
Cranial Neural Crest Specific Deletion of Alpl (TNAP) via P0-Cre Causes Abnormal Chondrocyte Maturation and Deficient Cranial Base Growth


Abstract
:1. Introduction
2. Results
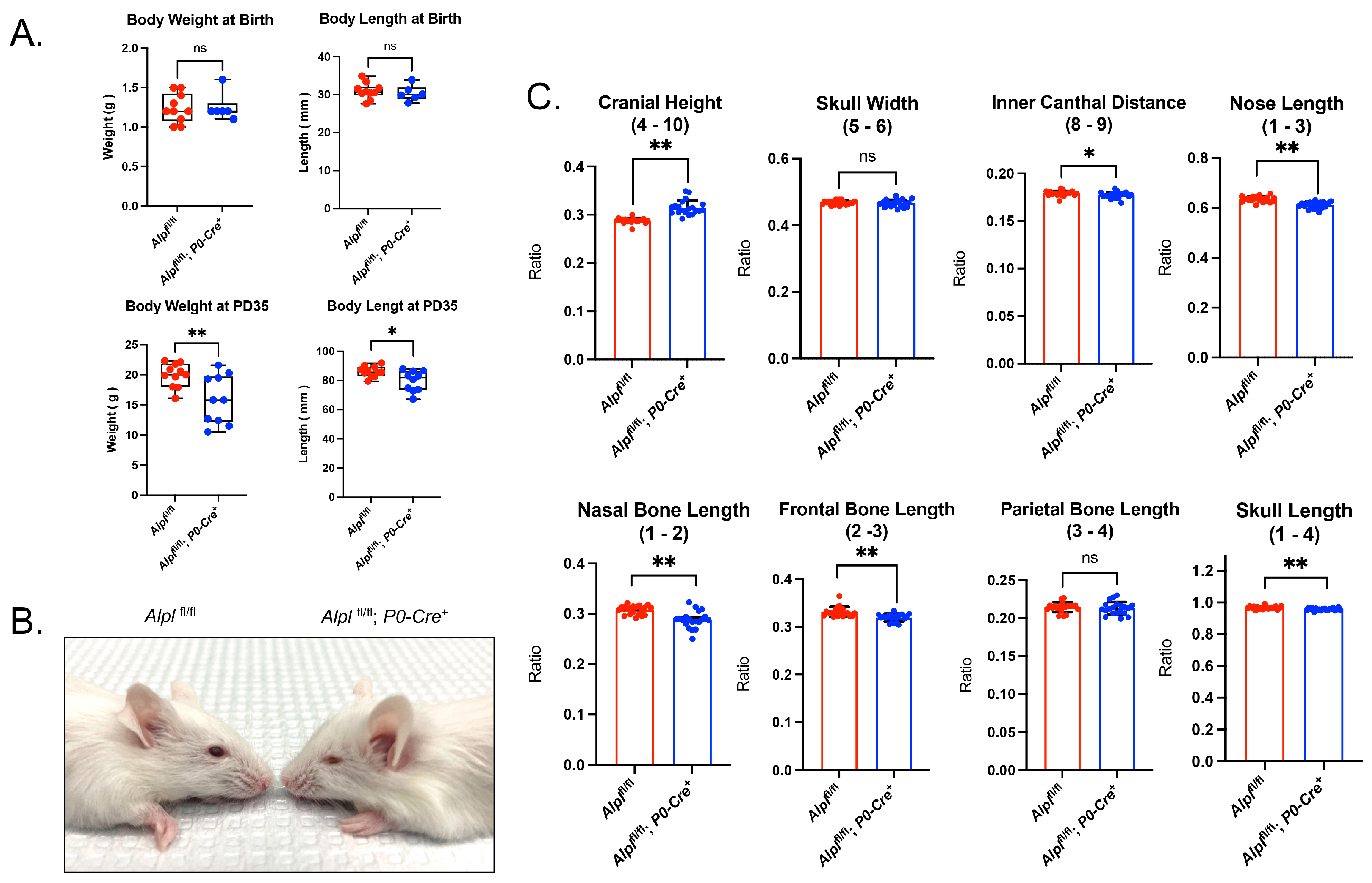
2.1. Alpl Deletion by P0-Cre Does Not Cause Craniosynostosis but Does Lead to Skull Shape Abnormalities
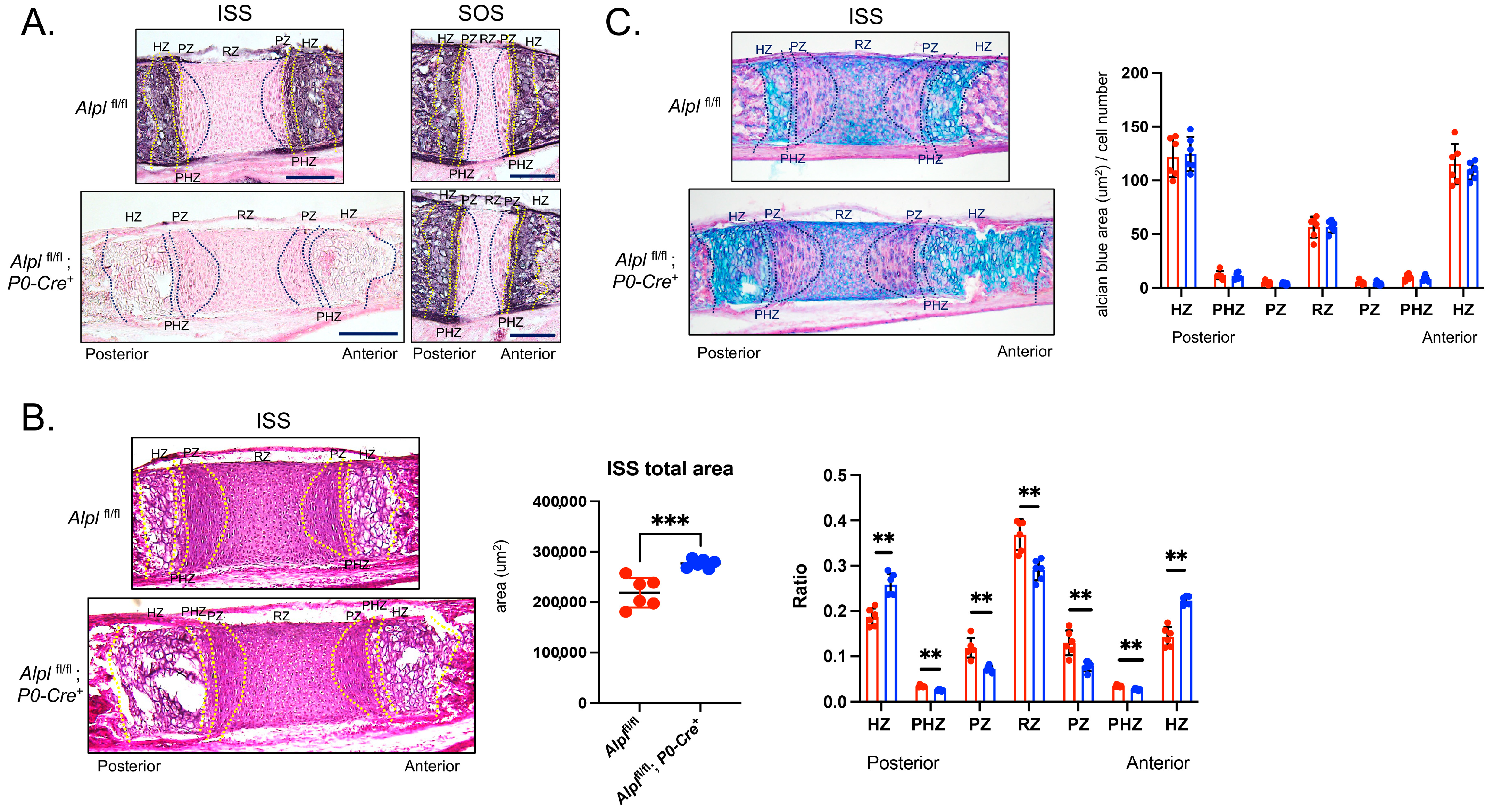
2.2. P0-Cre Alpl Deletion Causes Abnormal Development of the Anterior but Not the Posterior Cranial Base
2.3. P0-Cre Alpl Deletion Increases Chondrocyte Proliferation and Decreases Apoptosis in the Anterior Cranial Base Synchondrosis (ISS)
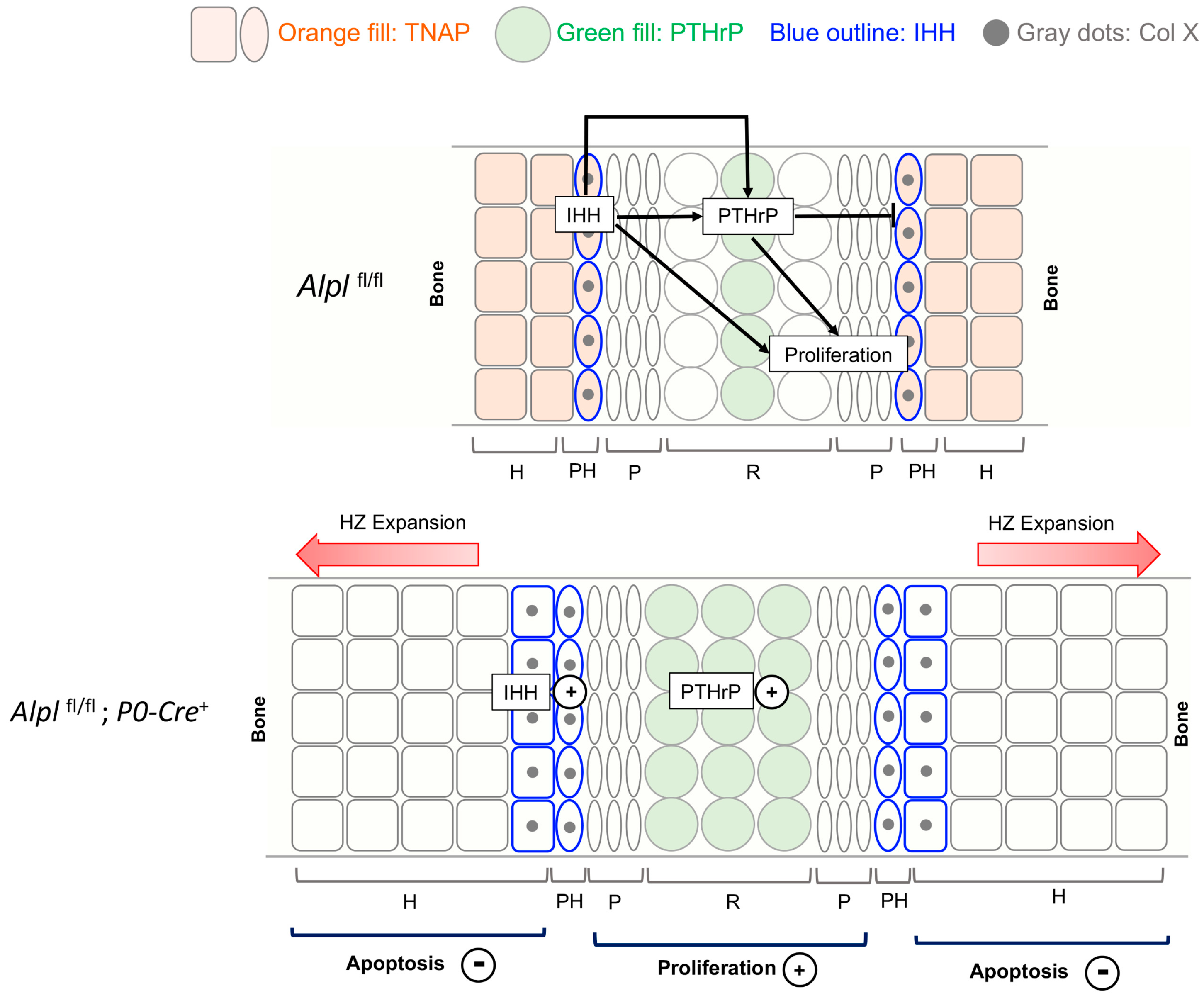
2.4. P0-Cre Alpl Deletion Increased PTH-Related Protein (PTHrP) and Indian Hedgehog (IHH) Expression in the Anterior Cranial Base Synchondrosis (ISS)
2.5. P0-Cre Alpl Deletion Promotes Premature Chondrocyte Differentiation in the Anterior Cranial Base Synchondrosis (ISS)
2.6. PPi and Pi Differentially Regulate PTHrP-IHH Signaling and Chondrocyte Maturation in Cultured Anterior Cranial Base
2.7. Alpl Deficient O9-1 Cells Influences the Stemness and Differentiation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Nano Computed Tomography
4.3. Linear Skull and Cranial Base Measurements
4.4. Histology and Histomorphometry
4.5. Immunofluoresence
4.6. O9-1 Cell Culture and Procedures
4.7. RNA Analysis
4.8. Organ Culture and Procedures
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Abad, V.; Meyers, J.L.; Weise, M.; Gafni, R.I.; Barnes, K.M.; Nilsson, O.; Bacher, J.D.; Baron, J. The role of the resting zone in growth plate chondrogenesis. Endocrinology 2002, 143, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, H.M. Developmental regulation of the growth plate. Nature 2003, 423, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Nie, X. Cranial base in craniofacial development: Developmental features, influence on facial growth, anomaly, and molecular basis. Acta Odontol. Scand. 2005, 63, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Funato, N. New Insights into Cranial Synchondrosis Development: A Mini Review. Front. Cell Dev. Biol. 2020, 8, 706. [Google Scholar] [CrossRef]
- McBratney-Owen, B.; Iseki, S.; Bamforth, S.D.; Olsen, B.R.; Morriss-Kay, G.M. Development and tissue origins of the mammalian cranial base. Dev. Biol. 2008, 322, 121–132. [Google Scholar] [CrossRef]
- Wei, X.; Hu, M.; Liu, F. Mid-facial developmental defects caused by the widely used LacZ reporter gene when expressed in neural crest-derived cells. Transgenic Res. 2018, 27, 551–558. [Google Scholar] [CrossRef]
- Hessle, L.; Johnson, K.A.; Anderson, H.C.; Narisawa, S.; Sali, A.; Goding, J.W.; Terkeltaub, R.; Millan, J.L. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc. Natl. Acad. Sci. USA 2002, 99, 9445–9449. [Google Scholar] [CrossRef]
- Murshed, M.; Harmey, D.; Millan, J.L.; McKee, M.D.; Karsenty, G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005, 19, 1093–1104. [Google Scholar] [CrossRef]
- Narisawa, S.; Frohlander, N.; Millan, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446. [Google Scholar] [CrossRef]
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millan, J.L.; MacGregor, G.R.; et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J. Bone Min. Res. 1999, 14, 2015–2026. [Google Scholar] [CrossRef]
- Whyte, M.P.; Zhang, F.; Wenkert, D.; McAlister, W.H.; Mack, K.E.; Benigno, M.C.; Coburn, S.P.; Wagy, S.; Griffin, D.M.; Ericson, K.L.; et al. Hypophosphatasia: Validation and expansion of the clinical nosology for children from 25 years experience with 173 pediatric patients. Bone 2015, 75, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Whyte, M.P. Hypophosphatasia—Aetiology, nosology, pathogenesis, diagnosis and treatment. Nat. Rev. Endocrinol. 2016, 12, 233–246. [Google Scholar] [CrossRef]
- Collmann, H.; Mornet, E.; Gattenlohner, S.; Beck, C.; Girschick, H. Neurosurgical aspects of childhood hypophosphatasia. Childs Nerv. Syst. 2009, 25, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nam, H.K.; Campbell, C.; Gasque, K.C.; Millan, J.L.; Hatch, N.E. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl(−/−) mouse model of infantile hypophosphatasia. Bone 2014, 67, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.K.; Sharma, M.; Liu, J.; Hatch, N.E. Tissue Nonspecific Alkaline Phosphatase (TNAP) Regulates Cranial Base Growth and Synchondrosis Maturation. Front. Physiol. 2017, 8, 161. [Google Scholar] [CrossRef]
- Chen, G.; Ishan, M.; Yang, J.; Kishigami, S.; Fukuda, T.; Scott, G.; Ray, M.K.; Sun, C.; Chen, S.Y.; Komatsu, Y.; et al. Specific and spatial labeling of P0-Cre versus Wnt1-Cre in cranial neural crest in early mouse embryos. Genesis 2017, 55, e23034. [Google Scholar] [CrossRef]
- St-Jacques, B.; Hammerschmidt, M.; McMahon, A.P. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999, 13, 2072–2086. [Google Scholar] [CrossRef]
- Wei, X.; Hu, M.; Mishina, Y.; Liu, F. Developmental Regulation of the Growth Plate and Cranial Synchondrosis. J. Dent. Res. 2016, 95, 1221–1229. [Google Scholar] [CrossRef]
- Beck, G.R., Jr.; Moran, E.; Knecht, N. Inorganic phosphate regulates multiple genes during osteoblast differentiation, including Nrf2. Exp. Cell Res. 2003, 288, 288–300. [Google Scholar] [CrossRef]
- Camalier, C.E.; Yi, M.; Yu, L.R.; Hood, B.L.; Conrads, K.A.; Lee, Y.J.; Lin, Y.; Garneys, L.M.; Bouloux, G.F.; Young, M.R.; et al. An integrated understanding of the physiological response to elevated extracellular phosphate. J. Cell Physiol. 2013, 228, 1536–1550. [Google Scholar] [CrossRef]
- Nam, H.K.; Liu, J.; Li, Y.; Kragor, A.; Hatch, N.E. Ectonucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1) protein regulates osteoblast differentiation. J. Biol. Chem. 2011, 286, 39059–39071. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Delaney, J.D.; Kirsch, T. The role of pyrophosphate/phosphate homeostasis in terminal differentiation and apoptosis of growth plate chondrocytes. Bone 2010, 47, 657–665. [Google Scholar] [CrossRef] [PubMed]
- McElwee, K.J.; Kissling, S.; Wenzel, E.; Huth, A.; Hoffmann, R. Cultured peribulbar dermal sheath cells can induce hair follicle development and contribute to the dermal sheath and dermal papilla. J. Investig. Dermatol. 2003, 121, 1267–1275. [Google Scholar] [CrossRef]
- Kermer, V.; Ritter, M.; Albuquerque, B.; Leib, C.; Stanke, M.; Zimmermann, H. Knockdown of tissue nonspecific alkaline phosphatase impairs neural stem cell proliferation and differentiation. Neurosci. Lett. 2010, 485, 208–211. [Google Scholar] [CrossRef]
- Yasuda, S.Y.; Tsuneyoshi, N.; Sumi, T.; Hasegawa, K.; Tada, T.; Nakatsuji, N.; Suemori, H. NANOG maintains self-renewal of primate ES cells in the absence of a feeder layer. Genes Cells 2006, 11, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, M.D.; Kardel, M.D.; Iosfina, I.; Youssef, D.; Lu, M.; Li, M.M.; Vercauteren, S.; Nagy, A.; Eaves, C.J. Alkaline phosphatase-positive colony formation is a sensitive, specific, and quantitative indicator of undifferentiated human embryonic stem cells. Stem Cells 2008, 26, 1109–1116. [Google Scholar] [CrossRef]
- Brambrink, T.; Foreman, R.; Welstead, G.G.; Lengner, C.J.; Wernig, M.; Suh, H.; Jaenisch, R. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell Stem Cell 2008, 2, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.H.; Ishii, M.; Maxson, R.E.; Wang, J. Culturing and Manipulation of O9-1 Neural Crest Cells. J. Vis. Exp. 2018, 140, e58346. [Google Scholar] [CrossRef]
- Whyte, M.P.; Greenberg, C.R.; Salman, N.J.; Bober, M.B.; McAlister, W.H.; Wenkert, D.; Van Sickle, B.J.; Simmons, J.H.; Edgar, T.S.; Bauer, M.L.; et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N. Engl. J. Med. 2012, 366, 904–913. [Google Scholar] [CrossRef]
- Fraser, D. Hypophosphatasia. Am. J. Med. 1957, 22, 730–746. [Google Scholar] [CrossRef]
- Mishina, Y.; Snider, T.N. Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res. 2014, 325, 138–147. [Google Scholar] [CrossRef]
- Jiang, X.; Iseki, S.; Maxson, R.E.; Sucov, H.M.; Morriss-Kay, G.M. Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 2002, 241, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Vivatbutsiri, P.; Morriss-Kay, G.; Saga, Y.; Iseki, S. Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 2008, 125, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Deckelbaum, R.A.; Holmes, G.; Zhao, Z.; Tong, C.; Basilico, C.; Loomis, C.A. Regulation of cranial morphogenesis and cell fate at the neural crest-mesoderm boundary by engrailed 1. Development 2012, 139, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.R.; Camci, E.D.; Cox, T.C. Postnatal Ontogeny of the Cranial Base and Craniofacial Skeleton in Male C57BL/6J Mice: A Reference Standard for Quantitative Analysis. Front. Physiol. 2015, 6, 417. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.K.; Vesela, I.; Siismets, E.; Hatch, N.E. Tissue nonspecific alkaline phosphatase promotes calvarial progenitor cell cycle progression and cytokinesis via Erk1,2. Bone 2018, 120, 125–136. [Google Scholar] [CrossRef]
- Nam, H.K.; Emmanouil, E.; Hatch, N.E. Deletion of the Pyrophosphate Generating Enzyme ENPP1 Rescues Craniofacial Abnormalities in the TNAP(−/−) Mouse Model of Hypophosphatasia and Reveals FGF23 as a Marker of Phenotype Severity. Front. Dent. Med. 2022, 3, 846962. [Google Scholar] [CrossRef]
- Ishii, M.; Merrill, A.E.; Chan, Y.S.; Gitelman, I.; Rice, D.P.; Sucov, H.M.; Maxson, R.E., Jr. Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development 2003, 130, 6131–6142. [Google Scholar] [CrossRef]
- Sun, J.; Ishii, M.; Ting, M.C.; Maxson, R. Foxc1 controls the growth of the murine frontal bone rudiment by direct regulation of a Bmp response threshold of Msx2. Development 2013, 140, 1034–1044. [Google Scholar] [CrossRef]
- Kaufman, M.H. The Atlas of Mouse Development; The Elsevier Academic Press: Amsterdam, The Netherlands; Boston, MA, USA; London, UK; New York, NY, USA, 2003. [Google Scholar]
- Yamauchi, Y.; Abe, K.; Mantani, A.; Hitoshi, Y.; Suzuki, M.; Osuzu, F.; Kuratani, S.; Yamamura, K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol. 1999, 212, 191–203. [Google Scholar] [CrossRef]
- Kulkarni, A.K.; Louie, K.W.; Yatabe, M.; Ruellas, A.C.O.; Mochida, Y.; Cevidanes, L.H.S.; Mishina, Y.; Zhang, H. A Ciliary Protein EVC2/LIMBIN Plays a Critical Role in the Skull Base for Mid-Facial Development. Front. Physiol. 2018, 9, 1484. [Google Scholar] [CrossRef]
- Ueharu, H.; Yang, J.; Komatsu, Y.; Mishina, Y. Isolation and Culture of Cranial Neural Crest Cells from the First Branchial Arch of Mice. Bio Protoc 2022, 12, e4371. [Google Scholar] [CrossRef] [PubMed]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef]
- Sabbagh, Y.; Carpenter, T.O.; Demay, M.B. Hypophosphatemia leads to rickets by impairing caspase-mediated apoptosis of hypertrophic chondrocytes. Proc. Natl. Acad. Sci. USA 2005, 102, 9637–9642. [Google Scholar] [CrossRef] [PubMed]
- Leung, V.Y.; Gao, B.; Leung, K.K.; Melhado, I.G.; Wynn, S.L.; Au, T.Y.; Dung, N.W.; Lau, J.Y.; Mak, A.C.; Chan, D.; et al. SOX9 governs differentiation stage-specific gene expression in growth plate chondrocytes via direct concomitant transactivation and repression. PLoS Genet. 2011, 7, e1002356. [Google Scholar] [CrossRef] [PubMed]
- Dy, P.; Wang, W.; Bhattaram, P.; Wang, Q.; Wang, L.; Ballock, R.T.; Lefebvre, V. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev. Cell 2012, 22, 597–609. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Chaboissier, M.C.; Martin, J.F.; Schedl, A.; de Crombrugghe, B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes Dev. 2002, 16, 2813–2828. [Google Scholar] [CrossRef]
- Vortkamp, A.; Lee, K.; Lanske, B.; Segre, G.V.; Kronenberg, H.M.; Tabin, C.J. Regulation of rate of cartilage differentiation by Indian hedgehog and PTH-related protein. Science 1996, 273, 613–622. [Google Scholar] [CrossRef]
- Lanske, B.; Karaplis, A.C.; Lee, K.; Luz, A.; Vortkamp, A.; Pirro, A.; Karperien, M.; Defize, L.H.; Ho, C.; Mulligan, R.C.; et al. PTH/PTHrP receptor in early development and Indian hedgehog-regulated bone growth. Science 1996, 273, 663–666. [Google Scholar] [CrossRef]
- Nie, X.; Luukko, K.; Kvinnsland, I.H.; Kettunen, P. Developmentally regulated expression of Shh and Ihh in the developing mouse cranial base: Comparison with Sox9 expression. Anat Rec. Part A Discov. Mol. Cell Evol. Biol. 2005, 286, 891–898. [Google Scholar] [CrossRef]
- Hallett, S.A.; Ono, W.; Franceschi, R.T.; Ono, N. Cranial Base Synchondrosis: Chondrocytes at the Hub. Int. J. Mol. Sci. 2022, 23, 7817. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Arias, A.C.; Liu, L.; Chen, Y.B.; Bronner, M.E.; Maxson, R.E. A stable cranial neural crest cell line from mouse. Stem Cells Dev. 2012, 21, 3069–3080. [Google Scholar] [CrossRef] [PubMed]
- Schock, E.N.; LaBonne, C. Sorting Sox: Diverse Roles for Sox Transcription Factors during Neural Crest and Craniofacial Development. Front. Physiol. 2020, 11, 606889. [Google Scholar] [CrossRef]
- Mori-Akiyama, Y.; Akiyama, H.; Rowitch, D.H.; de Crombrugghe, B. Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc. Natl. Acad. Sci. USA 2003, 100, 9360–9365. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tan, X.N.; Hu, S.; Liu, R.Q.; Peng, L.H.; Li, Y.M.; Wu, P. Molecular Mechanisms of Chondrocyte Proliferation and Differentiation. Front. Cell Dev. Biol. 2021, 9, 664168. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nam, H.K.; Wang, E.; Hatch, N.E. Further analysis of the Crouzon mouse: Effects of the FGFR2(C342Y) mutation are cranial bone-dependent. Calcif Tissue Int. 2013, 92, 451–466. [Google Scholar] [CrossRef]
- de Carlos, F.; Alvares-Suárez, A.; Costilla, A.; Noval, I.; Vega, J.A. 3D-μCT Cephalometric Measurements in Mice; InTech: London, UK, 2011. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Company | Cat. # | Type | Anti-Retrieval | Dilution |
|---|---|---|---|---|---|
| Ki67 | Abcam, Cambridge, UK | ab16667 | Rabbit Monoclonal | 0.5% Trypsin 37 °C 20 min | 200 |
| PTHrP | Invitrogen (Thermo Fisher Scientific, Waltham, MA, USA) | PA5-102455 | Rabbit Polyclonal | No | 500 |
| IHH | Abcam | ab39634 | Rabbit Polyclonal | No | 100 |
| Col 2 | Abcam | ab34712 | Rabbit Polyclonal | pH6.0 5.5 mM Citric Acid 90 °C 20 min, RT 20 min | 100 |
| Col X | Cosmo Bio USA, Carlsbad, CA, USA | LSL-LB-0092 | Rabbit Polyclonal | No | 100 |
| Sox9 | Abcam | ab185966 | Rabbit Monoclonal | No | 500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohkura, N.; Nam, H.K.; Liu, F.; Hatch, N. Cranial Neural Crest Specific Deletion of Alpl (TNAP) via P0-Cre Causes Abnormal Chondrocyte Maturation and Deficient Cranial Base Growth. Int. J. Mol. Sci. 2023, 24, 15401. https://doi.org/10.3390/ijms242015401
Ohkura N, Nam HK, Liu F, Hatch N. Cranial Neural Crest Specific Deletion of Alpl (TNAP) via P0-Cre Causes Abnormal Chondrocyte Maturation and Deficient Cranial Base Growth. International Journal of Molecular Sciences. 2023; 24(20):15401. https://doi.org/10.3390/ijms242015401
Chicago/Turabian StyleOhkura, Naoto, Hwa Kyung Nam, Fei Liu, and Nan Hatch. 2023. "Cranial Neural Crest Specific Deletion of Alpl (TNAP) via P0-Cre Causes Abnormal Chondrocyte Maturation and Deficient Cranial Base Growth" International Journal of Molecular Sciences 24, no. 20: 15401. https://doi.org/10.3390/ijms242015401