

Molecular Mechanisms of Craniofacial and Dental Abnormalities in Osteopetrosis

Abstract
:1. Introduction
2. Genotype and Clinical Phenotype of Osteopetrosis
2.1. Osteopetrosis, Autosomal Dominant 1
2.2. Osteopetrosis, Autosomal Dominant 2
2.3. Osteopetrosis, Autosomal Dominant 3
2.4. Osteopetrosis, Autosomal Recessive 1
2.5. Osteopetrosis, Autosomal Recessive 2
2.6. Osteopetrosis, Autosomal Recessive 3
2.7. Osteopetrosis, Autosomal Recessive 4
2.8. Osteopetrosis, Autosomal Recessive 5
2.9. Osteopetrosis, Autosomal Recessive 6
2.10. Osteopetrosis, Autosomal Recessive 7
2.11. Osteopetrosis, Autosomal Recessive 8
2.12. Osteopetrosis, Autosomal Recessive 9
2.13. Anhidrotic Ectodermal Dysplasia Associated with Immune Deficiency, Osteopetrosis, and Lymphedema
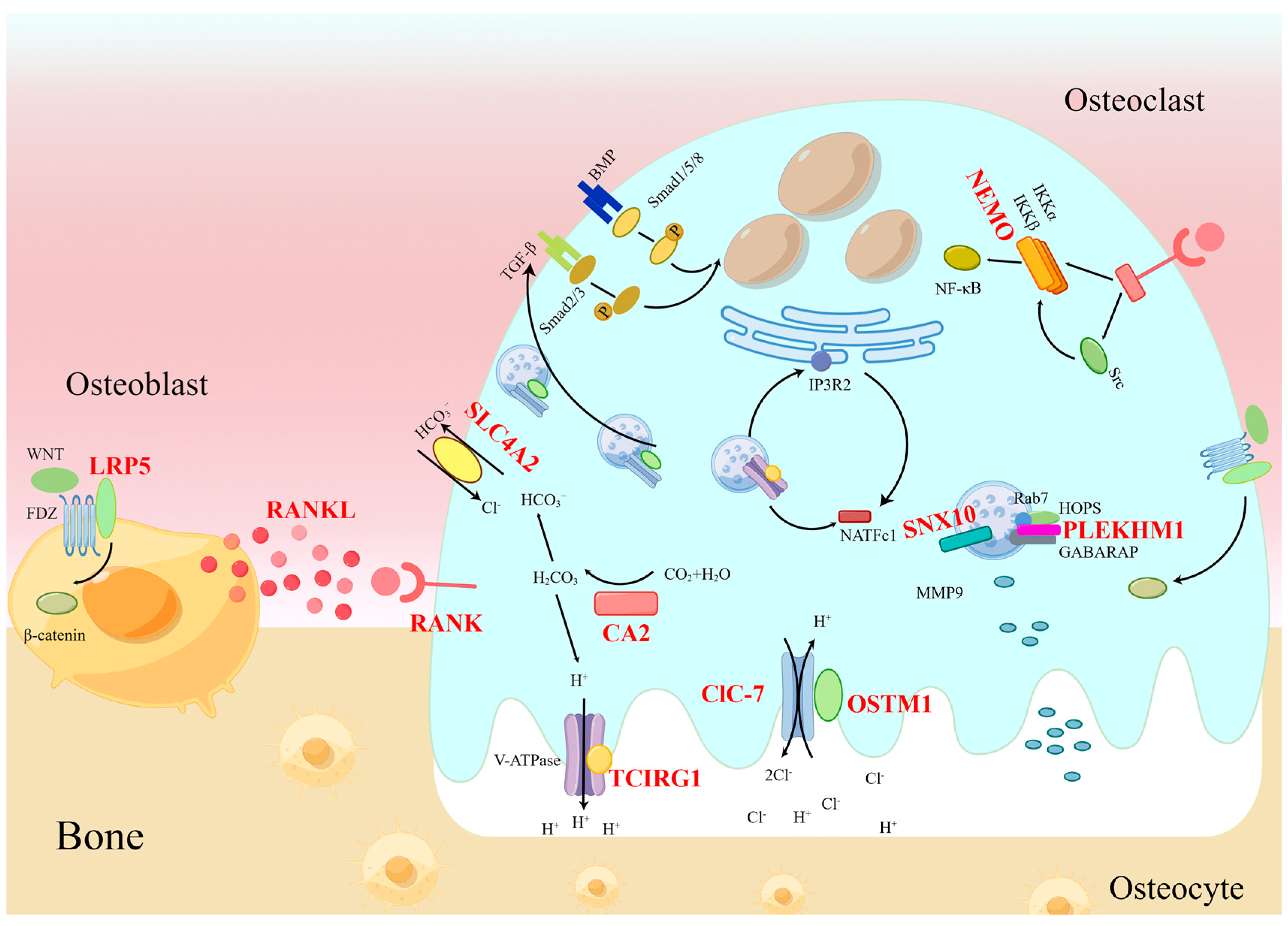
3. Molecular Pattern of Osteopetrosis
3.1. RANKL and RANK
3.2. TCIRG1
3.3. ClC-7
3.4. OSTM1
3.5. SNX10
3.6. PLEKHM1
3.7. LRP5
3.8. CA II
3.9. SLC4A2
3.10. IKBKG
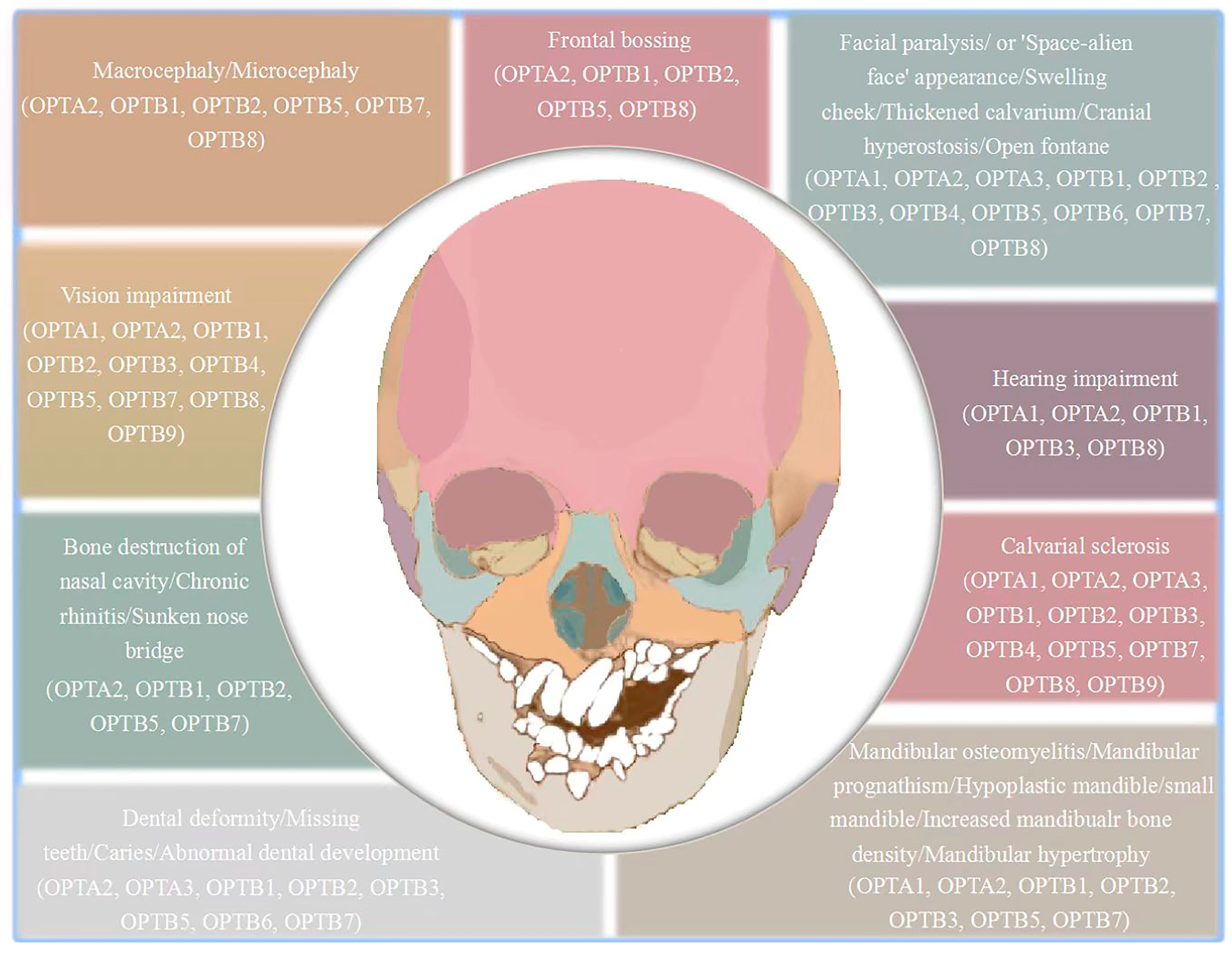
4. Clinical Craniofacial Bone and Tooth Phenotypes of Osteopetrosis Patients
4.1. Craniofacial Characteristics of Osteopetrosis Patients
4.2. Dental Characteristics of Osteopetrosis Patients
5. Molecular Mechanism of Craniofacial Bone and Tooth Phenotypes in Osteopetrosis
6. Conclusions

{kind=link}
{kind=link}
| Name | Abbreviation | OMIM | Inheritance | Main Characteristics | Craniofacial and Dental Characteristics | Mutation | Reference |
|---|---|---|---|---|---|---|---|
| Osteopetrosis, Autosomal Dominant 1 | OPTA1 | #607634 | AD |
|
| LRP5 | [4,102,103,104] |
| Osteopetrosis, Autosomal Dominant 2 | OPTA2 | #166600 | AD |
|
| CLCN7 | [7,9,98,105,106] |
| Osteopetrosis, Autosomal Dominant 3 | OPTA3 | #618107 | AD |
|
| PLEKHM1 | [20,21] |
| Osteopetrosis, Autosomal Recessive 1 | OPTB1 | #259700 | AR |
|
| TCIRG1 | [23,24,25] |
| Osteopetrosis, Autosomal Recessive 2 | OPTB2 | #259710 | AR |
|
| TNFSF11 | [32,33,34,54,107] |
| Osteopetrosis, Autosomal Recessive 3 | OPTB3 | #259730 | AR |
|
| CA2 | [40,41,42,108,109,110,111,112,113,114] |
| Osteopetrosis, Autosomal Recessive 4 | OPTB4 | #611490 | AR |
|
| CLCN7 | [6,45] |
| Osteopetrosis, Autosomal Recessive 5 | OPTB5 | #259720 | AR |
|
| OSTM1 | [46,47,48,49,51,115] |
| Osteopetrosis, Autosomal Recessive 6 | OPTB6 | #611497 | AR |
|
| PLEKHM1 | [53] |
| Osteopetrosis, Autosomal Recessive 7 | OPTB7 | #612301 | AR |
|
| TNFRSF11A | [54,55,56,116,117] |
| Osteopetrosis, Autosomal Recessive 8 | OPTB8 | #615085 | AR |
|
| SNX10 | [61,62,118] |
| Osteopetrosis, Autosomal Recessive 9 | OPTB9 | #620366 | AR |
|
| SLC4A2 | [66,67] |
| Anhidrotic ectodermal dysplasia associated with immune deficiency, osteopetrosis, and lymphedema | OLEDAID | #300301 | XLR |
|
| IKBKG | [2,68,69] |
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Palagano, E.; Menale, C.; Sobacchi, C.; Villa, A. Genetics of Osteopetrosis. Curr. Osteoporos. Rep. 2018, 16, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Döffinger, R.; Smahi, A.; Bessia, C.; Geissmann, F.; Feinberg, J.; Durandy, A.; Bodemer, C.; Kenwrick, S.; Dupuis-Girod, S.; Blanche, S.; et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet. 2001, 27, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ji, D.; Li, L.; Yang, S.; Zhang, H.; Duan, X. ClC-7 Regulates the Pattern and Early Development of Craniofacial Bone and Tooth. Theranostics 2019, 9, 1387–1400. [Google Scholar] [CrossRef] [PubMed]
- Van Hul, E.; Gram, J.; Bollerslev, J.; Van Wesenbeeck, L.; Mathysen, D.; Andersen, P.E.; Vanhoenacker, F.; Van Hul, W. Localization of the gene causing autosomal dominant osteopetrosis type I to chromosome 11q12-13. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2002, 17, 1111–1117. [Google Scholar] [CrossRef]
- Pangrazio, A.; Boudin, E.; Piters, E.; Damante, G.; Lo Iacono, N.; D’Elia, A.V.; Vezzoni, P.; Van Hul, W.; Villa, A.; Sobacchi, C. Identification of the first deletion in the LRP5 gene in a patient with autosomal dominant osteopetrosis type I. Bone 2011, 49, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Cleiren, E.; Bénichou, O.; Van Hul, E.; Gram, J.; Bollerslev, J.; Singer, F.R.; Beaverson, K.; Aledo, A.; Whyte, M.P.; Yoneyama, T.; et al. Albers-Schönberg disease (autosomal dominant osteopetrosis, type II) results from mutations in the ClCN7 chloride channel gene. Hum. Mol. Genet. 2001, 10, 2861–2867. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, X.; Wang, Y.; Fu, W.; Liu, Y.; Zhang, Z.; Wang, C. Natural History of Type II Autosomal Dominant Osteopetrosis: A Single Center Retrospective Study. Front. Endocrinol. 2022, 13, 819641. [Google Scholar] [CrossRef]
- Andersen, P.E., Jr.; Bollerslev, J. Heterogeneity of autosomal dominant osteopetrosis. Radiology 1987, 164, 223–225. [Google Scholar] [CrossRef]
- Waguespack, S.G.; Hui, S.L.; Dimeglio, L.A.; Econs, M.J. Autosomal dominant osteopetrosis: Clinical severity and natural history of 94 subjects with a chloride channel 7 gene mutation. J. Clin. Endocrinol. Metab. 2007, 92, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Caetano-Lopes, J.; Lessard, S.G.; Hann, S.; Espinoza, K.; Kang, K.S.; Lim, K.E.; Horan, D.J.; Noonan, H.R.; Hu, D.; Baron, R.; et al. Clcn7(F318L/+) as a new mouse model of Albers-Schönberg disease. Bone 2017, 105, 253–261. [Google Scholar] [CrossRef]
- Alam, I.; Gray, A.K.; Chu, K.; Ichikawa, S.; Mohammad, K.S.; Capannolo, M.; Capulli, M.; Maurizi, A.; Muraca, M.; Teti, A.; et al. Generation of the first autosomal dominant osteopetrosis type II (ADO2) disease models. Bone 2014, 59, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Weinert, S.; Jabs, S.; Supanchart, C.; Schweizer, M.; Gimber, N.; Richter, M.; Rademann, J.; Stauber, T.; Kornak, U.; Jentsch, T.J. Lysosomal pathology and osteopetrosis upon loss of H+-driven lysosomal Cl- accumulation. Science 2010, 328, 1401–1403. [Google Scholar] [CrossRef] [Green Version]
- Kornak, U.; Kasper, D.; Bösl, M.R.; Kaiser, E.; Schweizer, M.; Schulz, A.; Friedrich, W.; Delling, G.; Jentsch, T.J. Loss of the ClC-7 chloride channel leads to osteopetrosis in mice and man. Cell 2001, 104, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Rajan, I.; Read, R.; Small, D.L.; Perrard, J.; Vogel, P. An alternative splicing variant in Clcn7-/- mice prevents osteopetrosis but not neural and retinal degeneration. Vet. Pathol. 2011, 48, 663–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Pan, M.; Ni, J.; Zhang, Y.; Zhang, Y.; Gao, S.; Liu, J.; Wang, Z.; Zhang, R.; He, H.; et al. ClC-7 Deficiency Impairs Tooth Development and Eruption. Sci. Rep. 2016, 6, 19971. [Google Scholar] [CrossRef] [Green Version]
- Weinert, S.; Jabs, S.; Hohensee, S.; Chan, W.L.; Kornak, U.; Jentsch, T.J. Transport activity and presence of ClC-7/Ostm1 complex account for different cellular functions. EMBO Rep. 2014, 15, 784–791. [Google Scholar] [CrossRef] [Green Version]
- Alam, I.; McQueen, A.K.; Acton, D.; Reilly, A.M.; Gerard-O’Riley, R.L.; Oakes, D.K.; Kasipathi, C.; Huffer, A.; Wright, W.B.; Econs, M.J. Phenotypic severity of autosomal dominant osteopetrosis type II (ADO2) mice on different genetic backgrounds recapitulates the features of human disease. Bone 2017, 94, 34–41. [Google Scholar] [CrossRef]
- Wen, X.; Lacruz, R.S.; Paine, M.L. Dental and Cranial Pathologies in Mice Lacking the Cl(-) /H(+) -Exchanger ClC-7. Anat. Rec. 2015, 298, 1502–1508. [Google Scholar] [CrossRef] [Green Version]
- Lange, P.F.; Wartosch, L.; Jentsch, T.J.; Fuhrmann, J.C. ClC-7 requires Ostm1 as a beta-subunit to support bone resorption and lysosomal function. Nature 2006, 440, 220–223. [Google Scholar] [CrossRef] [Green Version]
- Bo, T.; Yan, F.; Guo, J.; Lin, X.; Zhang, H.; Guan, Q.; Wang, H.; Fang, L.; Gao, L.; Zhao, J.; et al. Characterization of a Relatively Malignant Form of Osteopetrosis Caused by a Novel Mutation in the PLEKHM1 Gene. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2016, 31, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Del Fattore, A.; Fornari, R.; Van Wesenbeeck, L.; de Freitas, F.; Timmermans, J.P.; Peruzzi, B.; Cappariello, A.; Rucci, N.; Spera, G.; Helfrich, M.H.; et al. A new heterozygous mutation (R714C) of the osteopetrosis gene, pleckstrin homolog domain containing family M (with run domain) member 1 (PLEKHM1), impairs vesicular acidification and increases TRACP secretion in osteoclasts. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2008, 23, 380–391. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Ye, S.; Castro-Gomes, T.; Winchell, C.G.; Andrews, N.W.; Voth, D.E.; Varughese, K.I.; Mackintosh, S.G.; Feng, Y.; Pavlos, N.; et al. PLEKHM1/DEF8/RAB7 complex regulates lysosome positioning and bone homeostasis. JCI Insight 2016, 1, e86330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.K.; Wallbrandt, P.; Zecca, L.; et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000, 25, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Li, N.; Yao, R.E.; Yu, T.; Ding, L.; Chen, J.; Wang, J. Clinical and molecular characterization of five Chinese patients with autosomal recessive osteopetrosis. Mol. Genet. Genom. Med. 2021, 9, e1815. [Google Scholar] [CrossRef] [PubMed]
- Zirngibl, R.A.; Wang, A.; Yao, Y.; Manolson, M.F.; Krueger, J.; Dupuis, L.; Mendoza-Londono, R.; Voronov, I. Novel c.G630A TCIRG1 mutation causes aberrant splicing resulting in an unusually mild form of autosomal recessive osteopetrosis. J. Cell. Biochem. 2019, 120, 17180–17193. [Google Scholar] [CrossRef]
- Palagano, E.; Muggeo, S.; Crisafulli, L.; Tourkova, I.L.; Strina, D.; Mantero, S.; Fontana, E.; Locatelli, S.L.; Monari, M.; Morenghi, E.; et al. Generation of an immunodeficient mouse model of tcirg1-deficient autosomal recessive osteopetrosis. Bone Rep. 2020, 12, 100242. [Google Scholar] [CrossRef]
- Bronckers, A.L.; Lyaruu, D.M.; Bervoets, T.J.; Medina, J.F.; DenBesten, P.; Richter, J.; Everts, V. Murine ameloblasts are immunonegative for Tcirg1, the v-H-ATPase subunit essential for the osteoclast plasma proton pump. Bone 2012, 50, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Blin-Wakkach, C.; Wakkach, A.; Sexton, P.M.; Rochet, N.; Carle, G.F. Hematological defects in the oc/oc mouse, a model of infantile malignant osteopetrosis. Leukemia 2004, 18, 1505–1511. [Google Scholar] [CrossRef] [Green Version]
- Blin-Wakkach, C.; Breuil, V.; Quincey, D.; Bagnis, C.; Carle, G.F. Establishment and characterization of new osteoclast progenitor cell lines derived from osteopetrotic and wild type mice. Bone 2006, 39, 53–60. [Google Scholar] [CrossRef]
- Kawamura, N.; Tabata, H.; Sun-Wada, G.H.; Wada, Y. Optic nerve compression and retinal degeneration in Tcirg1 mutant mice lacking the vacuolar-type H-ATPase a3 subunit. PLoS ONE 2010, 5, e12086. [Google Scholar] [CrossRef]
- Ochotny, N.; Flenniken, A.M.; Owen, C.; Voronov, I.; Zirngibl, R.A.; Osborne, L.R.; Henderson, J.E.; Adamson, S.L.; Rossant, J.; Manolson, M.F.; et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2011, 26, 1484–1493. [Google Scholar] [CrossRef]
- Kahler, S.G.; Burns, J.A.; Aylsworth, A.S. A mild autosomal recessive form of osteopetrosis. Am. J. Med. Genet. 1984, 17, 451–464. [Google Scholar] [CrossRef]
- Sharma, A.; Ingole, S.N.; Deshpande, M.D.; Kazi, N.; Meshram, D.; Ranadive, P. A Rare Case of Osteoclast-poor Osteopetrosis (RANKL Mutation) with Recurrent Osteomyelitis of Mandible: A Case Report. Int. J. Clin. Pediatr. Dent. 2020, 13, 717–721. [Google Scholar] [CrossRef]
- Sobacchi, C.; Frattini, A.; Guerrini, M.M.; Abinun, M.; Pangrazio, A.; Susani, L.; Bredius, R.; Mancini, G.; Cant, A.; Bishop, N.; et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat. Genet. 2007, 39, 960–962. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, R.S.; Cawley, K.M.; Gubrij, I.; Nookaew, I.; Onal, M.; O’Brien, C.A. Effective CRISPR interference of an endogenous gene via a single transgene in mice. Sci. Rep. 2019, 9, 17312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kurata, K.; Oh-Hora, M.; Feng, J.Q.; Bonewald, L.F.; Kodama, T.; Wutz, A.; Wagner, E.F.; et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat. Med. 2011, 17, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Fumoto, T.; Takeshita, S.; Ito, M.; Ikeda, K. Physiological functions of osteoblast lineage and T cell-derived RANKL in bone homeostasis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2014, 29, 830–842. [Google Scholar] [CrossRef]
- Huang, H.; Wang, J.; Zhang, Y.; Zhu, G.; Li, Y.P.; Ping, J.; Chen, W. Bone resorption deficiency affects tooth root development in RANKL mutant mice due to attenuated IGF-1 signaling in radicular odontoblasts. Bone 2018, 114, 161–171. [Google Scholar] [CrossRef]
- Sobacchi, C.; Abinun, M. Osteoclast-poor osteopetrosis. Bone 2022, 164, 116541. [Google Scholar] [CrossRef]
- Pang, Q.; Qi, X.; Jiang, Y.; Wang, O.; Li, M.; Xing, X.; Dong, J.; Xia, W. Two novel CAII mutations causing carbonic anhydrase II deficiency syndrome in two unrelated Chinese families. Metab. Brain Dis. 2015, 30, 989–997. [Google Scholar] [CrossRef]
- Yang, Y.; Tang, N.; Zhu, Y.; Zhang, L.; Cao, X.; Liu, L.; Xia, W.; Li, P.; Yang, Y. A novel homozygous nonsense mutation in the CA2 gene (c.368G>A, p.W123X) linked to carbonic anhydrase II deficiency syndrome in a Chinese family. Metab. Brain Dis. 2021, 36, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Fathallah, D.M.; Bejaoui, M.; Lepaslier, D.; Chater, K.; Sly, W.S.; Dellagi, K. Carbonic anhydrase II (CA II) deficiency in Maghrebian patients: Evidence for founder effect and genomic recombination at the CA II locus. Hum. Genet. 1997, 99, 634–637. [Google Scholar] [CrossRef]
- Margolis, D.S.; Szivek, J.A.; Lai, L.W.; Lien, Y.H. Phenotypic characteristics of bone in carbonic anhydrase II-deficient mice. Calcif. Tissue Int. 2008, 82, 66–76. [Google Scholar] [CrossRef]
- Lewis, S.E.; Erickson, R.P.; Barnett, L.B.; Venta, P.J.; Tashian, R.E. N-ethyl-N-nitrosourea-induced null mutation at the mouse Car-2 locus: An animal model for human carbonic anhydrase II deficiency syndrome. Proc. Natl. Acad. Sci. USA 1988, 85, 1962–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, C.W.; Tong, S.F.; Wong, K.; Luo, Y.F.; Tang, H.Y.; Ha, S.Y.; Chan, M.H. DNA-based diagnosis of malignant osteopetrosis by whole-genome scan using a single-nucleotide polymorphism microarray: Standardization of molecular investigations of genetic diseases due to consanguinity. J. Hum. Genet. 2007, 52, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Vacher, J. OSTM1 pleiotropic roles from osteopetrosis to neurodegeneration. Bone 2022, 163, 116505. [Google Scholar] [CrossRef]
- Quarello, P.; Forni, M.; Barberis, L.; Defilippi, C.; Campagnoli, M.F.; Silvestro, L.; Frattini, A.; Chalhoub, N.; Vacher, J.; Ramenghi, U. Severe malignant osteopetrosis caused by a GL gene mutation. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2004, 19, 1194–1199. [Google Scholar] [CrossRef]
- Mahmoud Adel, A.H.; Abdullah, A.A.; Eissa, F. Infantile osteopetrosis, craniosynostosis, and Chiari malformation type I with novel OSTEM1 mutation. J. Pediatr. Neurosci. 2013, 8, 34–37. [Google Scholar] [CrossRef] [Green Version]
- Khazen, N.E.; Faverly, D.; Vamos, E.; Van Regemorter, N.; Flament-Durand, J.; Carton, B.; Cremer-Perlmutter, N. Lethal osteopetrosis with multiple fractures in utero. Am. J. Med. Genet. 1986, 23, 811–819. [Google Scholar] [CrossRef]
- Pata, M.; Vacher, J. Ostm1 Bifunctional Roles in Osteoclast Maturation: Insights From a Mouse Model Mimicking a Human OSTM1 Mutation. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2018, 33, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Pangrazio, A.; Poliani, P.L.; Megarbane, A.; Lefranc, G.; Lanino, E.; Di Rocco, M.; Rucci, F.; Lucchini, F.; Ravanini, M.; Facchetti, F.; et al. Mutations in OSTM1 (grey lethal) define a particularly severe form of autosomal recessive osteopetrosis with neural involvement. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2006, 21, 1098–1105. [Google Scholar] [CrossRef]
- Bosman, E.A.; Estabel, J.; Ismail, O.; Podrini, C.; White, J.K.; Steel, K.P. Omi, a recessive mutation on chromosome 10, is a novel allele of Ostm1. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2013, 24, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Van Wesenbeeck, L.; Odgren, P.R.; Coxon, F.P.; Frattini, A.; Moens, P.; Perdu, B.; MacKay, C.A.; Van Hul, E.; Timmermans, J.P.; Vanhoenacker, F.; et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J. Clin. Investig. 2007, 117, 919–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Yu, X.; Huang, M. A novel mutation in TNFRSF11A gene causes pediatric osteopetrosis: Case report. BMC Surg. 2021, 21, 269. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, M.M.; Sobacchi, C.; Cassani, B.; Abinun, M.; Kilic, S.S.; Pangrazio, A.; Moratto, D.; Mazzolari, E.; Clayton-Smith, J.; Orchard, P.; et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am. J. Hum. Genet. 2008, 83, 64–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor-Miller, T.; Sivaprakasam, P.; Smithson, S.F.; Steward, C.G.; Burren, C.P. Challenges in long-term control of hypercalcaemia with denosumab after haematopoietic stem cell transplantation for TNFRSF11A osteoclast-poor autosomal recessive osteopetrosis. Bone Rep. 2021, 14, 100738. [Google Scholar] [CrossRef]
- Alonso, N.; Wani, S.; Rose, L.; Van’t Hof, R.J.; Ralston, S.H.; Albagha, O.M.E. Insertion Mutation in Tnfrsf11a Causes a Paget’s Disease-Like Phenotype in Heterozygous Mice and Osteopetrosis in Homozygous Mice. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2021, 36, 1376–1386. [Google Scholar] [CrossRef]
- Kapur, R.P.; Yao, Z.; Iida, M.H.; Clarke, C.M.; Doggett, B.; Xing, L.; Boyce, B.F. Malignant autosomal recessive osteopetrosis caused by spontaneous mutation of murine Rank. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2004, 19, 1689–1697. [Google Scholar] [CrossRef]
- Dougall, W.C.; Glaccum, M.; Charrier, K.; Rohrbach, K.; Brasel, K.; De Smedt, T.; Daro, E.; Smith, J.; Tometsko, M.E.; Maliszewski, C.R.; et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999, 13, 2412–2424. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef] [Green Version]
- Udupa, P.; Ghosh, D.K.; Kausthubham, N.; Shah, H.; Bartakke, S.; Dalal, A.; Girisha, K.M.; Bhavani, G.S. Genome sequencing identifies a large non-coding region deletion of SNX10 causing autosomal recessive osteopetrosis. J. Hum. Genet. 2023, 68, 287–290. [Google Scholar] [CrossRef]
- Mégarbané, A.; Pangrazio, A.; Villa, A.; Chouery, E.; Maarawi, J.; Sabbagh, S.; Lefranc, G.; Sobacchi, C. Homozygous stop mutation in the SNX10 gene in a consanguineous Iraqi boy with osteopetrosis and corpus callosum hypoplasia. Eur. J. Med. Genet. 2013, 56, 32–35. [Google Scholar] [CrossRef]
- Stein, M.; Barnea-Zohar, M.; Shalev, M.; Arman, E.; Brenner, O.; Winograd-Katz, S.; Gerstung, J.; Thalji, F.; Kanaan, M.; Elinav, H.; et al. Massive osteopetrosis caused by non-functional osteoclasts in R51Q SNX10 mutant mice. Bone 2020, 136, 115360. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Morse, L.R.; Zhang, L.; Sasaki, H.; Mills, J.C.; Odgren, P.R.; Sibbel, G.; Stanley, J.R.; Wong, G.; Zamarioli, A.; et al. Osteopetrorickets due to Snx10 deficiency in mice results from both failed osteoclast activity and loss of gastric acid-dependent calcium absorption. PLoS Genet. 2015, 11, e1005057. [Google Scholar] [CrossRef]
- Zhou, C.; You, Y.; Shen, W.; Zhu, Y.Z.; Peng, J.; Feng, H.T.; Wang, Y.; Li, D.; Shao, W.W.; Li, C.X.; et al. Deficiency of sorting nexin 10 prevents bone erosion in collagen-induced mouse arthritis through promoting NFATc1 degradation. Ann. Rheum. Dis. 2016, 75, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.Y.; Grigelioniene, G.; Wang, Z.; Nishimura, G.; Iida, A.; Matsumoto, N.; Tham, E.; Miyake, N.; Ikegawa, S.; Guo, L. SLC4A2 Deficiency Causes a New Type of Osteopetrosis. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2022, 37, 226–235. [Google Scholar] [CrossRef]
- Xue, J.Y.; Ikegawa, S.; Guo, L. SLC4A2, another gene involved in acid-base balancing machinery of osteoclasts, causes osteopetrosis. Bone 2023, 167, 116603. [Google Scholar] [CrossRef]
- Dupuis-Girod, S.; Corradini, N.; Hadj-Rabia, S.; Fournet, J.C.; Faivre, L.; Le Deist, F.; Durand, P.; Döffinger, R.; Smahi, A.; Israel, A.; et al. Osteopetrosis, lymphedema, anhidrotic ectodermal dysplasia, and immunodeficiency in a boy and incontinentia pigmenti in his mother. Pediatrics 2002, 109, e97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, C.M.; Angus, J.E.; Leach, I.H.; McDermott, E.M.; Walker, D.A.; Ravenscroft, J.C. A novel NEMO gene mutation causing osteopetrosis, lymphoedema, hypohidrotic ectodermal dysplasia and immunodeficiency (OL-HED-ID). Eur. J. Pediatr. 2010, 169, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Swarnkar, G.; Shim, K.; Nasir, A.M.; Seehra, K.; Chen, H.P.; Mbalaviele, G.; Abu-Amer, Y. Myeloid Deletion of Nemo Causes Osteopetrosis in Mice Owing to Upregulation of Transcriptional Repressors. Sci. Rep. 2016, 6, 29896. [Google Scholar] [CrossRef] [PubMed]
- Qiu, H.; Qin, A.; Cheng, T.; Chim, S.M.; Smithers, L.; Chen, K.; Song, D.; Liu, Q.; Zhao, J.; Wang, C.; et al. A missense mutation sheds light on a novel structure-function relationship of RANKL. J. Cell. Physiol. 2021, 236, 2800–2816. [Google Scholar] [CrossRef]
- Takeshita, S.; Fumoto, T.; Ito, M.; Ikeda, K. Serum CTX levels and histomorphometric analysis in Src versus RANKL knockout mice. J. Bone Miner. Metab. 2018, 36, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lin, L.; Yang, B.; Meng, Z.; Zhang, B. Knockdown of Tcirg1 inhibits large-osteoclast generation by down-regulating NFATc1 and IP3R2 expression. PLoS ONE 2020, 15, e0237354. [Google Scholar] [CrossRef]
- Peng, H.; He, H.B.; Wen, T. A Novel Variant in CLCN7 Regulates the Coupling of Angiogenesis and Osteogenesis. Front. Cell Dev. Biol. 2020, 8, 599826. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Zhang, B.; Zhou, J.; Li, T.; Liu, Z.; Li, Y.; Yang, M. Molecular insights into the human CLC-7/Ostm1 transporter. Sci. Adv. 2020, 6, eabb4747. [Google Scholar] [CrossRef]
- Feigin, M.E.; Malbon, C.C. OSTM1 regulates beta-catenin/Lef1 interaction and is required for Wnt/beta-catenin signaling. Cell. Signal. 2008, 20, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.H.; Morse, L.R.; Battaglino, R.A. SNX10 is required for osteoclast formation and resorption activity. J. Cell. Biochem. 2012, 113, 1608–1615. [Google Scholar] [CrossRef]
- Sultana, F.; Morse, L.R.; Picotto, G.; Liu, W.; Jha, P.K.; Odgren, P.R.; Battaglino, R.A. Snx10 and PIKfyve are required for lysosome formation in osteoclasts. J. Cell. Biochem. 2020, 121, 2927–2937. [Google Scholar] [CrossRef]
- Xu, J.; Qiu, H.; Zhao, J.; Pavlos, N.J. The molecular structure and function of sorting nexin 10 in skeletal disorders, cancers, and other pathological conditions. J. Cell. Physiol. 2021, 236, 4207–4215. [Google Scholar] [CrossRef]
- Barnea-Zohar, M.; Winograd-Katz, S.E.; Shalev, M.; Arman, E.; Reuven, N.; Roth, L.; Golani, O.; Stein, M.; Thalji, F.; Kanaan, M.; et al. An SNX10-dependent mechanism downregulates fusion between mature osteoclasts. J. Cell Sci. 2021, 134, jcs254979. [Google Scholar] [CrossRef]
- Maruzs, T.; Lakatos, E.; Feil-Börcsök, D.; Lőrincz, P.; Juhász, G. Isolation and characterization of novel plekhm1 and def8 mutant alleles in Drosophila. Biol. Futur. 2022, 73, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lathrop, W.; Weaver, D.; Qiu, Q.; Cini, J.; Bertolini, D.; Chen, D. Molecular cloning and characterization of LR3, a novel LDL receptor family protein with mitogenic activity. Biochem. Biophys. Res. Commun. 1998, 251, 784–790. [Google Scholar] [CrossRef]
- Gong, Y.; Slee, R.B.; Fukai, N.; Rawadi, G.; Roman-Roman, S.; Reginato, A.M.; Wang, H.; Cundy, T.; Glorieux, F.H.; Lev, D.; et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell 2001, 107, 513–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khrystoforova, I.; Shochat-Carvalho, C.; Harari, R.; Henke, K.; Woronowicz, K.; Harris, M.P.; Karasik, D. Zebrafish mutants reveal unexpected role of Lrp5 in osteoclast regulation. Front. Endocrinol. 2022, 13, 985304. [Google Scholar] [CrossRef]
- Sun, X.; Li, K.; Hase, M.; Zha, R.; Feng, Y.; Li, B.Y.; Yokota, H. Suppression of breast cancer-associated bone loss with osteoblast proteomes via Hsp90ab1/moesin-mediated inhibition of TGFβ/FN1/CD44 signaling. Theranostics 2022, 12, 929–943. [Google Scholar] [CrossRef]
- Alkhayal, Z.; Shinwari, Z.; Gaafar, A.; Alaiya, A. Carbonic Anhydrase II Activators in Osteopetrosis Treatment: A Review. Curr. Issues Mol. Biol. 2023, 45, 1373–1386. [Google Scholar] [CrossRef]
- Coury, F.; Zenger, S.; Stewart, A.K.; Stephens, S.; Neff, L.; Tsang, K.; Shull, G.E.; Alper, S.L.; Baron, R.; Aliprantis, A.O. SLC4A2-mediated Cl-/HCO3- exchange activity is essential for calpain-dependent regulation of the actin cytoskeleton in osteoclasts. Proc. Natl. Acad. Sci. USA 2013, 110, 2163–2168. [Google Scholar] [CrossRef] [Green Version]
- Dai, S.; Abu-Amer, W.; Karuppaiah, K.; Abu-Amer, Y. Evidence that the kinase-truncated c-Src regulates NF-κB signaling by targeting NEMO. J. Cell. Biochem. 2011, 112, 2463–2470. [Google Scholar] [CrossRef] [Green Version]
- Clohisy, J.C.; Yamanaka, Y.; Faccio, R.; Abu-Amer, Y. Inhibition of IKK activation, through sequestering NEMO, blocks PMMA-induced osteoclastogenesis and calvarial inflammatory osteolysis. J. Orthop. Res. Off. Publ. Orthop. Res. Soc. 2006, 24, 1358–1365. [Google Scholar] [CrossRef]
- Ohazama, A.; Courtney, J.M.; Sharpe, P.T. Opg, Rank, and Rankl in tooth development: Co-ordination of odontogenesis and osteogenesis. J. Dent. Res. 2004, 83, 241–244. [Google Scholar] [CrossRef]
- Alkhayal, Z.; Shinwari, Z.; Gaafar, A.; Alaiya, A. Proteomic Profiling of the First Human Dental Pulp Mesenchymal Stem/Stromal Cells from Carbonic Anhydrase II Deficiency Osteopetrosis Patients. Int. J. Mol. Sci. 2020, 22, 380. [Google Scholar] [CrossRef]
- Wu, J.; Glimcher, L.H.; Aliprantis, A.O. HCO3-/Cl- anion exchanger SLC4A2 is required for proper osteoclast differentiation and function. Proc. Natl. Acad. Sci. USA 2008, 105, 16934–16939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whyte, M.P.; McAlister, W.H.; Zhang, F.; Bijanki, V.N.; Nenninger, A.; Gottesman, G.S.; Lin, E.L.; Huskey, M.; Duan, S.; Dahir, K.; et al. New explanation for autosomal dominant high bone mass: Mutation of low-density lipoprotein receptor-related protein 6. Bone 2019, 127, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Klein, O.D. Molecular and cellular mechanisms of tooth development, homeostasis and repair. Development 2020, 147, dev184754. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.V.; Reichman, L. OSTEOPETROSIS WITH A COMPLICATING OSTEOMYELITIS OF THE MANDIBLE. REPORT OF A CASE. Oral Surg. Oral Med. Oral Pathol. 1965, 19, 462–465. [Google Scholar] [CrossRef] [PubMed]
- Di Zanni, E.; Palagano, E.; Lagostena, L.; Strina, D.; Rehman, A.; Abinun, M.; De Somer, L.; Martire, B.; Brown, J.; Kariminejad, A.; et al. Pathobiologic Mechanisms of Neurodegeneration in Osteopetrosis Derived From Structural and Functional Analysis of 14 ClC-7 Mutants. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2021, 36, 531–545. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Xu, T.; Fan, Y.; Ding, Y.; Qian, J. A novel compound heterozygous mutation of the CLCN7 gene is associated with autosomal recessive osteopetrosis. Front. Pediatr. 2023, 11, 978879. [Google Scholar] [CrossRef]
- Whyte, M.P.; McAlister, W.H.; Dhiman, V.; Gopinathan, N.R.; Bhadada, S.K. Drug-induced osteopetrosis. Bone 2023, 173, 116788. [Google Scholar] [CrossRef]
- Menale, C.; Campodoni, E.; Palagano, E.; Mantero, S.; Erreni, M.; Inforzato, A.; Fontana, E.; Schena, F.; Van’t Hof, R.; Sandri, M.; et al. Mesenchymal Stromal Cell-Seeded Biomimetic Scaffolds as a Factory of Soluble RANKL in Rankl-Deficient Osteopetrosis. Stem Cells Transl. Med. 2019, 8, 22–34. [Google Scholar] [CrossRef]
- Capo, V.; Penna, S.; Merelli, I.; Barcella, M.; Scala, S.; Basso-Ricci, L.; Draghici, E.; Palagano, E.; Zonari, E.; Desantis, G.; et al. Expanded circulating hematopoietic stem/progenitor cells as novel cell source for the treatment of TCIRG1 osteopetrosis. Haematologica 2021, 106, 74–86. [Google Scholar] [CrossRef] [Green Version]
- Bollerslev, J.; Mosekilde, L. Autosomal dominant osteopetrosis. Clin. Orthop. Relat. Res. 1993, 294, 45–51. [Google Scholar] [CrossRef]
- Janssens, K.; Van Hul, W. Molecular genetics of too much bone. Hum. Mol. Genet. 2002, 11, 2385–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Wesenbeeck, L.; Cleiren, E.; Gram, J.; Beals, R.K.; Bénichou, O.; Scopelliti, D.; Key, L.; Renton, T.; Bartels, C.; Gong, Y.; et al. Six novel missense mutations in the LDL receptor-related protein 5 (LRP5) gene in different conditions with an increased bone density. Am. J. Hum. Genet. 2003, 72, 763–771. [Google Scholar] [CrossRef] [Green Version]
- Bollerslev, J.; Andersen, P.E., Jr. Radiological, biochemical and hereditary evidence of two types of autosomal dominant osteopetrosis. Bone 1988, 9, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Bénichou, O.D.; Laredo, J.D.; de Vernejoul, M.C. Type II autosomal dominant osteopetrosis (Albers-Schönberg disease): Clinical and radiological manifestations in 42 patients. Bone 2000, 26, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Lertwilaiwittaya, P.; Suktitipat, B.; Khongthon, P.; Pongsapich, W.; Limwongse, C.; Pithukpakorn, M. Identification of novel mutation in RANKL by whole-exome sequencing in a Thai family with osteopetrosis; a case report and review of RANKL osteopetrosis. Mol. Genet. Genom. Med. 2021, 9, e1727. [Google Scholar] [CrossRef]
- Borthwick, K.J.; Kandemir, N.; Topaloglu, R.; Kornak, U.; Bakkaloglu, A.; Yordam, N.; Ozen, S.; Mocan, H.; Shah, G.N.; Sly, W.S.; et al. A phenocopy of CAII deficiency: A novel genetic explanation for inherited infantile osteopetrosis with distal renal tubular acidosis. J. Med. Genet. 2003, 40, 115–121. [Google Scholar] [CrossRef]
- Hamroun, A.; Maanaoui, M.; Lenain, R.; Lionet, A. Marble brain disease: A rare cause of renal tubular acidosis. J. Nephrol. 2021, 34, 1261–1262. [Google Scholar] [CrossRef]
- Vainsel, M.; Fondu, P.; Cadranel, S.; Rocmans, C.; Gepts, W. Osteopetrosis associated with proximal and distal tubular acidosis. Acta Paediatr. Scand. 1972, 61, 429–434. [Google Scholar] [CrossRef]
- Strisciuglio, P.; Sartorio, R.; Pecoraro, C.; Lotito, F.; Sly, W.S. Variable clinical presentation of carbonic anhydrase deficiency: Evidence for heterogeneity? Eur. J. Pediatr. 1990, 149, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, A.; Stark, G.; Sakati, N. Marble brain disease: Recessive osteopetrosis, renal tubular acidosis and cerebral calcification in three Saudi Arabian families. Dev. Med. Child Neurol. 1980, 22, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, S.; Yoshida, I.; Yoshino, M.; Kondo, M.; Sato, Y.; Noda, K.; Jo, R.; Okue, A.; Sai, N.; Yamashita, F. Carbonic anhydrase II deficiency in three unrelated Japanese patients. J. Inherit. Metab. Dis. 1993, 16, 982–990. [Google Scholar] [CrossRef]
- Al Rajeh, S.; el Mouzan, M.I.; Ahlberg, A.; Ozaksoy, D. The syndrome of osteopetrosis, renal acidosis and cerebral calcification in two sisters. Neuropediatrics 1988, 19, 162–165. [Google Scholar] [CrossRef]
- Alotaibi, Q.; Dighe, M. Managing challenging pain and irritability in OSTM1 mutation-related infantile malignant osteopetrosis. BMJ Case Rep. 2021, 14, e242498. [Google Scholar] [CrossRef]
- Chen, T.; Sun, J.; Liu, G.; Yin, C.; Liu, H.; Qu, L.; Fang, S.; Shifra, A.; Gilad, G. A Homozygous Mutation in 5’ Untranslated Region of TNFRSF11A Leading to Molecular Diagnosis of Osteopetrosis Coinheritance With Wiskott-Aldrich Syndrome. J. Pediatr. Hematol. Oncol. 2021, 43, e264–e267. [Google Scholar] [CrossRef]
- Pangrazio, A.; Cassani, B.; Guerrini, M.M.; Crockett, J.C.; Marrella, V.; Zammataro, L.; Strina, D.; Schulz, A.; Schlack, C.; Kornak, U.; et al. RANK-dependent autosomal recessive osteopetrosis: Characterization of five new cases with novel mutations. J. Bone Miner. Res. Off. J. Am. Soc. Bone Miner. Res. 2012, 27, 342–351. [Google Scholar] [CrossRef] [Green Version]
- Aker, M.; Rouvinski, A.; Hashavia, S.; Ta-Shma, A.; Shaag, A.; Zenvirt, S.; Israel, S.; Weintraub, M.; Taraboulos, A.; Bar-Shavit, Z.; et al. An SNX10 mutation causes malignant osteopetrosis of infancy. J. Med. Genet. 2012, 49, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, Y.; Xu, Y.; Zhang, Y.; Duan, X. Molecular Mechanisms of Craniofacial and Dental Abnormalities in Osteopetrosis. Int. J. Mol. Sci. 2023, 24, 10412. https://doi.org/10.3390/ijms241210412
Ma Y, Xu Y, Zhang Y, Duan X. Molecular Mechanisms of Craniofacial and Dental Abnormalities in Osteopetrosis. International Journal of Molecular Sciences. 2023; 24(12):10412. https://doi.org/10.3390/ijms241210412
Chicago/Turabian StyleMa, Yu, Yali Xu, Yanli Zhang, and Xiaohong Duan. 2023. "Molecular Mechanisms of Craniofacial and Dental Abnormalities in Osteopetrosis" International Journal of Molecular Sciences 24, no. 12: 10412. https://doi.org/10.3390/ijms241210412