
Brief Oxygen Exposure after Traumatic Brain Injury Hastens Recovery and Promotes Adaptive Chronic Endoplasmic Reticulum Stress Responses

Abstract
:1. Introduction
2. Results
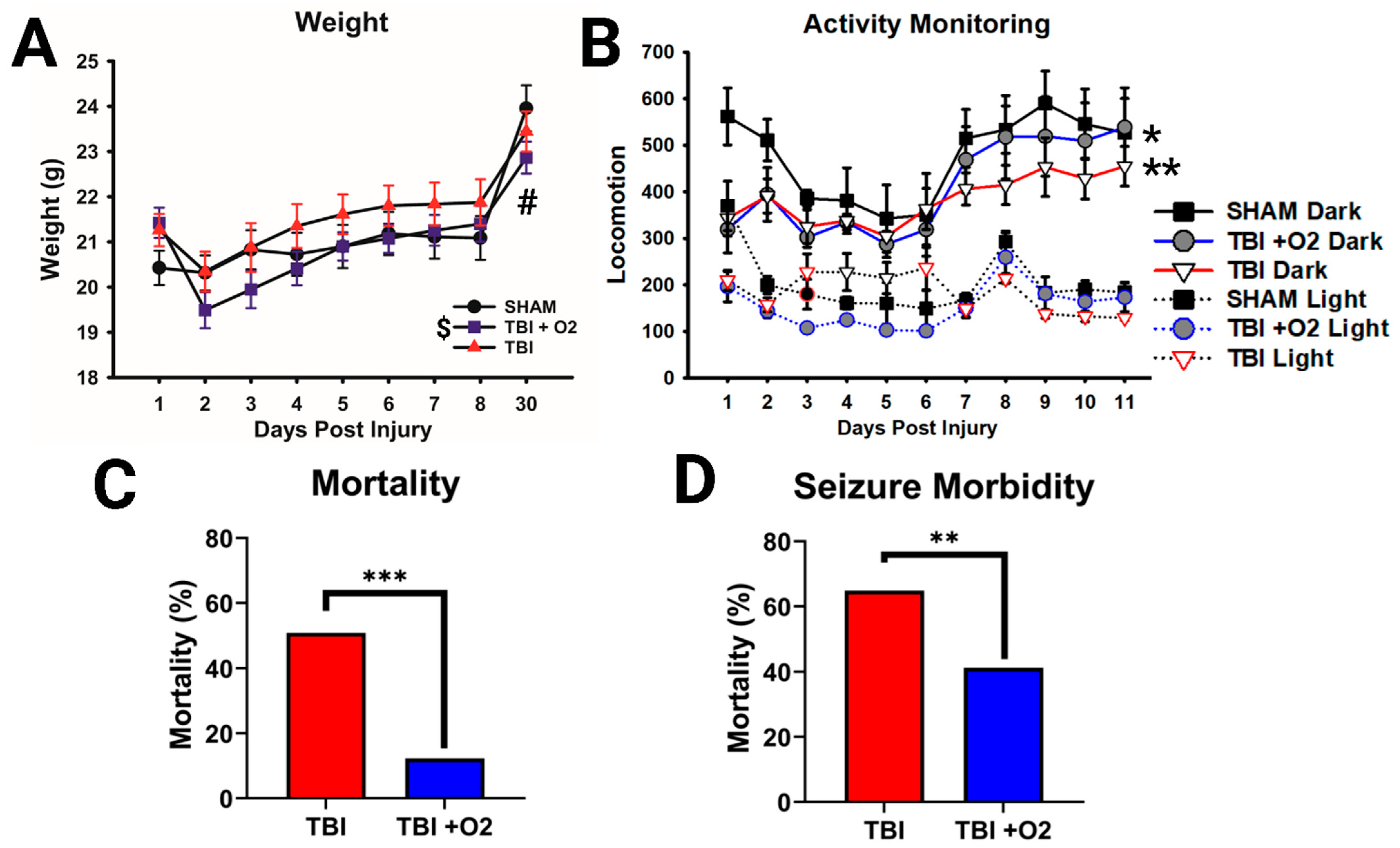
2.1. Weight, Morbidity, and Mortality following TBI in Adolescent Mice
2.2. Injured Mice Have a Blunted Optokinetic Response, and Oxygen Shows Partial Rescue Acutely
2.3. Oxygen Slows Acute RGC Loss but Does Not Ultimately Prevent Loss of RGCs
2.4. Oxygen Exposure Does Not Prevent Degeneration but May Accelerate Recovery
2.5. Astroglial Reactivity Is Significantly Reduced in the Brain following O2 Exposure
2.6. Oxygen Exposure Promotes IRE-1α Pathway Activation Acutely
2.7. The PERK Pathway Is Sub-Acutely Elevated after TBI, but Supplemental Oxygen Reduces Long-Term Expression of Pro-Apoptotic Markers
3. Discussion
3.1. Supplemental Oxygen Reduces Mortality
3.2. Optokinetic Response Outcomes
3.3. Histology
3.4. ER Stress
4. Materials and Methods
4.1. Animals
4.2. Traumatic Brain Injury/Traumatic Optic Neuropathy
4.3. Behavior
4.3.1. Home Cage Activity Monitoring
4.3.2. Optokinetic Response (OKR) and Visual Acuity
4.4. Histology
4.4.1. Tissue Collection
4.4.2. FluoroJade-C
4.4.3. Immunofluorescence
4.5. Image Analysis
4.6. Western Blots
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, J.H. The effect of associated injuries, blood loss, and oxygen debt on death and disability in blunt traumatic brain injury: The need for early physiologic predictors of severity. J. Neurotrauma 1995, 12, 579–590. [Google Scholar] [CrossRef] [PubMed]
- Jeremitsky, E.; Omert, L.; Dunham, C.M.; Protetch, J.; Rodriguez, A. Harbingers of poor outcome the day after severe brain injury: Hypothermia, hypoxia, and hypoperfusion. J. Trauma 2003, 54, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.J.; Youn, T.S.; Benson, D.; Mattick, H.; Andrade, N.; Harper, C.R.; Moore, C.B.; Madden, C.J.; Diaz-Arrastia, R.R. Physiologic and functional outcome correlates of brain tissue hypoxia in traumatic brain injury. Crit. Care Med. 2009, 37, 283–290. [Google Scholar] [CrossRef]
- Oddo, M.; Levine, J.M.; Mackenzie, L.; Frangos, S.; Feihl, F.; Kasner, S.E.; Katsnelson, M.; Pukenas, B.; Macmurtrie, E.; Maloney-Wilensky, E.; et al. Brain hypoxia is associated with short-term outcome after severe traumatic brain injury independently of intracranial hypertension and low cerebral perfusion pressure. Neurosurgery 2011, 69, 1037–1045; discussion 1045. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Carney, N.; Adelson, P.D.; Ashwal, S.; Bell, M.J.; Bratton, S.; Carson, S.; Chesnut, R.M.; Ghajar, J.; Goldstein, B.; et al. Guidelines for the acute medical management of severe traumatic brain injury in infants, children, and adolescents--second edition. Pediatr. Crit. Care Med. 2012, 13 (Suppl. 1), S1-82. [Google Scholar] [CrossRef] [PubMed]
- Yan, E.B.; Hellewell, S.C.; Bellander, B.-M.; Agyapomaa, D.A.; Morganti-Kossmann, M.C. Post-traumatic hypoxia exacerbates neurological deficit, neuroinflammation and cerebral metabolism in rats with diffuse traumatic brain injury. J. Neuroinflamm. 2011, 8, 147. [Google Scholar] [CrossRef] [Green Version]
- Hellewell, S.C.; Yan, E.B.; Agyapomaa, D.A.; Bye, N.; Morganti-Kossmann, M.C. Post-Traumatic Hypoxia Exacerbates Brain Tissue Damage: Analysis of Axonal Injury and Glial Responses. J. Neurotrauma 2010, 27, 1997–2010. [Google Scholar] [CrossRef]
- Hellewell, S.C.; Yan, E.B.; Alwis, D.S.; Bye, N.; Morganti-Kossmann, M.C. Erythropoietin improves motor and cognitive deficit, axonal pathology, and neuroinflammation in a combined model of diffuse traumatic brain injury and hypoxia, in association with upregulation of the erythropoietin receptor. J. Neuroinflamm. 2013, 10, 926. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Oda, Y.; Wei, E.P.; Povlishock, J.T. The Adverse Pial Arteriolar and Axonal Consequences of Traumatic Brain Injury Complicated by Hypoxia and Their Therapeutic Modulation with Hypothermia in Rat. J. Cereb. Blood Flow. Metab. 2009, 30, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Tanno, H.; Nockels, R.P.; Pitts, L.H.; Noble, L.J. Breakdown of the blood-brain barrier after fluid percussion brain injury in the rat: Part 2: Effect of hypoxia on permeability to plasma proteins. J. Neurotrauma 1992, 9, 335–347. [Google Scholar] [CrossRef]
- Hallam, T.M.; Floyd, C.L.; Folkerts, M.M.; Lee, L.L.; Gong, Q.Z.; Lyeth, B.G.; Muizelaar, J.P.; Berman, R.F. Comparison of Behavioral Deficits and Acute Neuronal Degeneration in Rat Lateral Fluid Percussion and Weight-Drop Brain Injury Models. J. Neurotrauma 2004, 21, 521–539. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, C.; Thelin, E.P. Secondary Insults in Experimental Traumatic Brain Injury: The Addition of Hypoxia. In Animal Models of Neurotrauma; Risling, M., Davidsson, J., Eds.; Humana: New York, NY, USA, 2019. [Google Scholar]
- Cansler, S.M.; Guilhaume-Correa, F.; Day, D.; Bedolla, A.; Evanson, N.K. Indirect traumatic optic neuropathy after head trauma in adolescent male mice is associated with behavioral visual impairment, neurodegeneration, and elevated endoplasmic reticulum stress markers at acute and subacute times. bioRxiv 2020. [Google Scholar] [CrossRef]
- Carlo, W.A.; Finer, N.N.; Walsh, M.C.; Rich, W.; Gantz, M.G.; Laptook, A.R.; Yoder, B.A.; Faix, R.G.; Das, A.; Poole, W.K.; et al. Target ranges of oxygen saturation in extremely preterm infants. N. Engl. J. Med. 2010, 362, 1959–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaus, M.; Meyer, B.P. Oxygen Therapy for the Newborn. Pediatr. Clin. N. Am. 1966, 13, 731–752. [Google Scholar] [CrossRef]
- Hellström, A.; Smith, L.E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457. [Google Scholar] [CrossRef] [Green Version]
- Brenner, M.; Stein, D.; Hu, P.; Kufera, J.; Wooford, M.; Scalea, T. Association Between Early Hyperoxia and Worse Outcomes After Traumatic Brain Injury. Arch. Surg. 2012, 147, 1042–1046. [Google Scholar] [CrossRef] [Green Version]
- Hetzer, S.M.; Guilhaume-Correa, F.; Day, D.; Bedolla, A.; Evanson, N.K. Traumatic Optic Neuropathy Is Associated with Visual Impairment, Neurodegeneration, and Endoplasmic Reticulum Stress in Adolescent Mice. Cells 2021, 10, 996. [Google Scholar] [CrossRef]
- Bolton Hall, A.N.; Joseph, B.; Brelsfoard, J.M.; Saatman, K.E. Repeated Closed Head Injury in Mice Results in Sustained Motor and Memory Deficits and Chronic Cellular Changes. PLoS ONE 2016, 11, e0159442. [Google Scholar] [CrossRef] [Green Version]
- Bashir, A.; Abebe, Z.A.; McInnes, K.A.; Button, E.B.; Tatarnikov, I.; Cheng, W.H.; Haber, M.; Wilkinson, A.; Barron, C.; Diaz-Arrastia, R.; et al. Increased severity of the CHIMERA model induces acute vascular injury, sub-acute deficits in memory recall, and chronic white matter gliosis. Exp. Neurol. 2020, 324, 113116. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Wu, Y.; Zhang, L.; Xu, J.; Li, T.; Ding, Y.; Zhu, L.; He, J. Rapamycin protects against apoptotic neuronal death and improves neurologic function after traumatic brain injury in mice via modulation of the mTOR-p53-Bax axis. J. Surg. Res. 2015, 194, 239–247. [Google Scholar] [CrossRef]
- Ding, K.; Wang, H.; Xu, J.; Lu, X.; Zhang, L.; Zhu, L. Melatonin reduced microglial activation and alleviated neuroinflammation induced neuron degeneration in experimental traumatic brain injury: Possible involvement of mTOR pathway. Neurochem. Int. 2014, 76, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Dorsett, C.R.; McGuire, J.L.; Niedzielko, T.L.; DePasquale, E.A.K.; Meller, J.; Floyd, C.L.; McCullumsmith, R.E. Traumatic Brain Injury Induces Alterations in Cortical Glutamate Uptake without a Reduction in Glutamate Transporter-1 Protein Expression. J. Neurotrauma 2017, 34, 220–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.; Rau, T.; Poulsen, A.; MacWilliams, Z.; Patterson, D.; Kelly, W.; Poulsen, D. Convulsive seizures and EEG spikes after lateral fluid-percussion injury in the rat. Epilepsy Res. 2018, 147, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Kayton, A.; Timoney, P.; Vargo, L.; Perez, J.A. A Review of Oxygen Physiology and Appropriate Management of Oxygen Levels in Premature Neonates. Adv. Neonatal Care 2018, 18, 98–104. [Google Scholar] [CrossRef]
- Nagatomo, F.; Fujino, H.; Kondo, H.; Ishihara, A. Oxygen Concentration-Dependent Oxidative Stress Levels in Rats. Oxidative Med. Cell. Longev. 2012, 2012, 381763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, S.; Beloncle, F.; Koch, A.; Radermacher, P.; Asfar, P. Hyperoxia in intensive care, emergency, and peri-operative medicine: Dr. Jekyll or Mr. Hyde? A 2015 update. Ann. Intensive Care 2015, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmucker, C.; Seeliger, M.; Humphries, P.; Biel, M.; Schaeffel, F. Grating Acuity at Different Luminances in Wild-Type Mice and in Mice Lacking Rod or Cone Function. Investig. Ophthalmol. Vis. Sci. 2005, 46, 398–407. [Google Scholar] [CrossRef]
- Wang, J.; Saul, A.; Smith, S.B. Activation of Sigma 1 Receptor Extends Survival of Cones and Improves Visual Acuity in a Murine Model of Retinitis Pigmentosa. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4397–4407. [Google Scholar] [CrossRef] [Green Version]
- Neufeld, A.H.; Gachie, E.N. The inherent, age-dependent loss of retinal ganglion cells is related to the lifespan of the species. Neurobiol. Aging 2003, 24, 167–172. [Google Scholar] [CrossRef]
- Cui, Q.; Ren, C.; Sollars, P.J.; Pickard, G.E.; So, K.F. The injury resistant ability of melanopsin-expressing intrinsically photosensitive retinal ganglion cells. Neuroscience 2015, 284, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Evanson, N.K.; Guilhaume-Correa, F.; Herman, J.P.; Goodman, M.D. Optic tract injury after closed head traumatic brain injury in mice: A model of indirect traumatic optic neuropathy. PLoS ONE 2018, 13, e0197346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetzer, S.M.; O’Connell, C.; Lallo, V.; Robson, M.; Evanson, N.K. Model matters: Differential outcomes in traumatic optic neuropathy pathophysiology between blunt and blast-wave mediated head injuries. bioRxiv 2023. [CrossRef]
- Conforti, L.; Gilley, J.; Coleman, M.P. Wallerian degeneration: An emerging axon death pathway linking injury and disease. Nat. Rev. Neurosci. 2014, 15, 394–409. [Google Scholar] [CrossRef]
- Hao, Y.; Frey, E.; Yoon, C.; Wong, H.; Nestorovski, D.; Holzman, L.B.; Giger, R.J.; DiAntonio, A.; Collins, C. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. eLife 2016, 5, e14048. [Google Scholar] [CrossRef] [PubMed]
- Llobet Rosell, A.; Neukomm, L.J. Axon death signalling in Wallerian degeneration among species and in disease. Open. Biol. 2019, 9, 190118. [Google Scholar] [CrossRef] [Green Version]
- Vargas, M.E.; Barres, B.A. Why Is Wallerian Degeneration in the CNS So Slow? Annu. Rev. Neurosci. 2007, 30, 153–179. [Google Scholar] [CrossRef]
- Almasieh, M.; Catrinescu, M.-M.; Binan, L.; Costantino, S.; Levin, L.A. Axonal Degeneration in Retinal Ganglion Cells Is Associated with a Membrane Polarity-Sensitive Redox Process. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 3824–3839. [Google Scholar] [CrossRef] [Green Version]
- Hetzer, S.M.; Shalosky, E.M.; Torrens, J.N.; Evanson, N.K. Chronic Histological Outcomes of Indirect Traumatic Optic Neuropathy in Adolescent Mice: Persistent Degeneration and Temporally Regulated Glial Responses. Cells 2021, 10, 3343. [Google Scholar] [CrossRef]
- Günther, A.; Küppers-Tiedt, L.; Schneider, P.-M.; Kunert, I.; Berrouschot, J.; Schneider, D.; Roßner, S. Differential effects on glial cell activation of hyperbaric oxygen treatment in permanent focal cerebral ischemia of rats. Eur. J. Neurosci. 2005, 21, 3189–3194. [Google Scholar] [CrossRef]
- Hui, J.; Zhang, Z.-J.; Zhang, X.; Shen, Y.; Gao, Y.-J. Repetitive Hyperbaric Oxygen Treatment Attenuates Complete Freund’s Adjuvant-Induced Pain and Reduces Glia-Mediated Neuroinflammation in the Spinal Cord. J. Pain. 2013, 14, 747–758. [Google Scholar] [CrossRef]
- Ding, Y.; Yao, P.; Hong, T.; Li, H.; Zhu, Y.; Han, Z.; Zhou, G. The analgesic effect of early hyperbaric oxygen treatment in chronic constriction injury rats and its influence on nNOS and iNOS expression and inflammatory factor production. Mol. Pain. 2018, 14, 1744806918765837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Kasymov, V.; Christie, I.; Sheikhbahaei, S.; Turovsky, E.; Marina, N.; Korsak, A.; Zwicker, J.; Teschemacher, A.G.; Ackland, G.L.; et al. Functional Oxygen Sensitivity of Astrocytes. J. Neurosci. 2015, 35, 10460–10473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigdeli, M.R.; Hajizadeh, S.; Froozandeh, M.; Heidarianpour, A.; Rasoulian, B.; Asgari, A.R.; Pourkhalili, K.; Khoshbaten, A. Normobaric hyperoxia induces ischemic tolerance and upregulation of glutamate transporters in the rat brain and serum TNF-α level. Exp. Neurol. 2008, 212, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275 Pt 3, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Rutkowski, D.T.; Kaufman, R.J. That which does not kill me makes me stronger: Adapting to chronic ER stress. Trends Biochem. Sci. 2007, 32, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Hood, K.N.; Zhao, J.; Redell, J.B.; Hylin, M.J.; Harris, B.; Perez, A.; Moore, A.N.; Dash, P.K. Endoplasmic Reticulum Stress Contributes to the Loss of Newborn Hippocampal Neurons after Traumatic Brain Injury. J. Neurosci. 2018, 38, 2372–2384. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Lan, T.; Lu, J.; Zhang, H.; Zhang, D.; Lou, T.; Xu, P.; Ren, J.; Zhao, D.; Sun, L.; et al. DiDang Tang Inhibits Endoplasmic Reticulum Stress-Mediated Apoptosis Induced by Oxygen Glucose Deprivation and Intracerebral Hemorrhage through Blockade of the GRP78-IRE1/PERK Pathways. Front. Pharmacol. 2018, 9, 1423. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Dixon, B.J.; Doycheva, D.M.; Li, B.; Zhang, Y.; Hu, Q.; He, Y.; Guo, Z.; Nowrangi, D.; Flores, J.; et al. IRE1α inhibition decreased TXNIP/NLRP3 inflammasome activation through miR-17-5p after neonatal hypoxic-ischemic brain injury in rats. J. Neuroinflamm. 2018, 15, 32. [Google Scholar] [CrossRef] [Green Version]
- Liew, H.K.; Hu, W.F.; Lin, P.B.; Wang, P.K.; Tsai, A.P.; Pang, C.Y.; Chen, T.Y. Over-Activated Proteasome Mediates Neuroinflammation on Acute Intracerebral Hemorrhage in Rats. Cells 2019, 8, 1326. [Google Scholar] [CrossRef] [Green Version]
- Meng, C.; Zhang, J.; Dang, B.; Li, H.; Shen, H.; Li, X.; Wang, Z. PERK Pathway Activation Promotes Intracerebral Hemorrhage Induced Secondary Brain Injury by Inducing Neuronal Apoptosis Both in Vivo and in Vitro. Front. Neurosci. 2018, 12, 111. [Google Scholar] [CrossRef]
- Tan, H.-P.; Guo, Q.; Hua, G.; Chen, J.-X.; Liang, J.-C. Inhibition of endoplasmic reticulum stress alleviates secondary injury after traumatic brain injury. Neural Regen. Res. 2018, 13, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Gao, C.; Chen, W.; Gao, Y.; Wang, H.C.; Meng, Y.; Luo, C.L.; Zhang, M.Y.; Chen, G.; Chen, X.P.; et al. Salubrinal offers neuroprotection through suppressing endoplasmic reticulum stress, autophagy and apoptosis in a mouse traumatic brain injury model. Neurobiol. Learn. Mem. 2019, 161, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Lassot, I.; Ségéral, E.; Berlioz-Torrent, C.; Durand, H.; Groussin, L.; Hai, T.; Benarous, R.; Margottin-Goguet, F. ATF4 degradation relies on a phosphorylation-dependent interaction with the SCF(betaTrCP) ubiquitin ligase. Mol. Cell. Biol. 2001, 21, 2192–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frank, C.L.; Ge, X.; Xie, Z.; Zhou, Y.; Tsai, L.H. Control of activating transcription factor 4 (ATF4) persistence by multisite phosphorylation impacts cell cycle progression and neurogenesis. J. Biol. Chem. 2010, 285, 33324–33337. [Google Scholar] [CrossRef] [Green Version]
- Park, S.M.; Kang, T.I.; So, J.S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef]
- van Helvoort, H.A.; Heijdra, Y.F.; Heunks, L.M.; Meijer, P.L.; Ruitenbeek, W.; Thijs, H.M.; Dekhuijzen, P.N. Supplemental oxygen prevents exercise-induced oxidative stress in muscle-wasted patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2006, 173, 1122–1129. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, C.; Crupi, R.; Calabrese, V.; Graziano, A.; Milone, P.; Pennisi, G.; Radak, Z.; Calabrese, E.J.; Cuzzocrea, S. Traumatic Brain Injury: Oxidative Stress and Neuroprotection. Antioxid. Redox Signal. 2013, 19, 836–853. [Google Scholar] [CrossRef]
- Chandran, R.; Mehta, S.L.; Vemuganti, R. Antioxidant Combo Therapy Protects White Matter after Traumatic Brain Injury. Neuromol. Med. 2021, 23, 344–347. [Google Scholar] [CrossRef]
- Frati, A.; Cerretani, D.; Fiaschi, A.I.; Frati, P.; Gatto, V.; La Russa, R.; Pesce, A.; Pinchi, E.; Santurro, A.; Fraschetti, F.; et al. Diffuse Axonal Injury and Oxidative Stress: A Comprehensive Review. Int. J. Mol. Sci. 2017, 18, 2600. [Google Scholar] [CrossRef] [Green Version]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol. Disord.-Drug. Targets-CNS Neurol. Disord. 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Chaudhari, N.; Talwar, P.; Parimisetty, A.; Lefebvre d'Hellencourt, C.; Ravanan, P. A molecular web: Endoplasmic reticulum stress, inflammation, and oxidative stress. Front. Cell. Neurosci. 2014, 8, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Appenzeller-Herzog, C. Updates on “endoplasmic reticulum redox”. Antioxid. Redox Signal. 2012, 16, 760–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benham, A.M. Endoplasmic Reticulum redox pathways: In sickness and in health. FEBS J. 2019, 286, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, M.; Nagata, K. Redox-Dependent Protein Quality Control in the Endoplasmic Reticulum: Folding to Degradation. Antioxid. Redox Signal. 2012, 16, 1119–1128. [Google Scholar] [CrossRef]
- Cansler, S.M.; Evanson, N.K. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI, and traumatic optic neuropathy. J. Neurosci. Res. 2020, 98, 571–574. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid. Redox Signal. 2014, 21, 396–413. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. Mitochondrial Dysfunction Indirectly Elevates ROS Production by the Endoplasmic Reticulum. Cell. Metab. 2013, 18, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Ozgur, R.; Turkan, I.; Uzilday, B.; Sekmen, A.H. Endoplasmic reticulum stress triggers ROS signalling, changes the redox state, and regulates the antioxidant defence of Arabidopsis thaliana. J. Exp. Bot. 2014, 65, 1377–1390. [Google Scholar] [CrossRef]
- Roy, S.; Trudeau, K.; Roy, S.; Tien, T.; Barrette, K.F. Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Diabetic Retinopathy: Mechanistic Insights into High Glucose-Induced Retinal Cell Death. Curr. Clin. Pharmacol. 2013, 8, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Siegenthaler, K.D. Redox Signaling through the Endoplasmic Reticulum Chaperone BIP; Cornell University, eCommons Cornell Univeristy Library: Ithaca, NY, USA, 2019. [Google Scholar]
- Siegenthaler, K.D.; Sevier, C.S. Working Together: Redox Signaling between the Endoplasmic Reticulum and Mitochondria. Chem. Res. Toxicol. 2019, 32, 342–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victor, P.; Sarada, D.; Ramkumar, K.M. Crosstalk between endoplasmic reticulum stress and oxidative stress: Focus on protein disulfide isomerase and endoplasmic reticulum oxidase 1. Eur. J. Pharmacol. 2021, 892, 173749. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell. Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enyedi, B.; Várnai, P.; Geiszt, M. Redox state of the endoplasmic reticulum is controlled by Ero1L-alpha and intraluminal calcium. Antioxid. Redox Signal. 2010, 13, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Zeeshan, H.M.; Lee, G.H.; Kim, H.R.; Chae, H.J. Endoplasmic Reticulum Stress and Associated ROS. Int. J. Mol. Sci. 2016, 17, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, S.; Chng, W.-J.; Zhou, J. Crosstalk between endoplasmic reticulum stress and oxidative stress: A dynamic duo in multiple myeloma. Cell. Mol. Life Sci. 2021, 78, 3883–3906. [Google Scholar] [CrossRef]
- Wang, J.; Pareja, K.A.; Kaiser, C.A.; Sevier, C.S. Redox signaling via the molecular chaperone BiP protects cells against endoplasmic reticulum-derived oxidative stress. elife 2014, 3, e03496. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Gustafson, J.; Gangidine, M.; Stepien, D.; Schuster, R.; Pritts, T.A.; Goodman, M.D.; Remick, D.G.; Lentsch, A.B. A murine model of mild traumatic brain injury exhibiting cognitive and motor deficits. J. Surg. Res. 2013, 184, 981–988. [Google Scholar] [CrossRef] [Green Version]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Concentration | Host Species | Molecular Weight (kDa) | Supplier | Cat # | RRID | Immunogen |
|---|---|---|---|---|---|---|---|
| GFAP | 1:2000 | Rbt | N/A | DAKO (Agilent) | Cat: Z033401-2 | AB_10013382 | Whole bovine GFAP, isolated from spinal cord |
| Cy3 AffiniPure Donkey anti Rabbit IgG (H+L) conjugated secondary | 1:500 | Donkey | N/A | Jackson Immuno Research | Cat: 711-165-152 | AB_2307443 | Gamma immunoglobulins, heavy and light chains |
| ATF4 + p-ATF4 | 1:1500 | Rbt Poly | 48, 70 | Fisher | Cat: 10835-1-AP | AB_2058600 | UniProt AG1279 |
| CHOP/GADD153 | 1:1000 | Rbt Poly | 23 | Novus Bio | Cat: NBP2-13172 | UniProt P35638 | |
| eIF2α | 1:2500 | Rbt Poly | 38 | CST | Cat: 5324 | AB_10692650 | UniProt P05198 |
| P-eIF2α | 1:500 | Rbt Poly | 38 | CST | Cat: 3398 | AB_2096481 | UniProt P05198 |
| ERO1L α | 1:2500 | Rbt Mono | 50 | Fisher | Cat: 702709 | AB_2716886 | Protein corresponding to human ERO1L (aa22-aa468) |
| GADD34 | 1:750 | Rbt Poly | 73 | ProteinTech | Cat: 10449-1-AP | AB_2168724 | GADD34 fusion protein Ag0578 |
| IRE1α | 1:1000 | Rbt Poly | 100 | Novus Bio | Cat: NB100-2324SS | AB_10000972 | Swiss-Prot #O75460 |
| PERK | 1:1000 | Rbt Poly | 140 | CST | Cat: 5683 | AB_10841299 | UniProt Q9NZJ5 |
| XBP1U | 1:500 | Rbt Poly | 38 | Fisher | Cat: 25977 | AB_2880326 | XBP1 Fusion Protein Ag21714 (167-261 aa encoded by BC000938) |
| XBP1s | 1:500 | Rbt Poly | 45/55 | ProteinTech | Cat: 24868-1-AP | AB_2879766 | 24868-1-AP is XBP1S-specific Fusion Protein expressed in E. coli |
| anti-Rbt HRP secondary | 1:500–1:2500 | goat | N/A | CST | Cat: 7074 | AB_2099233 |
| Measure | Comparison | Test Statistic | p Value |
|---|---|---|---|
| Results Section 2.1 Weight, Morbidity, & Mortality following TBI in adolescent mice | |||
| Weight | 2-Way ANOVA Main effect of Injury | F2,221 = 5.95 | 0.009 |
| 2-Way ANOVA Main Effect of Day | F8,221 = 62.38 | <0.001 | |
| 2-Way ANOVA Interaction | F16,221 = 23.28 | <0.001 | |
| Activity | 3-Way ANOVA Main effect of Injury | F2,65 = 5.99 | 0.005 |
| 3-Way ANOVA Main Effect of Day | F10,65 = 3.02 | 0.004 | |
| 3-Way ANOVA Main Effect of Light | F1,65 = 163.41 | <0.001 | |
| Mortality x Time in Oxygen | Chi Square | X2 = 10.47 | 0.001 |
| Agonal Breathing | Student’s t-test | t = 0.22 | 0.82 |
| Mean time TBI room air | 1.3 min | ||
| Mean time TBI + O2 | 1.4 min | ||
| Righting Time | Student’s t-test | t = 1.48 | 0.16 |
| Mean time TBI room air | 11.2 min | ||
| Mean time TBI + O2 | 11.4 min | ||
| Oxygen x Righting Time | Correlation | r = 0.15 | 0.53 |
| Mortality x Seizures | Chi Square | X2 = 12.22 | 0.007 |
| Results Section 2.3. Oxygen slows acute RGC loss but does not ultimately prevent loss of RGCs. | |||
| Retinal Cell Loss Day 7 | One-Way ANOVA Peripheral | F2,42 = 14.89 | <0.001 |
| One-Way ANOVA Mid-Peripheral | F2,42 = 4.71 | 0.01 | |
| One-Way ANOVA Central | F2,42 = 12.84 | <0.001 | |
| Retinal Cell Loss Day 30 | One-Way ANOVA Peripheral | F2,42 = 12.01 | <0.001 |
| One-Way ANOVA Mid-Peripheral | F2,42 = 14.58 | <0.001 | |
| One-Way ANOVA Central | F2,42 = 0.58 | 0.5 | |
| Results from Section 2.4. Oxygen exposure does not prevent degeneration but may accelerate recovery. | |||
| FJ-C Degeneration | 2-Way ANOVA Main effect of Injury | F2,51 = 52.66 | <0.001 |
| 2-Way ANOVA Interaction | F2,52 = 11.97 | <0.001 | |
| Results from Section 2.5. Astroglial reactivity is significantly reduced in the brain following O2 exposure | |||
| GFAP Mean Fluorescence Intensity Day 7 | One-Way ANOVA | F2,33 = 97.4 | <0.001 |
| GFAP Mean Fluorescence Intensity Day 30 | One-Way ANOVA | F2,22 = 4.27 | 0.03 |
| Results from Section 2.6. Oxygen exposure promotes IRE-1α pathway activation acutely | |||
| IRE1α 7 Day | One-Way ANOVA | F2,20 = 3.47 | 0.04 |
| XBP1-U 7 Day | One-Way ANOVA | F2,20 = 0.75 | 0.5 |
| XBP1s 7 Day | One-Way ANOVA | F2,20 = 0.79 | 0.47 |
| XBP1s:XBP1-U 7 Day | One-Way ANOVA | F2,20 = 0.5 | 0.6 |
| IRE1α 30 Day | One-Way ANOVA | F2,19 = 7.3 | 0.004 |
| XBP1-U 30 Day | One-Way ANOVA | F2,28 = 1.5 | 0.24 |
| XBP1s 30 Day | One-Way ANOVA | F2,28 = 6.39 | 0.006 |
| XBP1s:XBP1-U 30 Day | One-Way ANOVA | F2,28 = 7.68 | 0.002 |
| Results from Section 2.7. The PERK pathway is sub-acutely elevated after TBI, but supplemental oxygen reduces long-term expression of pro-apoptotic markers. | |||
| PERK 7 Day | One-Way ANOVA | F2,21 = 0.66 | 0.5 |
| p-PERK 7 Day | One-Way ANOVA | F2,21 = 0.22 | 0.8 |
| PERK:p-PERK 7 Day | One-Way ANOVA | F2,21 = 2.35 | 0.12 |
| eIF2α 7 Day | One-Way ANOVA | F2,21 = 0.52 | 0.6 |
| p-eIF2α 7 Day | One-Way ANOVA | F2,21 = 0.52 | 0.6 |
| eIF2 α:p-eIF2α 7 Day | One-Way ANOVA | F2,23 = 2.8 | 0.08 |
| ATF4 7 Day | One-Way ANOVA | F2,21 = 2.87 | 0.08 |
| p-ATF4 7 Day | One-Way ANOVA | F2,21 = 6.24 | 0.007 |
| ATF4:p-ATF4 7 Day | One-Way ANOVA | F2,22 = 6.37 | 0.007 |
| CHOP 7 Day | One-Way ANOVA | F2,15 = 7.55 | 0.007 |
| ERO1Lα 7 Day | One-Way ANOVA | F2,23 = 0.67 | 0.52 |
| PERK 30 Day | One-Way ANOVA | F2,33 = 2.71 | <0.001 |
| p-PERK 30 Day | One-Way ANOVA | F2,32 = 0.14 | 0.87 |
| PERK:p-PERK 30 Day | One-Way ANOVA | F2,28 = 1.56 | 0.23 |
| eIF2α 30 Day | One-Way ANOVA | F2,31 = 7.9 | 0.002 |
| p-eIF2α 30 Day | One-Way ANOVA | F2,30 = 11.21 | <0.001 |
| eIF2 α:p-eIF2α 30 Day | One-Way ANOVA | F2,29 = 2.36 | 0.11 |
| ATF4 30 Day | One-Way ANOVA | F2,17 = 1.46 | 0.26 |
| p-ATF4 30 Day | One-Way ANOVA | F2,17 = 3.2 | 0.06 |
| ATF4:p-ATF4 30 Day | One-Way ANOVA | F2,19 = 1.12 | 0.35 |
| CHOP 30 Day | One-Way ANOVA | F2,8 = 9.4 | 0.18 |
| ERO1Lα 30 Day | One-Way ANOVA | F2,18 = 4.19 | 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torrens, J.N.; Hetzer, S.M.; Evanson, N.K. Brief Oxygen Exposure after Traumatic Brain Injury Hastens Recovery and Promotes Adaptive Chronic Endoplasmic Reticulum Stress Responses. Int. J. Mol. Sci. 2023, 24, 9831. https://doi.org/10.3390/ijms24129831
Torrens JN, Hetzer SM, Evanson NK. Brief Oxygen Exposure after Traumatic Brain Injury Hastens Recovery and Promotes Adaptive Chronic Endoplasmic Reticulum Stress Responses. International Journal of Molecular Sciences. 2023; 24(12):9831. https://doi.org/10.3390/ijms24129831
Chicago/Turabian StyleTorrens, Jordyn N., Shelby M. Hetzer, and Nathan K. Evanson. 2023. "Brief Oxygen Exposure after Traumatic Brain Injury Hastens Recovery and Promotes Adaptive Chronic Endoplasmic Reticulum Stress Responses" International Journal of Molecular Sciences 24, no. 12: 9831. https://doi.org/10.3390/ijms24129831