

Single-Molecule Analysis of the Improved Variants of the G-Quadruplex Recognition Protein G4P



Abstract
:1. Introduction
2. Results
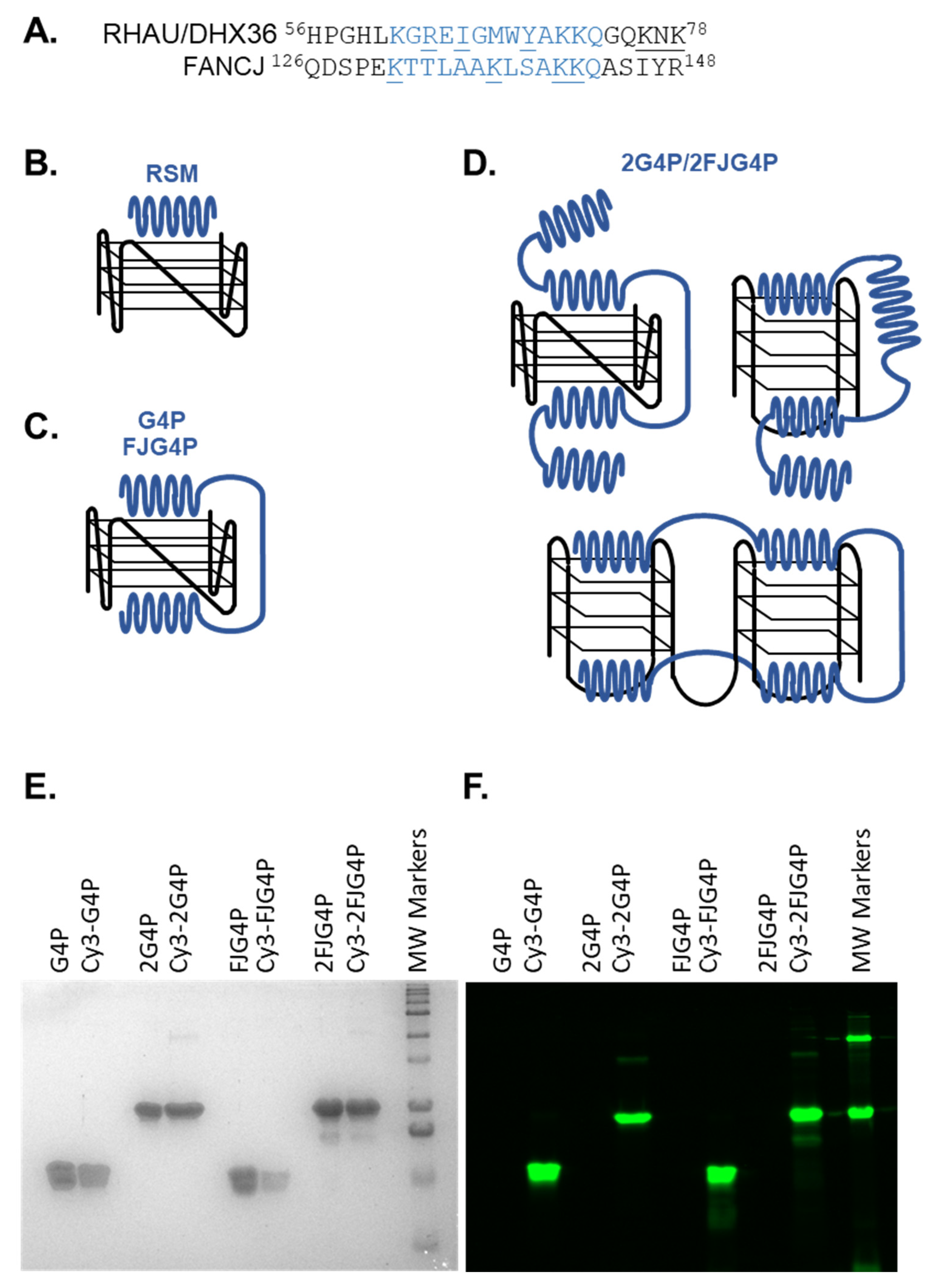
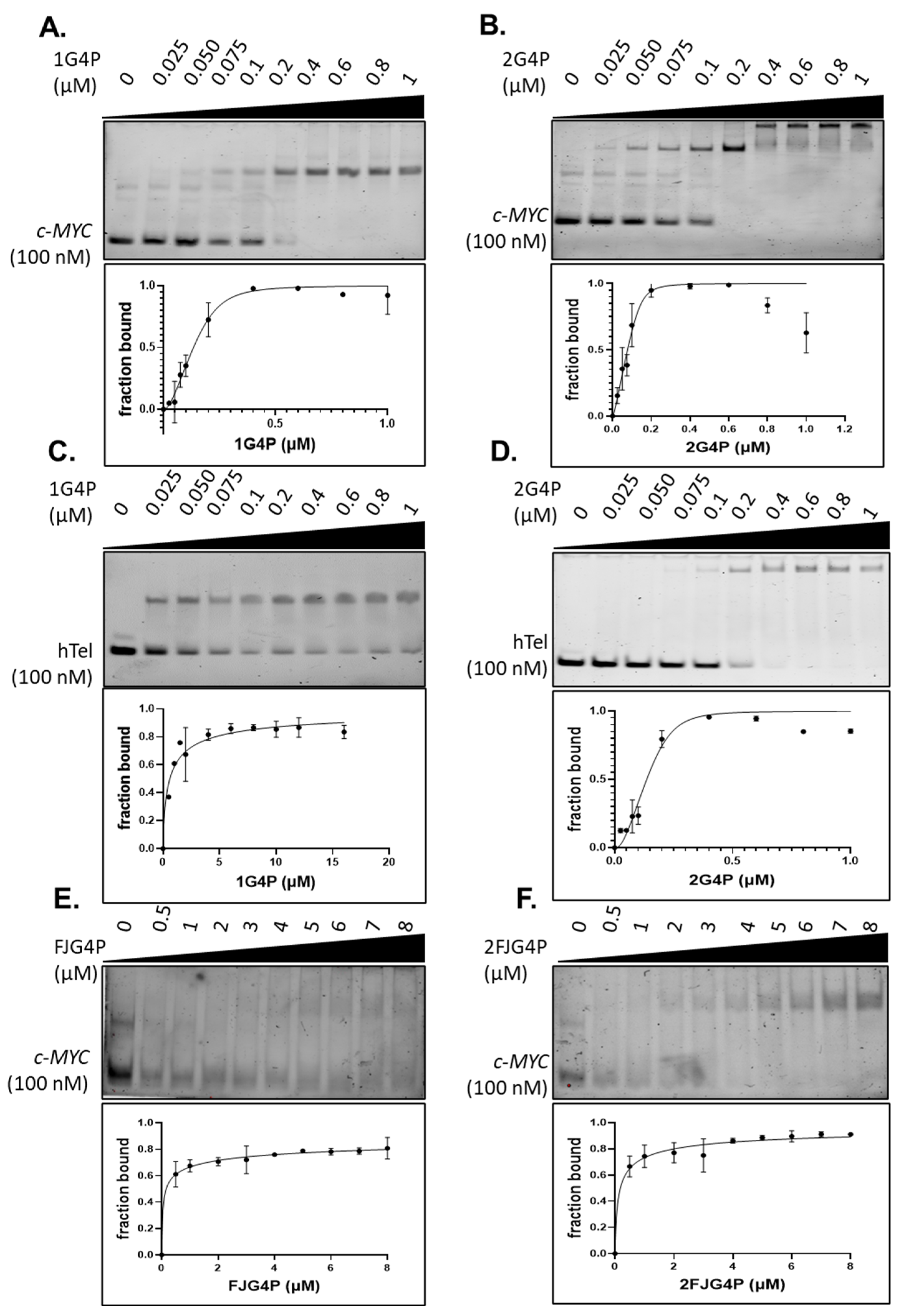
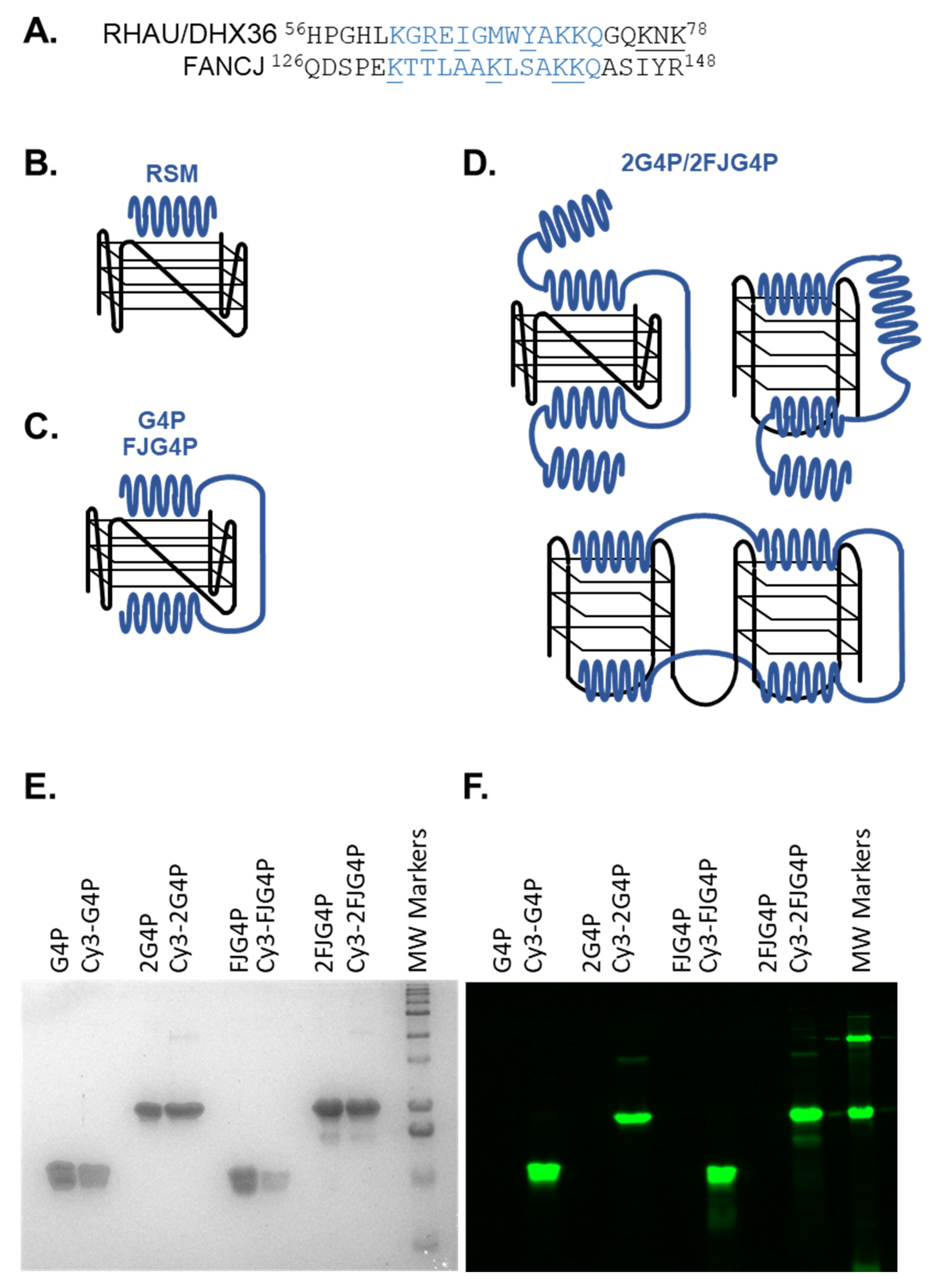
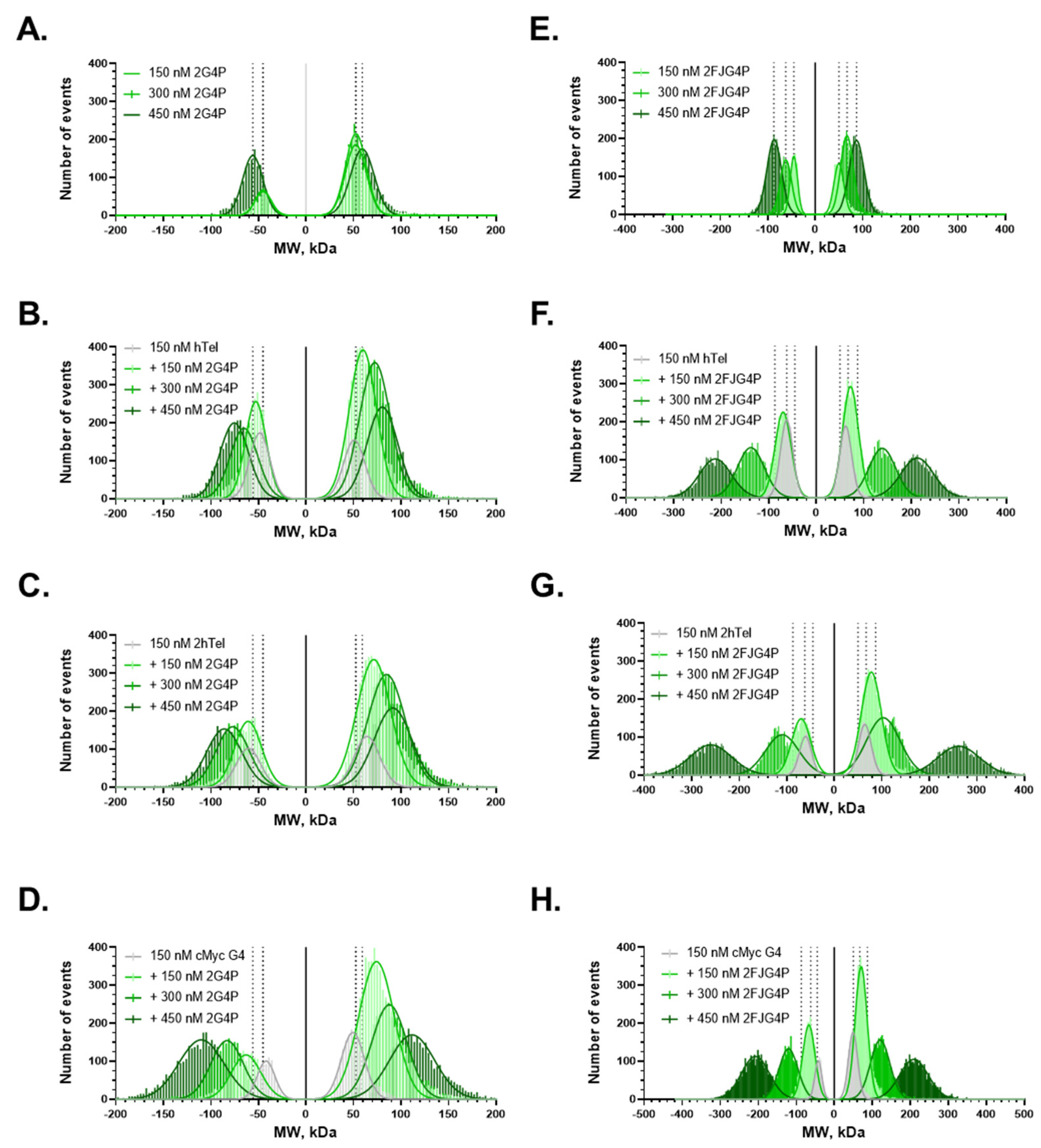
2.1. Affinity of the G4P Protein for G-Quadruplexes Can Be Improved by Increasing the Number of G4-Recognition Elements
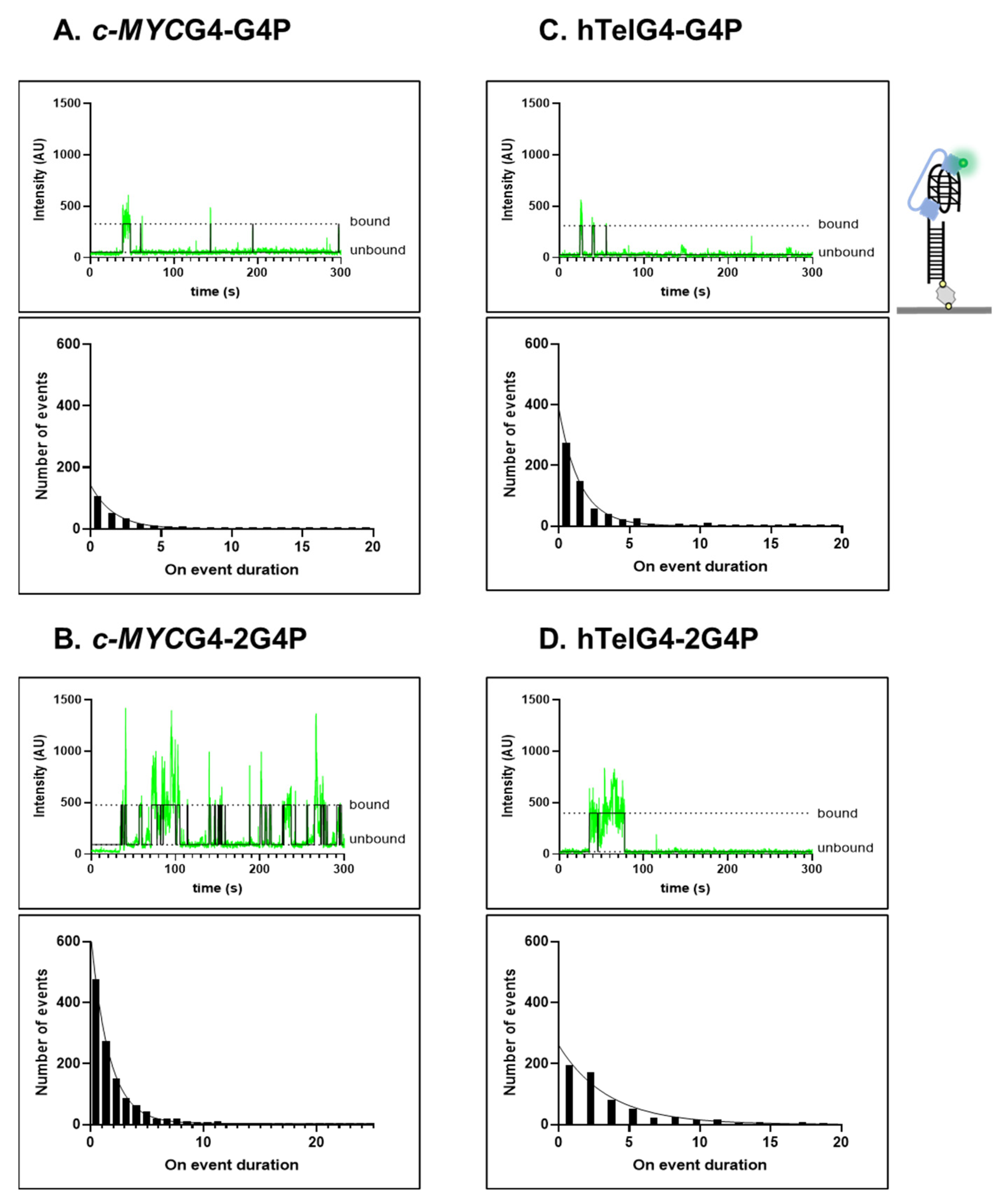
2.2. Association Kinetics Plays an Important Role in the G4P-hTelG4 Complex Formation
2.3. G4P Binding Does Not Interfere with the G4 Structure
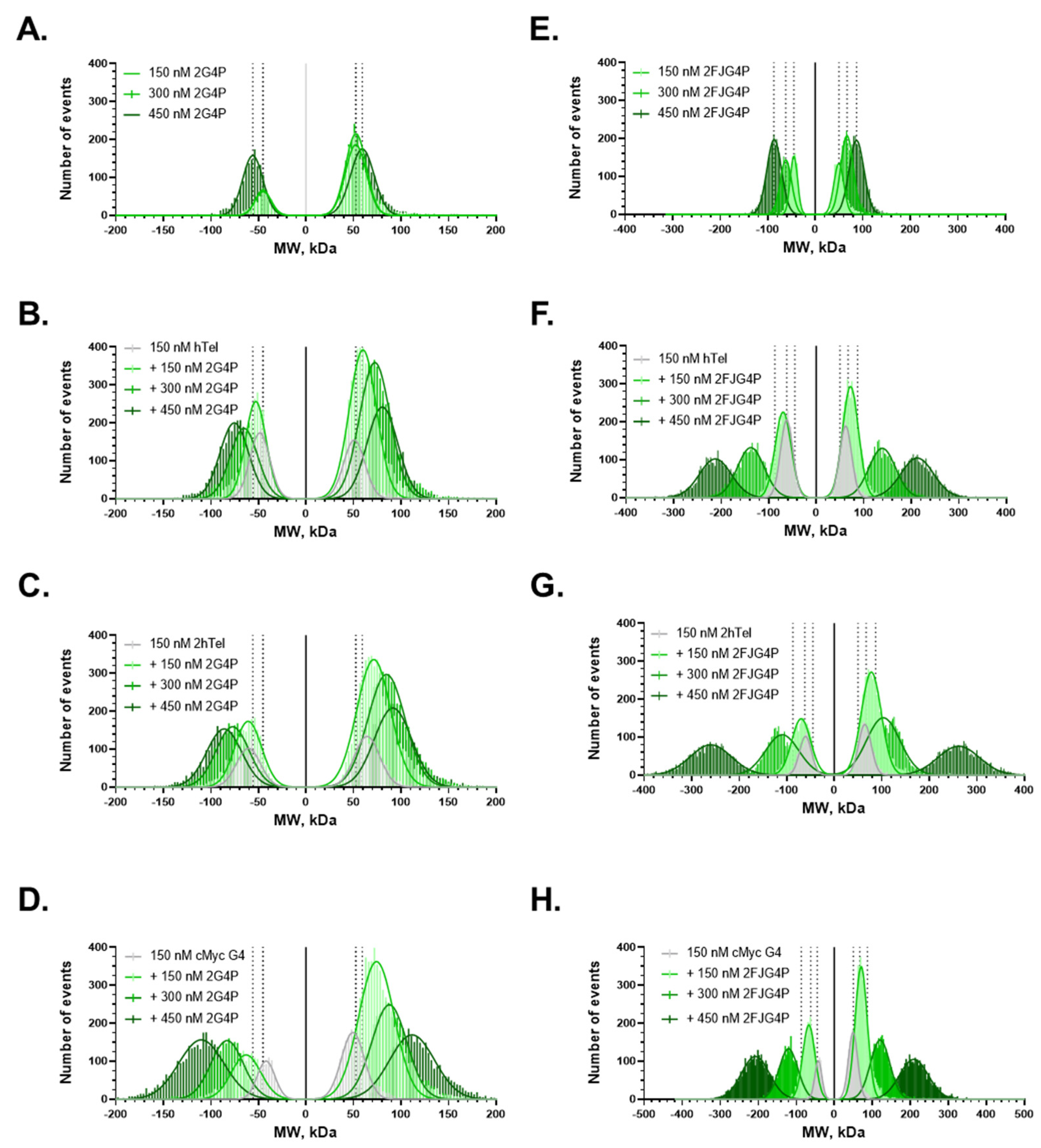
2.4. Increasing the Number of G4s Increases Valency of the Interactions
3. Discussion
4. Materials and Methods
4.1. Construction of Plasmids
4.2. DNA Oligos
4.3. Purification and Fluorescent Labeling of G4P, 2G4P, FJG4P and 2FJG4P
4.4. Electrophoretic Mobility Shift Assay
4.5. Mass Photometry
4.6. Single-Molecule Total Internal Reflection Microscopy
4.7. Surface Tethered DNA Single-Molecule Experiments
4.8. Surface Tethered G4P Single-Molecule Experiments
4.9. Single-Molecule Data Analysis
4.10. Single-Molecule FRET Experiments
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mirkin, S.M. Discovery of alternative DNA structures: A heroic decade (1979–1989). Front. Biosci. A J. Virtual Libr. 2008, 13, 1064–1071. [Google Scholar] [CrossRef]
- Wang, G.; Vasquez, K.M. Impact of alternative DNA structures on DNA damage, DNA repair, and genetic instability. DNA Repair. 2014, 19, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.; Gilbert, W. Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 1988, 334, 364–366. [Google Scholar] [CrossRef]
- Chen, L.; Dickerhoff, J.; Sakai, S.; Yang, D. DNA G-Quadruplex in Human Telomeres and Oncogene Promoters: Structures, Functions, and Small Molecule Targeting. Acc. Chem. Res. 2022, 55, 2628–2646. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef] [Green Version]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef] [Green Version]
- Hansel-Hertsch, R.; Beraldi, D.; Lensing, S.V.; Marsico, G.; Zyner, K.; Parry, A.; Di Antonio, M.; Pike, J.; Kimura, H.; Narita, M.; et al. G-quadruplex structures mark human regulatory chromatin. Nat. Genet. 2016, 48, 1267–1272. [Google Scholar] [CrossRef] [Green Version]
- Hansel-Hertsch, R.; Antonio, M.; Balasubramanian, S. DNA G-734 quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Bochman, M.L.; Paeschke, K.; Zakian, V.A. DNA secondary structures: Stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012, 13, 770–780. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Burrows, C.J. 8-Oxo-7,8-dihydroguanine, friend and foe: Epigenetic-like regulator versus initiator of mutagenesis. DNA Repair. 2017, 56, 75–83. [Google Scholar] [CrossRef]
- Bain, F.E.; Wu, C.G.; Spies, M. Single-molecule sorting of DNA helicases. Methods 2016, 108, 14–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, A.M.; Burrows, C.J. Oxidative stress-mediated epigenetic regulation by G-quadruplexes. NAR Cancer 2021, 3, zcab038. [Google Scholar] [CrossRef] [PubMed]
- Bielskutė, S.; Plavec, J.; Podbevšek, P. Oxidative lesions modulate G-quadruplex stability and structure in the human BCL2 promoter. Nucleic Acids Res. 2021, 49, 2346–2356. [Google Scholar] [CrossRef]
- Prorok, P.; Artufel, M.; Aze, A.; Coulombe, P.; Peiffer, I.; Lacroix, L.; Guédin, A.; Mergny, J.-L.; Damaschke, J.; Schepers, A.; et al. Involvement of G-quadruplex regions in mammalian replication origin activity. Nat. Commun. 2019, 10, 3274. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, A.V.; Kubareva, E.A.; Monakhova, M.V.; Zvereva, M.I.; Dolinnaya, N.G. Impact of G-Quadruplexes on the Regulation of Genome Integrity, DNA Damage and Repair. Biomolecules 2021, 11, 1284. [Google Scholar] [CrossRef] [PubMed]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Mao, S.Q.; Ghanbarian, A.T.; Spiegel, J.; Martínez Cuesta, S.; Beraldi, D.; Di Antonio, M.; Marsico, G.; Hänsel-Hertsch, R.; Tannahill, D.; Balasubramanian, S. DNA G-quadruplex structures mold the DNA methylome. Nat. Struct. Mol. Biol. 2018, 25, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Zyner, K.G.; Simeone, A.; Flynn, S.M.; Doyle, C.; Marsico, G.; Adhikari, S.; Portella, G.; Tannahill, D.; Balasubramanian, S. G-quadruplex DNA structures in human stem cells and differentiation. Nat. Commun. 2022, 13, 142. [Google Scholar] [CrossRef]
- Linke, R.; Limmer, M.; Juranek, S.A.; Heine, A.; Paeschke, K. The Relevance of G-Quadruplexes for DNA Repair. Int. J. Mol. Sci. 2021, 22, 12599. [Google Scholar] [CrossRef]
- Ying, L.; Green, J.J.; Li, H.; Klenerman, D.; Balasubramanian, S. Studies on the structure and dynamics of the human telomeric G quadruplex by single-molecule fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2003, 100, 14629–14634. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Okumus, B.; Kim, D.S.; Ha, T. Extreme conformational diversity in human telomeric DNA. Proc. Natl. Acad. Sci. USA 2005, 102, 18938–18943. [Google Scholar] [CrossRef] [Green Version]
- Heddi, B.; Phan, A.T. Structure of human telomeric DNA in crowded solution. J. Am. Chem. Soc. 2011, 133, 9824–9833. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Patel, D.J. Solution structure of the human telomeric repeat d[AG3(T2AG3)3] G-tetraplex. Structure 1993, 1, 263–282. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.; Costa, P.J.; Félix, V.; Williamson, M.P.; Thomas, J.A. Structural studies on dinuclear ruthenium(II) complexes that bind diastereoselectively to an antiparallel folded human telomere sequence. J. Med. Chem. 2013, 56, 8674–8683. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; Miller, J.; Howat, W.J.; Balasubramanian, S. Elevated levels of G-quadruplex formation in human stomach and liver cancer tissues. PLoS ONE 2014, 9, e102711. [Google Scholar] [CrossRef] [Green Version]
- Ambrus, A.; Chen, D.; Dai, J.; Jones, R.A.; Yang, D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry 2005, 44, 2048–2058. [Google Scholar] [CrossRef]
- Agrawal, P.; Lin, C.; Mathad, R.I.; Carver, M.; Yang, D. The major G-quadruplex formed in the human BCL-2 proximal promoter adopts a parallel structure with a 13-nt loop in K+ solution. J. Am. Chem. Soc. 2014, 136, 1750–1753. [Google Scholar] [CrossRef]
- Raiber, E.A.; Kranaster, R.; Lam, E.; Nikan, M.; Balasubramanian, S. A non-canonical DNA structure is a binding motif for the transcription factor SP1 in vitro. Nucleic Acids Res. 2012, 40, 1499–1508. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Martin, V.; Lopez-Pujante, C.; Soriano-Rodriguez, M.; Garcia-Salcedo, J.A. An Updated Focus on Quadruplex Structures as Potential Therapeutic Targets in Cancer. Int. J. Mol. Sci. 2020, 21, 8900. [Google Scholar] [CrossRef]
- Sun, D.Y.; Guo, K.X.; Rusche, J.J.; Hurley, L.H. Facilitation of a structural transition in the polypurine/polypyrimidine tract within the proximal promoter region of the human VEGF gene by the presence of potassium and G-quadruplex-interactive agents. Nucleic Acids Res. 2005, 33, 6070–6080. [Google Scholar] [CrossRef]
- Wang, Y.H.; Yang, Q.F.; Lin, X.; Chen, D.; Wang, Z.Y.; Chen, B.; Han, H.Y.; Chen, H.D.; Cai, K.C.; Li, Q.; et al. G4LDB 2.2: A database for discovering and studying G-quadruplex and i-Motif ligands. Nucleic Acids Res. 2022, 50, D150–D160. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xiang, J.F.; Yang, Q.F.; Sun, H.X.; Guan, A.J.; Tang, Y.L. G4LDB: A database for discovering and studying G-quadruplex ligands. Nucleic Acids Res. 2013, 41, D1115–D1123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tauchi, T.; Shin-ya, K.; Sashida, G.; Sumi, M.; Okabe, S.; Ohyashiki, J.H.; Ohyashiki, K. Telomerase inhibition with a novel G-quadruplex-interactive agent, telomestatin: In Vitro and In Vivo studies in acute leukemia. Oncogene 2006, 25, 5719–5725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin-ya, K.; Wierzba, K.; Matsuo, K.; Ohtani, T.; Yamada, Y.; Furihata, K.; Hayakawa, Y.; Seto, H. Telomestatin, a novel telomerase inhibitor from Streptomyces anulatus. J. Am. Chem. Soc. 2001, 123, 1262–1263. [Google Scholar] [CrossRef]
- Muller, S.; Kumari, S.; Rodriguez, R.; Balasubramanian, S. Small-molecule-mediated G-quadruplex isolation from human cells. Nat. Chem. 2010, 2, 1095–1098. [Google Scholar] [CrossRef]
- Wheelhouse, R.T.; Sun, D.K.; Han, H.Y.; Han, F.X.G.; Hurley, L.H. Cationic porphyrins as telomerase inhibitors: The interaction of tetra-(N-methyl-4-pyridyl)porphine with quadruplex DNA. J. Am. Chem. Soc. 1998, 120, 3261–3262. [Google Scholar] [CrossRef]
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 40. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef] [Green Version]
- Fry, M. Tetraplex DNA and its interacting proteins. Front. Biosci. A J. Virtual Libr. 2007, 12, 4336–4351. [Google Scholar] [CrossRef]
- Brázda, V.; Hároníková, L.; Liao, J.C.; Fojta, M. DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 2014, 15, 17493–17517. [Google Scholar] [CrossRef] [Green Version]
- Zheng, K.W.; Zhang, J.Y.; He, Y.D.; Gong, J.Y.; Wen, C.J.; Chen, J.N.; Hao, Y.H.; Zhao, Y.; Tan, Z. Detection of genomic G-quadruplexes in living cells using a small artificial protein. Nucleic Acids Res. 2020, 48, 11706–11720. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, J.P.; Creacy, S.D.; Routh, E.D.; Joyner-Butt, C.; Jenkins, G.S.; Pauli, S.; Nagamine, Y.; Akman, S.A. The DEXH protein product of the DHX36 gene is the major source of tetramolecular quadruplex G4-DNA resolving activity in HeLa cell lysates. J. Biol. Chem. 2005, 280, 38117–38120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heddi, B.; Cheong, V.V.; Martadinata, H.; Phan, A.T. Insights into G-quadruplex specific recognition by the DEAH-box helicase RHAU: Solution structure of a peptide-quadruplex complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9608–9613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hossain, K.A.; Jurkowski, M.; Czub, J.; Kogut, M. Mechanism of recognition of parallel G-quadruplexes by DEAH/RHAU helicase DHX36 explored by molecular dynamics simulations. Comput. Struct. Biotec 2021, 19, 2526–2536. [Google Scholar] [CrossRef] [PubMed]
- Lattmann, S.; Giri, B.; Vaughn, J.P.; Akman, S.A.; Nagamine, Y. Role of the amino terminal RHAU-specific motif in the recognition and resolution of guanine quadruplex-RNA by the DEAH-box RNA helicase RHAU. Nucleic Acids Res. 2010, 38, 6219–6233. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.C.; Tippana, R.; Demeshkina, N.A.; Murat, P.; Balasubramanian, S.; Myong, S.; Ferré-D’Amaré, A.R. Structural basis of G-quadruplex unfolding by the DEAH/RHA helicase DHX36. Nature 2018, 558, 465–469. [Google Scholar] [CrossRef]
- Wu, C.G.; Spies, M. G-quadruplex recognition and remodeling by the FANCJ helicase. Nucleic Acids Res. 2016, 44, 8742–8753. [Google Scholar] [CrossRef] [Green Version]
- Hon, J.; Gonzalez, R.L., Jr. Bayesian-Estimated Hierarchical HMMs Enable Robust Analysis of Single-Molecule Kinetic Heterogeneity. Biophys. J. 2019, 116, 1790–1802. [Google Scholar] [CrossRef]
- Tibbs, J.; Ghoneim, M.; Caldwell, C.C.; Buzynski, T.; Bowie, W.; Boehm, E.M.; Washington, M.T.; Tabei, S.M.A.; Spies, M. KERA: Analysis tool for multi-process, multi-state single-molecule data. Nucleic Acids Res. 2021, 49, e53. [Google Scholar] [CrossRef]
- Young, G.; Hundt, N.; Cole, D.; Fineberg, A.; Andrecka, J.; Tyler, A.; Olerinyova, A.; Ansari, A.; Marklund, E.G.; Collier, M.P.; et al. Quantitative mass imaging of single biological macromolecules. Science 2018, 360, 423–427. [Google Scholar] [CrossRef] [Green Version]
- Cole, D.; Young, G.; Weigel, A.; Sebesta, A.; Kukura, P. Label-Free Single-Molecule Imaging with Numerical-Aperture-Shaped Interferometric Scattering Microscopy. ACS Photonics 2017, 4, 211–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiekh, S.; Mustafa, G.; Kodikara, S.G.; Hoque, M.E.; Yokie, E.; Portman, J.J.; Balci, H. Emerging accessibility patterns in long telomeric overhangs. Proc. Natl. Acad. Sci. USA 2022, 119, e2202317119. [Google Scholar] [CrossRef] [PubMed]
- Biffi, G.; Tannahill, D.; McCafferty, J.; Balasubramanian, S. Quantitative visualization of DNA G-quadruplex structures in human cells. Nat. Chem. 2013, 5, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Wild, A.T.; Wu, W.H.; Shah, R.; Danussi, C.; Riggins, G.J.; Kannan, K.; Sulman, E.P.; Chan, T.A.; et al. G-quadruplex DNA drives genomic instability and represents a targetable molecular abnormality in ATRX-deficient malignant glioma. Nat. Commun. 2019, 10, 943. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, B.; Li, T.; Liu, R.; Xiao, Y.; Geng, X.; Li, G.; Liu, Q.; Price, C.M.; Liu, Y.; et al. Mammalian CST averts replication failure by preventing G-quadruplex accumulation. Nucleic Acids Res. 2019, 47, 5243–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshney, D.; Cuesta, S.M.; Herdy, B.; Abdullah, U.B.; Tannahill, D.; Balasubramanian, S. RNA G-quadruplex structures control ribosomal protein production. Sci. Rep. 2021, 11, 22735. [Google Scholar] [CrossRef]
- Hatzakis, E.; Okamoto, K.; Yang, D. Thermodynamic stability and folding kinetics of the major G-quadruplex and its loop isomers formed in the nuclease hypersensitive element in the human c-Myc promoter: Effect of loops and flanking segments on the stability of parallel-stranded intramolecular G-quadruplexes. Biochemistry 2010, 49, 9152–9160. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.X.; Carver, M.; Hurley, L.H.; Yang, D.Z. Solution Structure of a 2:1 Quindoline-c-MYC G-Quadruplex: Insights into G-Quadruplex-Interactive Small Molecule Drug Design. J. Am. Chem. Soc. 2011, 133, 17673–17680. [Google Scholar] [CrossRef] [Green Version]
- Zacheja, T.; Toth, A.; Harami, G.M.; Yang, Q.; Schwindt, E.; Kovacs, M.; Paeschke, K.; Burkovics, P. Mgs1 protein supports genome stability via recognition of G-quadruplex DNA structures. FASEB J. 2020, 34, 12646–12662. [Google Scholar] [CrossRef]
- Bain, F.E.; Fischer, L.A.; Chen, R.; Wold, M.S. Single-Molecule Analysis of Replication Protein A-DNA Interactions. Methods Enzym. 2018, 600, 439–461. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein-DNA | State 1 (kon) * s−1 M−1 | State 2 (koff) s−1 | State 2 (τ) s | Kd = koff/kon (nM) | |
|---|---|---|---|---|---|
| 1. | G4P-c-MYCG4 | (2.16 ± 0.2) × 109 s−1 M−1 | 0.61 ± 0.010 | 1.64 ± 0.03 | 0.22 ± 0.02 |
| 2. | 2G4P-c-MYCG4 | (3.9 ± 0.2) × 109 s−1 M−1 | 0.62 ± 0.010 | 1.62 ± 0.03 | 0.16 ± 0.01 |
| 3. | G4P-hTelG4 | (0.43 ± 0.036) × 109 s−1 M−1 | 0.67 ± 0.015 | 1.5 ± 0.03 | 1.6 ± 0.1 |
| 4. | 2G4P-hTelG4 | (0.0845 ± 0.007) × 109 s−1 M−1 | 0.29 ± 0.014 | 3.5 ± 0.2 | 3.4 ± 0.3 |
| 5. | FJG4P-c-MYCG4 | (3.45 ± 0.39) × 109 s−1 M−1 | 0.94 ± 0.010 | 1.1 ± 0.01 | 0.27 ± 0.03 |
| 6. | 2FJG4P-c-MYCG4 | (4.7 ± 0.26) × 109 s−1 M−1 | 1.0 ± 0.023 | 1 ± 0.02 | 0.20 ± 0.01 |
| 7. | FJG4P-hTelG4 | (0.01 ± 0.001428) × 109 s−1 M−1 | 0.33 ± 0.011 | 3.1 ± 0.1 | 32.7 ± 4.8 |
| 8. | 2FJG4P-hTelG4 | (0.0339 ± 0.005357) × 109 s−1 M−1 | 0.4 ± 0.008 | 2.8 ± 0.06 | 10.7 ± 1.7 |
| 9. | G4P-2hTelG4 | (0.209 ± 0.1545) × 109 s−1 M−1 | 0.4 ± 0.02 | 2.5 ± 0.1 | 1.9 ± 1.4 |
| 10. | 2G4P-2hTelG4 (3 states) | (0.0845 ± 0.00704) × 109 s−1 M−1 | NA | NA | NA |
| 11. | FJG4P-2hTelG4 | (0.0238 ± 0.003571) × 109 s−1 M−1 | 0.35 ± 0.01 | 2.9 ± 0.09 | 14.5 ± 2.2 |
| 12. | 2FJG4P-2hTelG4 | (0.0642 ± 0.00714) × 109 s−1 M−1 | 0.3 ± 0.01 | 3.35 ± 0.137 | 4.6 ± 0.55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaur, P.; Bain, F.E.; Honda, M.; Granger, S.L.; Spies, M. Single-Molecule Analysis of the Improved Variants of the G-Quadruplex Recognition Protein G4P. Int. J. Mol. Sci. 2023, 24, 10274. https://doi.org/10.3390/ijms241210274
Gaur P, Bain FE, Honda M, Granger SL, Spies M. Single-Molecule Analysis of the Improved Variants of the G-Quadruplex Recognition Protein G4P. International Journal of Molecular Sciences. 2023; 24(12):10274. https://doi.org/10.3390/ijms241210274
Chicago/Turabian StyleGaur, Paras, Fletcher E. Bain, Masayoshi Honda, Sophie L. Granger, and Maria Spies. 2023. "Single-Molecule Analysis of the Improved Variants of the G-Quadruplex Recognition Protein G4P" International Journal of Molecular Sciences 24, no. 12: 10274. https://doi.org/10.3390/ijms241210274
APA StyleGaur, P., Bain, F. E., Honda, M., Granger, S. L., & Spies, M. (2023). Single-Molecule Analysis of the Improved Variants of the G-Quadruplex Recognition Protein G4P. International Journal of Molecular Sciences, 24(12), 10274. https://doi.org/10.3390/ijms241210274