
Weak, Broken, but Working—Intramolecular Hydrogen Bond in 2,2′-bipyridine


Abstract
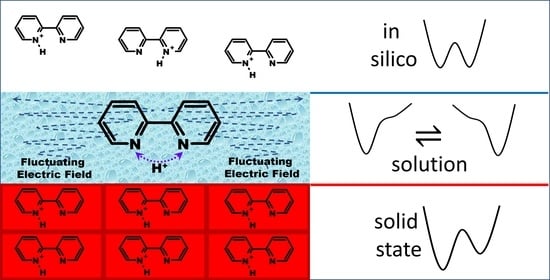
1. Introduction
2. Results and Discussion
2.1. Hydrogen Bond Geometry and Energy in BiPyH+ and PhenH+
2.1.1. Experimental 15N NMR Chemical Shifts
2.1.2. Calculated 15N NMR Chemical Shifts
2.1.3. Hydrogen Bond Energy
2.2. Intramolecular Proton Transfer in BiPyH+ and PhenH+
Proton Transfer Pathway
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hynes, J.T.; Klinman, J.P.; Limbach, H.-H.; Schowen, R.L. Hydrogen-Transfer Reachtions; WILEY-VCH Verlag: Weinheim, Germany, 2007. [Google Scholar]
- Kohen, A.; Limbach, H.-H. Isotope Effects in Chemistry and Biology; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Howard, S.T. Conformers, Energetics, and Basicity of 2,2‘-Bipyridine. J. Am. Chem. Soc. 1996, 118, 10269–10274. [Google Scholar] [CrossRef]
- Merritt, L.L., Jr.; Schroeder, E.D. The crystal structure of 2,2′-bipyridine. Acta Crystallogr. 1956, 9, 801–804. [Google Scholar] [CrossRef]
- Chisholm, M.H.; Huffman, J.C.; Rothwell, I.P.; Bradley, P.G.; Kress, N.; Woodruff, W.H. Bis(2,2′-bipyridyl)diisopropoxymolybdenum(II). Structural and Spectroscopic Evidence for Molybdenum-to-Bipyridyl π* Bonding. J. Am. Chem. Soc. 1981, 103, 4945–4947. [Google Scholar] [CrossRef]
- Kühn, F.E.; Groarke, M.; Bencze, E.; Herdtweck, E.; Prazeres, A.; Santos, A.M.; Calhorda, M.J.; Romão, C.C.; Gonçalves, I.S.; Lopes, A.D.; et al. Octahedral Bipyridine and Bipyrimidine Dioxomolybdenum(vi) Complexes:Characterization, Application in Catalytic Epoxidation, and DensityFunctional Mechanistic Study. Chem. Eur. J. 2002, 8, 2370–2383. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.L.; Murthy, D.S.N.; Ritchie, G.L.D. Molecular conformations from magnetic anisotropies. J. Chem. Soc. Faraday Trans. 1972, 68, 1679–1690. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Kamdem, N.; Mauder, D.; Denisov, G.S.; Shenderovich, I.G. Studies of Adsorption of 2,2′-Bipyridyl on the Surface of Highly Regulated Silica Matrix of the MCM-41 Type by Means of 15N NMR Spectroscopy. Russ. J. Gen. Chem. 2010, 80, 2027–2031. [Google Scholar] [CrossRef]
- Lesnichin, S.B.; Tolstoy, P.M.; Limbach, H.-H.; Shenderovich, I.G. Counteranion-Dependent Mechanisms of Intramolecular Proton Transfer in Aprotic Solution. Phys. Chem. Chem. Phys. 2010, 12, 10373–10379. [Google Scholar] [CrossRef]
- Bakshi, P.K.; Cameron, T.S.; Knop, O. Crystal chemistry of tetraradial species. Part 8. Mix and match: Cation geometry, ion packing, hydrogen bonding, and π–π interactions in cis-2,2′-bipyridinium(1+) and 1,10-phenanthrolinium(1+) tetraphenylborates—And what about proton sponges? Can. J. Chem. 1996, 74, 201–220. [Google Scholar] [CrossRef]
- Schone, S.; Radoske, T.; Marz, J.; Stumpf, T.; Ikeda-Ohno, A. Synthesis and Characterization of Heterometallic Iron–Uranium Complexes with a Bidentate N-Donor Ligand (2,2′-Bipyridine or 1,10-Phenanthroline). Inorg. Chem. 2018, 57, 13318–13329. [Google Scholar] [CrossRef]
- Kavitha, S.J.; Panchanatheswaran, K.; Low, N.J.; Glidewell, C. 2-(2-Pyridyl)pyridinium perchlorate, redetermined at 120 K: Complex hydrogen-bonded sheets. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2005, 61, o473–o474. [Google Scholar] [CrossRef]
- Kadarkaraisamy, M.; Engelhart, D.P.; Basa, P.N.; Sykes, A.G. Hexafluorophosphate salts of bis and tetrakis(2,2′-bipyridine)lead(II) complexes. J. Coord. Chem. 2010, 63, 2261–2267. [Google Scholar] [CrossRef]
- Milani, B.; Anzilutti, A.; Vicentini, L.; Sessanta o Santi, A.; Zangrando, E.; Geremia, S.; Mestroni, G. Bis-Chelated Palladium(II) Complexes with Nitrogen-Donor Chelating Ligands Are Efficient Catalyst Precursors for the CO/Styrene Copolymerization Reaction. Organometallics 1997, 16, 5064–5075. [Google Scholar] [CrossRef]
- Kraus, F.; Breu, J. Arene-arene stacking in the revised structure of 2,2′-bipyridinium hexafluorophosphate. Acta Crystallogr. Sect. C Cryst. Struct. Commun. 2002, 58, o254–o256. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, N.; Yamazaki, S.; Kato, N.; Kubota, A.; Sawai, M.; Noda, K.; Kanehisa, N.; Inoue, T.; Nakata, E.; Takashima, H. Syntheses, X-Ray Crystal Structures, Emission Properties and DFT Calculations of Monoprotonated Polypyridines. Chem. Sel. 2019, 4, 59–65. [Google Scholar] [CrossRef]
- Buttery, J.H.; Effendy; Junk,, P.C.; Mutrofin, S.; Skelton, B.W.; Whitaker, C.R.; White, A.H. The Structural Systematics of Protonation of Some Important Nitrogen-base Ligands. IV. Some Ethane-1,2-diaminium Univalent Anion Salt/1,10-Phenanthroline (Hydrate) Arrays. Z. Anorg. Allg. Chem. 2006, 632, 1326–1339. [Google Scholar] [CrossRef]
- Jagan, R.; Sathya, D.; Sivakumar, K. A dihydrogen phosphate anionic network as a host lattice for cations in 1-methyl piperazine-1,4-diium bis (di hydrogen phosphate) and 2-(pyridin-2-yl)pyridinium di hydrogen phosphate-ortho phosphoric acid (1/1). Acta Crystallogr. Sect. C Cryst. Struct. Chem. 2015, 71, 374–380. [Google Scholar] [CrossRef]
- Ivanova, B.; Spiteller, M. On the chemical identification and determination of flavonoids in solid-state. Talanta 2012, 94, 9–21. [Google Scholar] [CrossRef]
- Charushnikova, I.A.; Den Auwer, C. 2,2′-Bipyridyl-2-ium aqua(2,2′-bipyridyl-κ2N,N’)-tetakis-(nitrato-κ2O,O’) -praseodymium(III) 2,2′-bipyridyl hemisolvate. Acta Crystallogr. Sect. E Struct. Rep. Online 2004, 60, m1775–m1777. [Google Scholar] [CrossRef]
- Thuery, P. Sulfonate Complexes of Actinide Ions: Structural Diversity in Uranyl Complexes with 2-Sulfobenzoate. Inorg. Chem. 2013, 52, 435–447. [Google Scholar] [CrossRef]
- Mrvos-Sermek, D.; Popovic, Z.; Matkovic-Calogovic, D. Hydrogen Bonding in the 2,2′-Bipyridinium Salt of 1,2,4,5-Benzenetetracarboxylic Acid (Pyromellitic Acid). Acta Crystallogr. Sect. C Cryst. Struct. Commun. 1996, 52, 2538–2541. [Google Scholar] [CrossRef]
- Thuery, P. 2,2′-Bipyridine and 1,10-Phenanthroline as Coligands or Structure-Directing Agents in Uranyl–Organic Assemblies with Polycarboxylic Acids. Eur. J. Inorg. Chem. 2013, 2013, 4563–4573. [Google Scholar] [CrossRef]
- Jurowska, A.; Szklarzewicz, J.; Basta, N.; Hodorowicz, M. Structural and physicochemical characterization of ion-pair salts with dipyridyl cations and [Fe(CN)6]4− and [Fe(CN)5(NO)]2− anions. Inorg. Chim. Acta 2022, 539, 121033. [Google Scholar] [CrossRef]
- Balabanova, S.P.; Buikin, P.A.; Ilyukhin, A.B.; Rudenko, A.Y.; Dorovatovskii, P.V.; Korlyukov, A.A.; Kotov, V.Y. Crystal Structure and Optical Properties of New Hybrid Halobismuthates of 2,2′-Bipyridinium Derivatives. Russ. J. Inorg. Chem. 2022, 67, 1018–1024. [Google Scholar] [CrossRef]
- Jurowska, A.; Szklarzewicz, J.; Hodorowicz, M. Ion pair charge-transfer salts based on protonated bipyridines and [W(CN)6(bpy)]2− anion-structure and properties. J. Mol. Struct. 2022, 1261, 132931. [Google Scholar] [CrossRef]
- Summers, L.A. The Bipyridines. Adv. Heterocycl. Chem. 1984, 35, 281–374. [Google Scholar] [CrossRef]
- Vogler, A.; Shenderovich, I.G. Photochemistry of deprotonated rhenium(I) (3,3′-dihydroxy-2,2′-bipyridine) tricarbonyl chloride. Photoisomerization at the chelate in basic solution. Inorg. Chim. Acta 2014, 421, 496–499. [Google Scholar] [CrossRef]
- Nishigaki, S.; Yoshioka, H.; Nakatsu, K. The crystal and molecular structure of o-phenanthroline. Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1978, 34, 875–879. [Google Scholar] [CrossRef]
- Gembicky, M. The Cambridge Structural Database Communication ID OPENAN01. University of Buffalo: Buffalo, NY, USA, 2020. [Google Scholar]
- Rojas, Y.; Tadrosse, M.; Assaf, C.; Bernal, I.; Lalancette, R.A. Preparation and structural studies of manganese polychloride anions in three metal charge states [(II), (III), (IV)]—Their stereochemistries. Struct. Chem. 2020, 31, 1309–1316. [Google Scholar] [CrossRef]
- Oswald, I.W.H.; Ahn, H.; Neilson, J.R. Influence of organic cation planarity on structural templating in hybrid metal-halides. Dalton Trans. 2019, 48, 16340–16349. [Google Scholar] [CrossRef]
- Gel’mbol’dt, V.O.; Anisimov, V.Y.; Fonar’, M.S. Interaction between hydrofluorosilicic acid and 1,10-phenanthroline: Hydrolytic stability of chelate complexes of silicon tetrafluoride with bidentate N-donors. Russ. J. Inorg. Chem. 2016, 61, 822–825. [Google Scholar] [CrossRef]
- Ma, G.; Ilyukhin, A.; Glaser, J. 2,2′-Bipyridinium bis(perchlorate). Acta Crystallogr. Sect. C Cryst. Struct. Chem. 2000, C56, 1473–1475. [Google Scholar] [CrossRef] [PubMed]
- Bujak, M.; Frank, W. Crystal structure of 1,10-phenanthrolindiium bis(triiodide) monohydrate, C12H12I6N2O. Z. Kristallogr. -New Cryst. Struct. 2014, 229, 379–380. [Google Scholar] [CrossRef]
- Li, F.; Yin, H.; Simpson, J. catena-Poly[1,10-phen anthroline-1,10-diium [[dichlorido bismuthate(III)]-di-μ-chlorido]]. Acta Crystallogr. Sect. E Struct. Rep. Online 2007, 63, m1746. [Google Scholar] [CrossRef]
- Pelties, S.; Maier, T.; Herrmann, D.; de Bruin, B.; Rebreyend, C.; Gärtner, S.; Shenderovich, I.G.; Wolf, R. Selective P4 Activation by a Highly Reduced Cobaltate: Synthesis of Dicobalt Tetraphosphido Complexes. Chem. Eur. J. 2017, 23, 6094–6102. [Google Scholar] [CrossRef] [PubMed]
- Solum, M.S.; Altmann, K.L.; Strohmeier, M.; Berges, D.A.; Zhang, Y.; Facelli, J.C.; Pugmire, R.J.; Grant, D.M. 15N Chemical Shift Principal Values in Nitrogen Heterocycles. J. Am. Chem. Soc. 1997, 119, 9804–9809. [Google Scholar] [CrossRef]
- Lorente, P.; Shenderovich, I.G.; Golubev, N.S.; Denisov, G.S.; Buntkowsky, G.; Limbach, H.-H. 1H/15N NMR Chemical Shielding, Dipolar 15N, 2H Coupling and Hydrogen Bond Geometry Correlations in a Novel Series of Hydrogen-Bonded Acid–Base Complexes of Collidine with Carboxylic Acids. Magn. Reson. Chem. 2001, 39, S18–S29. [Google Scholar] [CrossRef]
- Brilmayer, R.; Brodrecht, M.; Kaiser, C.; Breitzke, H.; Kumari, B.; Wachtveitl, J.; Buntkowsky, G.; Andrieu-Brunsen, A. The Interplay of Nanoconfinement and pH from the Perspective of a Dye-Reporter Molecule. ChemNanoMat 2020, 6, 1843–1853. [Google Scholar] [CrossRef]
- Sharif, S.; Denisov, G.S.; Toney, M.D.; Limbach, H.-H. NMR Studies of Solvent-Assisted Proton Transfer in a Biologically Relevant Schiff Base: Toward a Distinction of Geometric and Equilibrium H-Bond Isotope Effects. J. Am. Chem. Soc. 2006, 128, 3375–3387. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Sigalov, M.V.; Tolstoy, P.M. Simultaneous Estimation of Two Coupled Hydrogen Bond Geometries from Pairs of Entangled NMR Parameters: The Test Case of 4-Hydroxypyridine Anion. Molecules 2022, 27, 3923. [Google Scholar] [CrossRef]
- Andreeva, D.V.; Ip, B.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Limbach, H.-H. Geometrical Features of Hydrogen Bonded Complexes Involving Sterically Hindered Pyridines. J. Phys. Chem. A 2006, 110, 10872–10879. [Google Scholar] [CrossRef]
- Li, Z.J.; Abramov, Y.; Bordner, J.; Leonard, J.; Medek, A.; Trask, A.V. Solid-State Acid−Base Interactions in Complexes of Heterocyclic Bases with Dicarboxylic Acids: Crystallography, Hydrogen Bond Analysis, and 15N NMR Spectroscopy. J. Am. Chem. Soc. 2006, 128, 8199–8210. [Google Scholar] [CrossRef] [PubMed]
- Gorobets, N.; Yermolayev, S.A.; Gurley, T.; Gurinov, A.A.; Tolstoy, P.M.; Shenderovich, I.G.; Leadbeater, N.E. Difference between 1H NMR signals of primary amide protons as a simple spectral index of the amide intramolecular hydrogen bond strength. J. Phys. Org. Chem. 2012, 25, 287–295. [Google Scholar] [CrossRef]
- Hartman, J.D.; Harper, J.K. Improving the accuracy of GIPAW chemical shielding calculations with cluster and fragment corrections. Solid State Nucl. Magn. Reson. 2022, 122, 101832. [Google Scholar] [CrossRef]
- Stevens, J.S.; Byard, S.J.; Seaton, C.C.; Sadiq, G.; Daveya, R.J.; Schroeder, S.L.M. Proton transfer and hydrogen bonding in the organic solid state: A combined XRD/XPS/ssNMR study of 17 organic acid–base complexes. Phys. Chem. Chem. Phys. 2014, 16, 1150–1160. [Google Scholar] [CrossRef]
- Tatton, A.S.; Blade, H.; Brown, S.P.; Hodgkinson, P.; Hughes, L.P.; Lill, S.O.N.; Yates, J.R. Improving Confidence in Crystal Structure Solutions Using NMR Crystallography: The Case of β-Piroxicam. Cryst. Growth Des. 2018, 18, 3339–3351. [Google Scholar] [CrossRef]
- Chernyshov, I.Y.; Vener, M.V.; Shenderovich, I.G. Local-structure effects on 31P NMR chemical shift tensors in solid state. J. Chem. Phys. 2019, 150, 144706. [Google Scholar] [CrossRef] [PubMed]
- Hartman, J.D.; Kudla, R.A.; Day, G.M.; Mueller, L.J.; Beran, G.J.O. Benchmark fragment-based 1H, 13C, 15N and 17O chemical shift predictions in molecular crystals. Phys. Chem. Chem. Phys. 2016, 18, 21686–21709. [Google Scholar] [CrossRef] [PubMed]
- Kleemiss, F.; Dolomanov, O.V.; Bodensteiner, M.; Peyerimhoff, N.; Midgley, L.; Bourhis, L.J.; Genoni, A.; Malaspina, L.A.; Jayatilaka, D.; Spencer, J.L.; et al. Accurate crystal structures and chemical properties from NoSpherA2. Chem. Sci. 2021, 12, 1675–1692. [Google Scholar] [CrossRef]
- Ludwig, M.; Himmel, D.; Hillebrecht, H. GIAO versus GIPAW: Comparison of Methods To Calculate 11B NMR Shifts of Icosahedral Closo-Heteroboranes toward Boron-Rich Borides. J. Phys. Chem. A 2020, 124, 2173–2185. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Qualitative Analysis of the Geometry of the Hydrogen Bond in the Homoconjugated Pyridine Ion. Russ. J. Gen. Chem. 2007, 77, 620–625. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Experimentally Established Benchmark Calculations of 31P NMR Quantities. Chem.—Methods 2021, 1, 61–70. [Google Scholar] [CrossRef]
- Light, M.E.; Murphy, B. The Cambridge Structural Database Communication ID ZUTDAT01; University of Southampton: Southampton, UK, 2018. [Google Scholar]
- Gurinov, A.A.; Lesnichin, S.B.; Limbach, H.-H.; Shenderovich, I.G. How short is the Strongest Hydrogen Bond in the Proton-Bound Homodimers of Pyridine Derivatives? J. Phys. Chem. A 2014, 118, 10804–10812. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Borissova, A.O.; Lesnichin, S.B.; Hartl, M.; Daemen, L.L.; Eckert, J.; Antipin, M.Y.; Shenderovich, I.G. Geometry and Spectral Properties of the Protonated Homodimer of Pyridine in the Liquid and Solid States. A Combined NMR, X-ray Diffraction and Inelastic Neutron Scattering Study. J. Phys. Chem. A 2011, 115, 8041–8048. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.; Del Bene, J.E.; Radom, L. What factors determine whether a proton-bound homodimer has a symmetric or an asymmetric hydrogen bond? Mol. Phys. 2009, 107, 1095–1105. [Google Scholar] [CrossRef]
- Mori, Y.; Takano, K. Location of protons in N–H⋯N hydrogen-bonded systems: A theoretical study on intramolecular pyridine–dihydropyridine and pyridine–pyridinium pairs. Phys. Chem. Chem. Phys. 2012, 14, 11090–11098. [Google Scholar] [CrossRef]
- Melikova, S.M.; Rutkowski, K.S.; Gurinov, A.A.; Denisov, G.S.; Rospenk, M.; Shenderovich, I.G. FTIR study of the hydrogen bond symmetry in protonated homodimers of pyridine and collidine in solution. J. Mol. Struct. 2012, 1018, 39–44. [Google Scholar] [CrossRef]
- Hakkert, S.B.; Erdėlyi, M. Halogen bond symmetry: The N-X-N bond. J. Phys. Org. Chem. 2015, 28, 226–233. [Google Scholar] [CrossRef]
- Mori, Y.; Takano, K. Effect of solvent on proton location and dynamic behavior in short intramolecular hydrogen bonds studied by molecular dynamics simulations and NMR experiments. Chem. Phys. 2015, 458, 18–29. [Google Scholar] [CrossRef]
- Gurinov, A.A.; Denisov, G.S.; Borissova, A.O.; Goloveshkin, A.S.; Greindl, J.; Limbach, H.-H.; Shenderovich, I.G. NMR Study of Solvation Effect on the Geometry of Proton-Bound Homodimers of Increasing Size. J. Phys. Chem. A 2017, 121, 8697–8705. [Google Scholar] [CrossRef]
- Reiersølmoen, A.C.; Battaglia, S.; Øien-Ødegaard, S.; Gupta, A.K.; Fiksdahl, A.; Lindh, R.; Erdėlyi, M. Symmetry of three-center, four-electron bonds. Chem. Sci. 2020, 11, 7979–7990. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen Bond and Other Lewis Acid-Lewis Base Interactions as Preliminary Stages of Chemical Reactions. Molecules 2020, 25, 4668. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. Adduct under Field—A Qualitative Approach to Account for Solvent Effect on Hydrogen Bonding. Molecules 2020, 25, 436. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G. Actual Symmetry of Symmetric Molecular Adducts in the Gas Phase, Solution and in the Solid State. Symmetry 2021, 13, 756. [Google Scholar] [CrossRef]
- Steiner, T. Lengthening of the Covalent X-H Bond in Heteronuclear Hydrogen Bonds Quantified from Organic and Organometallic Neutron Crystal Structures. J. Phys. Chem. A 1998, 102, 7041–7052. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Limbach, H.-H.; Tolstoy, P.M.; Pérez-Hernández, N.; Guo, J.; Shenderovich, I.G.; Denisov, G.S. OHO Hydrogen Bond Geometries and NMR Chemical Shifts: From Equilibrium Structures to Geometric H/D Isotope Effects, with Applications for Water, Protonated Water, and Compressed Ice. Israel J. Chem. 2009, 49, 199–216. [Google Scholar] [CrossRef]
- Shibl, M.F.; Pietrzak, M.; Limbach, H.-H.; Kühn, O. Geometric H/D isotope effects and cooperativity of the hydrogen bonds in porphycene. ChemPhysChem 2007, 8, 315–321. [Google Scholar] [CrossRef]
- Yamamoto, K.; Kanematsu, Y.; Nagashima, U.; Ueda, A.; Mori, H.; Tachikawa, M. Theoretical study of the H/D isotope effect on phase transition of hydrogen-bonded organic conductor κ-H3(Cat-EDT-TTF)2. Phys. Chem. Chem. Phys. 2016, 18, 29673–29680. [Google Scholar] [CrossRef]
- Golubev, N.S.; Melikova, S.M.; Shchepkin, D.N.; Shenderovich, I.G.; Tolstoy, P.M.; Denisov, G.S. Interpretation of H/D Isotope Effects on NMR Chemical Shifts of [FHF]− Ion Based on Calculations of Nuclear Magnetic Shielding Tensor Surface. Z. Phys. Chem. 2003, 217, 1549–1563. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further conventions for NMR shielding and chemical shifts IUPAC recommendations 2008. Solid State Nucl. Magn. Reson. 2008, 33, 41–56. [Google Scholar] [CrossRef]
- Bertani, P.; Raya, J.; Bechinger, B. 15N chemical shift referencing in solid state NMR. Solid State Nucl. Magn. Reson. 2014, 61–62, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09; Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Segmented Contracted Basis Sets Optimized for Nuclear Magnetic Shielding. J. Chem. Theory Comput. 2015, 11, 132–138. [Google Scholar] [CrossRef]
- Pritchard, B.P.; Altarawy, D.; Didier, B.; Gibson, T.D.; Windus, T.L. New Basis Set Exchange: An Open, Up-to-Date Resource for the Molecular Sciences Community. J. Chem. Inf. Model. 2019, 59, 4814–4820. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3093. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Simplified Calculation Approaches Designed to Reproduce the Geometry of Hydrogen Bonds in Molecular Complexes in Aprotic Solvents. J. Chem. Phys. 2018, 148, 124313. [Google Scholar] [CrossRef]
- Bryce, D.L.; Bernard, G.M.; Gee, M.; Lumsden, M.D.; Eichele, K.; Wasylishen, R.E. Practical Aspects of Modern Routine Solid-State Multinuclear Magnetic Resonance Spectroscopy: One-Dimensional Experiments. Can. J. Anal. Sci. Spectrosc. 2001, 46, 46–82. Available online: http://anorganik.uni-tuebingen.de/klaus/nmr/practical/practical.pdf (accessed on 4 June 2023). [CrossRef]
- Shenderovich, I.G.; Limbach, H.-H. Solid State NMR for Nonexperts: An Overview of Simple but General Practical Methods. Solids 2021, 2, 139–154. [Google Scholar] [CrossRef]
- Shenderovich, I.G. An Experimentally Established 15N NMR Absolute Shielding Scale for Theoretical Calculations. J. Phys. Chem. A 2023. [Google Scholar] [CrossRef]
- Shenderovich, I.G. 1,3,5-Triaza-7-Phosphaadamantane (PTA) as a 31P NMR Probe for Organometallic Transition Metal Complexes in Solution. Molecules 2021, 26, 1390. [Google Scholar] [CrossRef]
- Shenderovich, I.G. Electric field effect on 31P NMR magnetic shielding. J. Chem. Phys. 2020, 153, 184501. [Google Scholar] [CrossRef]
- Sheldrick, G.M. ShelXT-Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. Olex2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Bourhis, L.J.; Dolomanov, O.V.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. The Anatomy of a Comprehensive Constrained, Restrained, Refinement Program for the Modern Computing Environment—Olex2 Disected. Acta Crystallogr. Sect. A Found. Adv. 2015, A71, 59–71. [Google Scholar] [CrossRef]
- Ananyev, I.V.; Lyssenko, K.A. Conjugation effects in a para-nitroaniline molecule and its structural non-rigidity. Russ. Chem. Bull. 2014, 63, 1270–1282. [Google Scholar] [CrossRef]
- Borissova, A.O.; Lyssenko, K.A.; Gurinov, A.A.; Shenderovich, I.G. Energy Analysis of Competing Non-Covalent Interaction in 1:1 and 1:2 Adducts of Collidine with Benzoic Acids by Means of X-Ray Diffraction. Z. Phys. Chem. 2013, 227, 775–790. [Google Scholar] [CrossRef]
- Vologzhanina, A.V.; Lyssenko, K.A. Interatomic interactions in the crystal of (C4H11N2)2(C4H12N2)[Mo(CN)8]: Comparison of two approaches. Russ. Chem. Bull. 2013, 64, 1786–1792. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Glukhov, I.V.; Antipin, M.Y.; Lyssenko, K.A. Higher density does not mean higher stability” mystery of paracetamol finally unraveled. Chem. Commun. 2010, 46, 3469–3471. [Google Scholar] [CrossRef]
- Nelyubina, Y.V.; Antipin, M.Y.; Lyssenko, K.A. Hydrogen Bonds between Zwitterions: Intermediate between Classical and Charge-Assisted Ones. A Case Study. J. Phys. Chem. A 2009, 113, 3615–3620. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. Modeling of the Response of Hydrogen Bond Properties on an External Electric Field: Geometry, NMR Chemical Shift, Spin-Spin Scalar Coupling. Molecules 2021, 26, 4967. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Denisov, G.S. Modeling of Solute-Solvent Interactions Using an External Electric Field—From Tautomeric Equilibrium in Nonpolar Solvents to the Dissociation of Alkali Metal Halides. Molecules 2021, 26, 1283. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substance | State | , ppm |
|---|---|---|
| BiPy | Polycrystalline | −72.7 |
| BiPy | In tetrahydrofuran | −71.65 |
| BiPyH+ [ClO4]− | Polycrystalline | −82.0 & −193.4 |
| BiPyH+ [B(C6H5)4]− | Polycrystalline | −85.0 & −195.5 |
| Phen | Polycrystalline | −68.6 & −72.2 |
| Phen | In tetrahydrofuran | −66.74 |
| PhenH+ [B(C6H5)4]− | Polycrystalline | −88.3 & −208.3 |
| Structure | r1, Å | r2, Å | r3, Å | φ 1, ° | , ppm |
|---|---|---|---|---|---|
| BiPy_Tq_W_ωt | − | − | 3.61234 | 180 | −70 |
| BiPy_Tq_W_ωq | − | − | 3.61234 | 180 | −70 |
| BiPy_ωq_W_ωt | − | − | 3.60001 | 180 | −75 |
| BiPy_ωq_W_ωq | − | − | 3.60001 | 180 | −74 |
| BiPycis_Tq_W_ωt | − | − | 2.72141 | 0 | −63 |
| BiPyH+_Tq_W_ωt | 1.02483 | 2.05149 | 2.58497 | 0 | −82 & −199 |
| BiPyHClO4_Tq_W_ωt | 1.02871 | 2.24853 | 2.66167 | 14.6 | −71 & −195 |
| ZUTDAT_Tq_W_ωt | 1.02365 | 2.08824 | 2.61085 | 4.3 | −76 & −198 |
| ZUTDAT01_Tq_W_ωt | 1.02376 | 2.08657 | 2.60115 | 4.7 | −82 & −202 |
| Structure | r1, Å | r2, Å | r3, Å | , ppm |
|---|---|---|---|---|
| Phen_Tq_W_ωt | − | − | 2.77022 | −67 |
| Phen_Tq_W_ωq | − | − | 2.77022 | −68 |
| Phen_Tq_W_ωS | − | − | 2.77022 | −67 |
| OPENAN_Tt_W_ωt | − | − | 2.72215 2.72614 | −65 −73 |
| OPENAN01_Tt_W_ωt | − | − | 2.73995 2.74499 | −65 −63 |
| PhenH+_Tq_W_ωt | 1.02021 | 2.26272 | 2.70884 | −84 & −207 |
| ZUTDEX_Tq_W_ωt | 1.01968 | 2.26295 | 2.70682 | −81 & −204 |
| Substance | r2, Å | r3, Å | q(H), e | q(Nf), e | ε 1, e2/Å | ΔE, kJ/mol |
|---|---|---|---|---|---|---|
| BiPyH+_wt_W | 2.079695 | 2.595704 | 0.293915 | −0.394282 | −0.0557 | −46 ± 3 |
| BiPyH+_Tq_W | 2.051480 | 2.584967 | 0.255385 | −0.362354 | −0.0451 | −25 ± 8 |
| BiPyH+_wq_W | 2.080350 | 2.595114 | 0.237234 | −0.446997 | −0.0510 | −27 ± 4 |
| PhenH+_wt_W | 2.226297 | 2.688838 | 0.288186 | −0.390619 | −0.0506 | −37 ± 3 |
| PhenH+_Tq_W | 2.262717 | 2.708843 | 0.259246 | −0.351045 | −0.0402 | −11 ± 8 |
| PhenH+_wq_W | 2.273517 | 2.710015 | 0.246195 | −0.424726 | −0.0460 | −14 ± 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shenderovich, I.G. Weak, Broken, but Working—Intramolecular Hydrogen Bond in 2,2′-bipyridine. Int. J. Mol. Sci. 2023, 24, 10390. https://doi.org/10.3390/ijms241210390
Shenderovich IG. Weak, Broken, but Working—Intramolecular Hydrogen Bond in 2,2′-bipyridine. International Journal of Molecular Sciences. 2023; 24(12):10390. https://doi.org/10.3390/ijms241210390
Chicago/Turabian StyleShenderovich, Ilya G. 2023. "Weak, Broken, but Working—Intramolecular Hydrogen Bond in 2,2′-bipyridine" International Journal of Molecular Sciences 24, no. 12: 10390. https://doi.org/10.3390/ijms241210390
APA StyleShenderovich, I. G. (2023). Weak, Broken, but Working—Intramolecular Hydrogen Bond in 2,2′-bipyridine. International Journal of Molecular Sciences, 24(12), 10390. https://doi.org/10.3390/ijms241210390