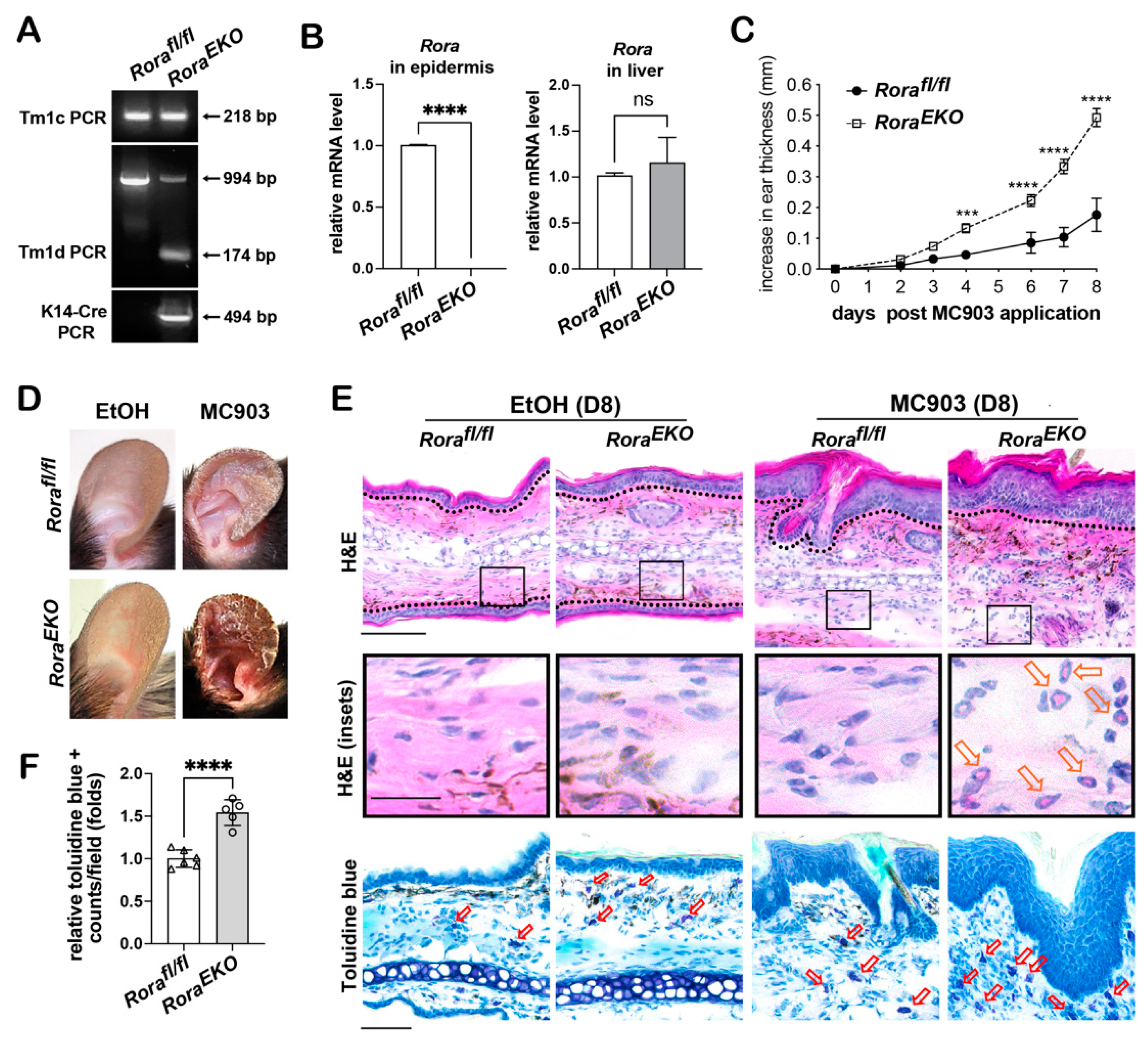
2.2. Epidermal Rora Ablation Exaggerates MC903-Induced Ear Thickening
2.3. Epidermal Rora Deletion Aggravates MC903-Induced AD-like Symptoms
2.4. Epidermal Loss of RORα Accelerates MC903-Induced Dermal Activation of Dendritic Cells and CD4+ T Cells
2.5. Epidermal Rora Deletion Increases Tslp Gene Transcription
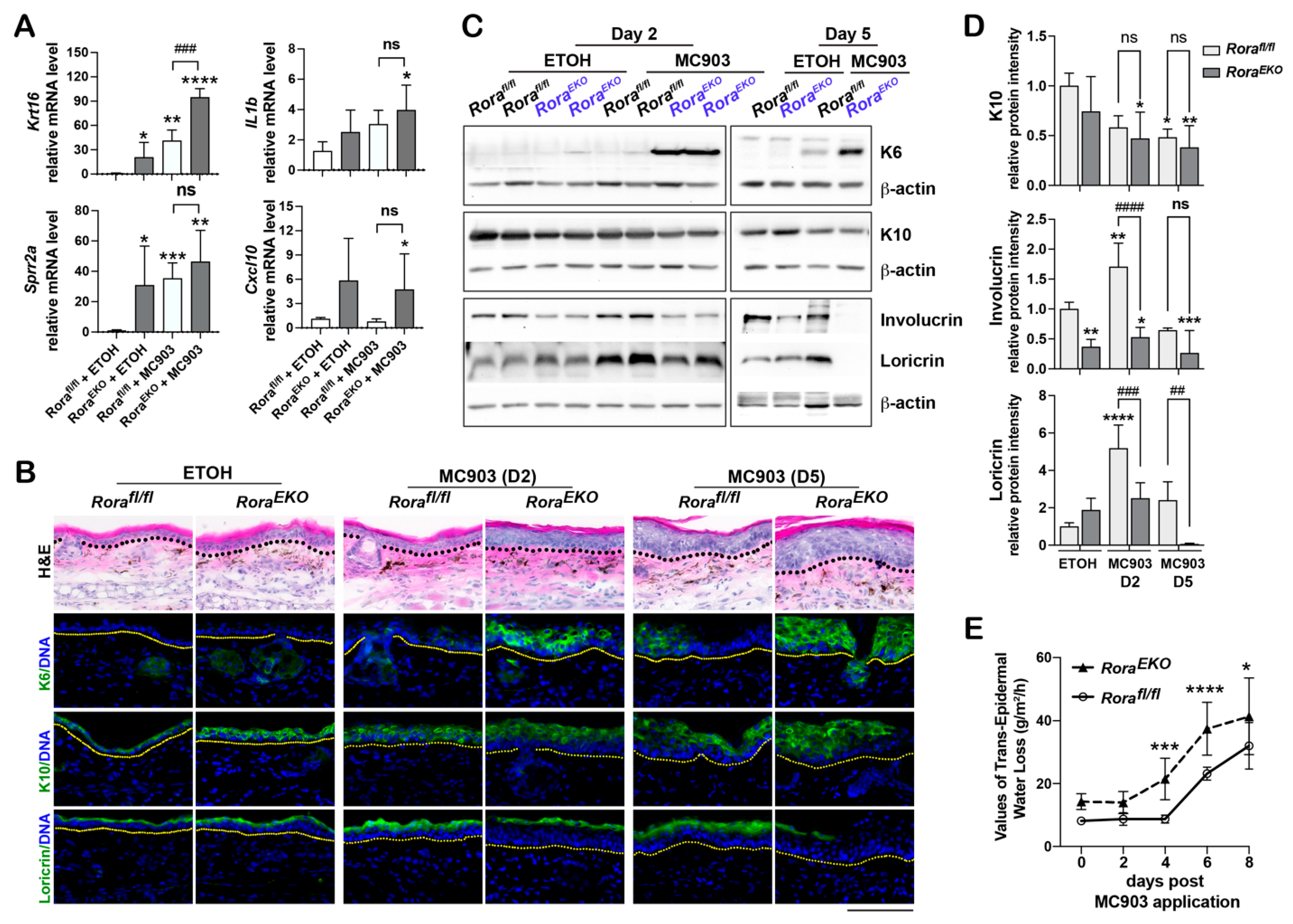
2.6. Epidermal Loss of RORα Deregulates Keratinocyte Differentiation and Accelerates Skin Barrier Disruption in Response to MC903
3. Discussion
4. Materials and Methods
4.1. Mice
4.2. Genotyping
4.3. MC903-Induced AD-like Model
4.4. X-Gal Staining
4.5. Histology and Immunofluorescence
4.6. Quantitative Real-Time (RT)-PCR
4.7. ELISA
4.8. Transepidermal Water Loss Measurements
4.9. Western Blot Analysis
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic dermatitis. Nat. Rev. Dis. Prim. 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drislane, C.; Irvine, A.D. The role of filaggrin in atopic dermatitis and allergic disease. Ann. Allergy Asthma Immunol. 2020, 124, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosbrugger-Martinz, V.; Leprince, C.; Méchin, M.C.; Simon, M.; Blunder, S.; Gruber, R.; Dubrac, S. Revisiting the Roles of Filaggrin in Atopic Dermatitis. Int. J. Mol. Sci. 2022, 23, 5318. [Google Scholar] [CrossRef]
- Leung, D.Y.M.; Berdyshev, E.; Goleva, E. Cutaneous barrier dysfunction in allergic diseases. J. Allergy Clin. Immunol. 2020, 145, 1485–1497. [Google Scholar] [CrossRef]
- Elias, P.M.; Hatano, Y.; Williams, M.L. Basis for the barrier abnormality in atopic dermatitis: Outside-inside-outside pathogenic mechanisms. J. Allergy Clin. Immunol. 2008, 121, 1337–1343. [Google Scholar] [CrossRef] [Green Version]
- Blanpain, C.; Fuchs, E. Epidermal homeostasis: A balancing act of stem cells in the skin. Nat. Rev. Mol. Cell Biol. 2009, 10, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, T.; Oka, T.; Demehri, S. Alarmin Cytokines as Central Regulators of Cutaneous Immunity. Front. Immunol. 2022, 13, 876515. [Google Scholar] [CrossRef]
- Pandey, A.; Ozaki, K.; Baumann, H.; Levin, S.D.; Puel, A.; Farr, A.G.; Ziegler, S.F.; Leonard, W.J.; Lodish, H.F. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat. Immunol. 2000, 1, 59–64. [Google Scholar] [CrossRef]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef] [Green Version]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestle, F.O.; Di Meglio, P.; Qin, J.Z.; Nickoloff, B.J. Skin immune sentinels in health and disease. Nat. Rev. Immunol. 2009, 9, 679–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chieosilapatham, P.; Kiatsurayanon, C.; Umehara, Y.; Trujillo-Paez, J.V.; Peng, G.; Yue, H.; Nguyen, L.T.H.; Niyonsaba, F. Keratinocytes: Innate immune cells in atopic dermatitis. Clin. Exp. Immunol. 2021, 204, 296–309. [Google Scholar] [CrossRef] [PubMed]
- Jetten, A.M. Retinoid-related orphan receptors (RORs): Critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl. Recept. Signal 2009, 7, e003. [Google Scholar] [CrossRef] [Green Version]
- Jetten, A.M.; Joo, J.H. Retinoid-related Orphan Receptors (RORs): Roles in Cellular Differentiation and Development. Adv. Dev. Biol. 2006, 16, 313–355. [Google Scholar]
- Giguere, V.; Tini, M.; Flock, G.; Ong, E.; Evans, R.M.; Otulakowski, G. Isoform-specific amino-terminal domains dictate DNA-binding properties of ROR alpha, a novel family of orphan hormone nuclear receptors. Genes Dev. 1994, 8, 538–553. [Google Scholar] [CrossRef] [Green Version]
- Hadj-Sahraoui, N.; Frederic, F.; Zanjani, H.; Delhaye-Bouchaud, N.; Herrup, K.; Mariani, J. Progressive atrophy of cerebellar Purkinje cell dendrites during aging of the heterozygous staggerer mouse (Rora(+/sg)). Brain Res. Dev. Brain Res. 2001, 126, 201–209. [Google Scholar] [CrossRef]
- Hamilton, B.A.; Frankel, W.N.; Kerrebrock, A.W.; Hawkins, T.L.; FitzHugh, W.; Kusumi, K.; Russell, L.B.; Mueller, K.L.; van Berkel, V.; Birren, B.W.; et al. Disruption of the nuclear hormone receptor RORalpha in staggerer mice. Nature 1996, 379, 736–739. [Google Scholar] [CrossRef]
- Meyer, T.; Kneissel, M.; Mariani, J.; Fournier, B. In vitro and in vivo evidence for orphan nuclear receptor RORalpha function in bone metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 9197–9202. [Google Scholar] [CrossRef] [Green Version]
- Dzhagalov, I.; Giguere, V.; He, Y.W. Lymphocyte development and function in the absence of retinoic acid-related orphan receptor alpha. J. Immunol. 2004, 173, 2952–2959. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, A.C.F.; Szeto, A.C.H.; Heycock, M.W.D.; Clark, P.A.; Walker, J.A.; Crisp, A.; Barlow, J.L.; Kitching, S.; Lim, A.; Gogoi, M.; et al. RORalpha is a critical checkpoint for T cell and ILC2 commitment in the embryonic thymus. Nat. Immunol. 2021, 22, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Fitzsimmons, R.L.; Raichur, S.; Wang, S.C.; Lechtken, A.; Muscat, G.E. The orphan nuclear receptor, RORalpha, regulates gene expression that controls lipid metabolism: Staggerer (SG/SG) mice are resistant to diet-induced obesity. J. Biol. Chem. 2008, 283, 18411–18421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, X.; Jin, W.; Bai, X.; Zhao, X.; Shao, J.; Li, J.; Sun, Q.; Su, B.; Wang, X.; Yang, X.O.; et al. RORalpha is critical for mTORC1 activity in T cell-mediated colitis. Cell Rep. 2021, 36, 109682. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.K.; Kim, D.; Kim, K.; Boo, K.; Yu, Y.S.; Kim, I.S.; Jeon, Y.; Im, S.K.; Lee, S.H.; Lee, J.M.; et al. RORα is crucial for attenuated inflammatory response to maintain intestinal homeostasis. Proc. Natl. Acad. Sci. USA 2019, 116, 21140–21149. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, N.; Leyva-Castillo, J.M.; Jadhav, U.; Barreiro, O.; Kam, C.; O’Neill, N.K.; Meylan, F.; Chambon, P.; von Andrian, U.H.; Siegel, R.M.; et al. RORα-expressing T regulatory cells restrain allergic skin inflammation. Sci. Immunol. 2018, 3, eaao6923. [Google Scholar] [CrossRef] [Green Version]
- Akashi, M.; Takumi, T. The orphan nuclear receptor RORalpha regulates circadian transcription of the mammalian core-clock Bmal1. Nat. Struct. Mol. Biol. 2005, 12, 441–448. [Google Scholar] [CrossRef]
- Sato, T.K.; Panda, S.; Miraglia, L.J.; Reyes, T.M.; Rudic, R.D.; McNamara, P.; Naik, K.A.; FitzGerald, G.A.; Kay, S.A.; Hogenesch, J.B. A functional genomics strategy reveals Rora as a component of the mammalian circadian clock. Neuron 2004, 43, 527–537. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Walker, J.A.; Jolin, H.E.; Drynan, L.F.; Hams, E.; Camelo, A.; Barlow, J.L.; Neill, D.R.; Panova, V.; Koch, U.; et al. Transcription factor RORalpha is critical for nuocyte development. Nat. Immunol. 2012, 13, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Salimi, M.; Barlow, J.L.; Saunders, S.P.; Xue, L.; Gutowska-Owsiak, D.; Wang, X.; Huang, L.C.; Johnson, D.; Scanlon, S.T.; McKenzie, A.N.; et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med. 2013, 210, 2939–2950. [Google Scholar] [CrossRef]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, J.; Brooks, Y.; Lefort, K.; Getsios, S.; Dotto, G.P. The retinoid-related orphan receptor RORalpha promotes keratinocyte differentiation via FOXN1. PLoS ONE 2013, 8, e70392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slominski, A.; Fischer, T.W.; Zmijewski, M.A.; Wortsman, J.; Semak, I.; Zbytek, B.; Slominski, R.M.; Tobin, D.J. On the role of melatonin in skin physiology and pathology. Endocrine 2005, 27, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Hanyu, O.; Nakae, H.; Miida, T.; Higashi, Y.; Fuda, H.; Endo, M.; Kohjitani, A.; Sone, H.; Strott, C.A. Cholesterol sulfate induces expression of the skin barrier protein filaggrin in normal human epidermal keratinocytes through induction of RORalpha. Biochem. Biophys. Res. Commun. 2012, 428, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Steinmayr, M.; Andre, E.; Conquet, F.; Rondi-Reig, L.; Delhaye-Bouchaud, N.; Auclair, N.; Daniel, H.; Crepel, F.; Mariani, J.; Sotelo, C.; et al. Staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 3960–3965. [Google Scholar] [CrossRef] [Green Version]
- Tsujihana, K.; Tanegashima, K.; Santo, Y.; Yamada, H.; Akazawa, S.; Nakao, R.; Tominaga, K.; Saito, R.; Nishito, Y.; Hata, R.I.; et al. Circadian protection against bacterial skin infection by epidermal CXCL14-mediated innate immunity. Proc. Natl. Acad. Sci. USA 2022, 119, e2116027119. [Google Scholar] [CrossRef]
- Brożyna, A.A.; Żmijewski, M.A.; Linowiecka, K.; Kim, T.K.; Slominski, R.M.; Slominski, A.T. Disturbed expression of vitamin D and retinoic acid-related orphan receptors α and γ and of megalin in inflammatory skin diseases. Exp. Dermatol. 2022, 31, 781–788. [Google Scholar] [CrossRef]
- Guttman-Yassky, E.; Suárez-Fariñas, M.; Chiricozzi, A.; Nograles, K.E.; Shemer, A.; Fuentes-Duculan, J.; Cardinale, I.; Lin, P.; Bergman, R.; Bowcock, A.M.; et al. Broad defects in epidermal cornification in atopic dermatitis identified through genomic analysis. J. Allergy Clin. Immunol. 2009, 124, 1235–1244.e58. [Google Scholar] [CrossRef]
- Coleman, J.L.; Brennan, K.; Ngo, T.; Balaji, P.; Graham, R.M.; Smith, N.J. Rapid Knockout and Reporter Mouse Line Generation and Breeding Colony Establishment Using EUCOMM Conditional-Ready Embryonic Stem Cells: A Case Study. Front. Endocrinol. 2015, 6, 105. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T.; et al. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Walsh, C.M.; Hill, R.Z.; Schwendinger-Schreck, J.; Deguine, J.; Brock, E.C.; Kucirek, N.; Rifi, Z.; Wei, J.; Gronert, K.; Brem, R.B.; et al. Neutrophils promote CXCR3-dependent itch in the development of atopic dermatitis. eLife 2019, 8, e48448. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hener, P.; Zhang, Z.; Kato, S.; Metzger, D.; Chambon, P. Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis. Proc. Natl. Acad. Sci. USA 2006, 103, 11736–11741. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hener, P.; Zhang, Z.; Ganti, K.P.; Metzger, D.; Chambon, P. Induction of thymic stromal lymphopoietin expression in keratinocytes is necessary for generating an atopic dermatitis upon application of the active vitamin D3 analogue MC903 on mouse skin. J. Investig. Dermatol. 2009, 129, 498–502. [Google Scholar] [CrossRef]
- Liu, Y.J. TSLP in epithelial cell and dendritic cell cross talk. Adv. Immunol. 2009, 101, 1–25. [Google Scholar] [PubMed] [Green Version]
- Liu, Y.J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.H.; Malefyt Rde, W.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Strugnell, S.A.; DeLuca, H.F. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar] [CrossRef] [Green Version]
- Carregaro, F.; Stefanini, A.C.; Henrique, T.; Tajara, E.H. Study of small proline-rich proteins (SPRRs) in health and disease: A review of the literature. Arch. Dermatol. Res. 2013, 305, 857–866. [Google Scholar] [CrossRef]
- Scharschmidt, T.C.; Man, M.Q.; Hatano, Y.; Crumrine, D.; Gunathilake, R.; Sundberg, J.P.; Silva, K.A.; Mauro, T.M.; Hupe, M.; Cho, S.; et al. Filaggrin deficiency confers a paracellular barrier abnormality that reduces inflammatory thresholds to irritants and haptens. J. Allergy Clin. Immunol. 2009, 124, 496–506.e6. [Google Scholar] [CrossRef] [Green Version]
- Muhandes, L.; Chapsa, M.; Pippel, M.; Behrendt, R.; Ge, Y.; Dahl, A.; Yi, B.; Dalpke, A.; Winkler, S.; Hiller, M.; et al. Low Threshold for Cutaneous Allergen Sensitization but No Spontaneous Dermatitis or Atopy in FLG-Deficient Mice. J. Investig. Dermatol. 2021, 141, 2611–2619.e2. [Google Scholar] [CrossRef]
- Wang, F.; Chen, S.; Liu, H.B.; Parent, C.A.; Coulombe, P.A. Keratin 6 regulates collective keratinocyte migration by altering cell-cell and cell-matrix adhesion. J. Cell Biol. 2018, 217, 4314–4330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yin, M.; Zhang, L.J. Keratin 6, 16 and 17-Critical Barrier Alarmin Molecules in Skin Wounds and Psoriasis. Cells 2019, 8, 807. [Google Scholar] [CrossRef] [Green Version]
- Jensen, J.M.; Fölster-Holst, R.; Baranowsky, A.; Schunck, M.; Winoto-Morbach, S.; Neumann, C.; Schütze, S.; Proksch, E. Impaired sphingomyelinase activity and epidermal differentiation in atopic dermatitis. J. Investig. Dermatol. 2004, 122, 1423–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kypriotou, M.; Boéchat, C.; Huber, M.; Hohl, D. Spontaneous atopic dermatitis-like symptoms in a/a ma ft/ma ft/J flaky tail mice appear early after birth. PLoS ONE 2013, 8, e67869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallon, P.G.; Sasaki, T.; Sandilands, A.; Campbell, L.E.; Saunders, S.P.; Mangan, N.E.; Callanan, J.J.; Kawasaki, H.; Shiohama, A.; Kubo, A.; et al. A homozygous frameshift mutation in the mouse Flg gene facilitates enhanced percutaneous allergen priming. Nat. Genet. 2009, 41, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Koch, P.J.; de Viragh, P.A.; Scharer, E.; Bundman, D.; Longley, M.A.; Bickenbach, J.; Kawachi, Y.; Suga, Y.; Zhou, Z.; Huber, M.; et al. Lessons from loricrin-deficient mice: Compensatory mechanisms maintaining skin barrier function in the absence of a major cornified envelope protein. J. Cell Biol. 2000, 151, 389–400. [Google Scholar] [CrossRef]
- Segre, J.A.; Bauer, C.; Fuchs, E. Klf4 is a transcription factor required for establishing the barrier function of the skin. Nat. Genet. 1999, 22, 356–360. [Google Scholar] [CrossRef]
- Suárez-Fariñas, M.; Tintle, S.J.; Shemer, A.; Chiricozzi, A.; Nograles, K.; Cardinale, I.; Duan, S.; Bowcock, A.M.; Krueger, J.G.; Guttman-Yassky, E. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J. Allergy Clin. Immunol. 2011, 127, 954–964.e4. [Google Scholar] [CrossRef] [Green Version]
- Leitch, C.S.; Natafji, E.; Yu, C.; Abdul-Ghaffar, S.; Madarasingha, N.; Venables, Z.C.; Chu, R.; Fitch, P.M.; Muinonen-Martin, A.J.; Campbell, L.E.; et al. Filaggrin-null mutations are associated with increased maturation markers on Langerhans cells. J. Allergy Clin. Immunol. 2016, 138, 482–490.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, M.D.; Kim, B.E.; Gao, P.; Grant, A.V.; Boguniewicz, M.; Debenedetto, A.; Schneider, L.; Beck, L.A.; Barnes, K.C.; Leung, D.Y. Cytokine modulation of atopic dermatitis filaggrin skin expression. J. Allergy Clin. Immunol. 2007, 120, 150–155. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.E.; Leung, D.Y.; Boguniewicz, M.; Howell, M.D. Loricrin and involucrin expression is down-regulated by Th2 cytokines through STAT-6. Clin. Immunol. 2008, 126, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Kim, H.; Sung, G.Y. An Interleukin-4 and Interleukin-13 Induced Atopic Dermatitis Human Skin Equivalent Model by a Skin-On-A-Chip. Int. J. Mol. Sci. 2022, 23, 2116. [Google Scholar] [CrossRef] [PubMed]
- Brauweiler, A.M.; Leung, D.Y.M.; Goleva, E. The Transcription Factor p63 Is a Direct Effector of IL-4- and IL-13-Mediated Repression of Keratinocyte Differentiation. J. Investig. Dermatol. 2021, 141, 770–778. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Choo, M.K.; Park, J.M.; Fisher, D.E. Topical ROR Inverse Agonists Suppress Inflammation in Mouse Models of Atopic Dermatitis and Acute Irritant Dermatitis. J. Investig. Dermatol. 2017, 137, 2523–2531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solt, L.A.; Kumar, N.; Nuhant, P.; Wang, Y.; Lauer, J.L.; Liu, J.; Istrate, M.A.; Kamenecka, T.M.; Roush, W.R.; Vidovic, D.; et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011, 472, 491–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solt, L.A.; Burris, T.P. Action of RORs and their ligands in (patho)physiology. Trends Endocrinol. Metab. 2012, 23, 619–627. [Google Scholar] [CrossRef] [Green Version]
- Park, K.C.; Kim, J.; Lee, A.; Lim, J.S.; Kim, K.I. Alleviation of imiquimod-induced psoriasis-like symptoms in Rorα-deficient mouse skin. BMB Rep. 2023, 56, 296. [Google Scholar] [CrossRef] [PubMed]
- Flutter, B.; Nestle, F.O. TLRs to cytokines: Mechanistic insights from the imiquimod mouse model of psoriasis. Eur. J. Immunol. 2013, 43, 3138–3146. [Google Scholar] [CrossRef]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Hayashi, S.; Lewis, P.; Pevny, L.; McMahon, A.P. Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Gene Expr. Patterns 2002, 2, 93–97. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hua, X.; Blosch, C.D.; Dorsey, H.; Ficaro, M.K.; Wallace, N.L.; Hsung, R.P.; Dai, J. Epidermal Loss of RORα Enhances Skin Inflammation in a MC903-Induced Mouse Model of Atopic Dermatitis. Int. J. Mol. Sci. 2023, 24, 10241. https://doi.org/10.3390/ijms241210241
Hua X, Blosch CD, Dorsey H, Ficaro MK, Wallace NL, Hsung RP, Dai J. Epidermal Loss of RORα Enhances Skin Inflammation in a MC903-Induced Mouse Model of Atopic Dermatitis. International Journal of Molecular Sciences. 2023; 24(12):10241. https://doi.org/10.3390/ijms241210241
Chicago/Turabian StyleHua, Xiangmei, Conrad Dean Blosch, Hannah Dorsey, Maria K. Ficaro, Nicole L. Wallace, Richard P. Hsung, and Jun Dai. 2023. "Epidermal Loss of RORα Enhances Skin Inflammation in a MC903-Induced Mouse Model of Atopic Dermatitis" International Journal of Molecular Sciences 24, no. 12: 10241. https://doi.org/10.3390/ijms241210241
APA StyleHua, X., Blosch, C. D., Dorsey, H., Ficaro, M. K., Wallace, N. L., Hsung, R. P., & Dai, J. (2023). Epidermal Loss of RORα Enhances Skin Inflammation in a MC903-Induced Mouse Model of Atopic Dermatitis. International Journal of Molecular Sciences, 24(12), 10241. https://doi.org/10.3390/ijms241210241