2.1. CL PES
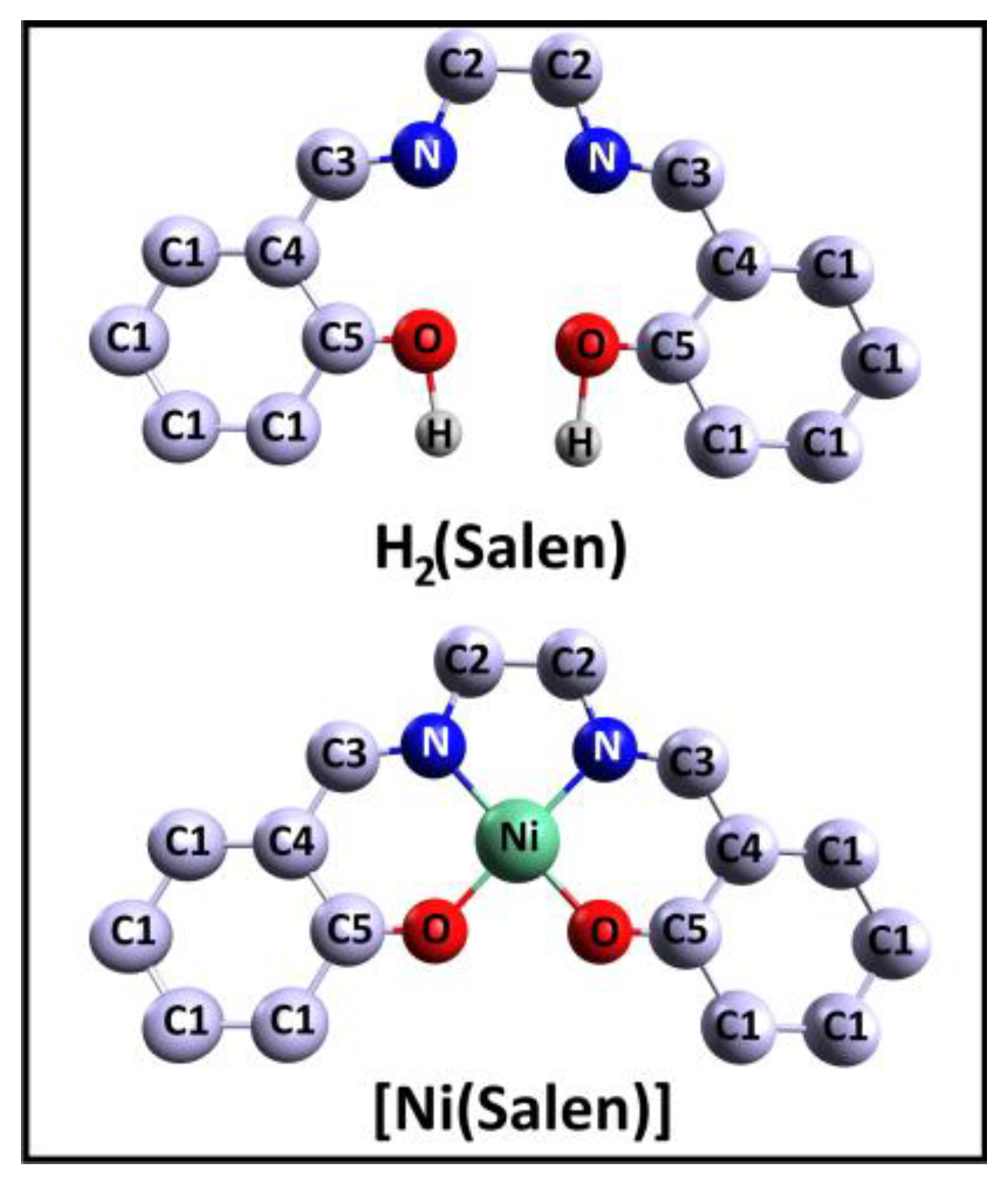
Let us begin the discussion of the results obtained by considering the conclusions of a comparative study of the corresponding CL PE spectra of the H
2(Salen) molecule and the [Ni(Salen)] complex.
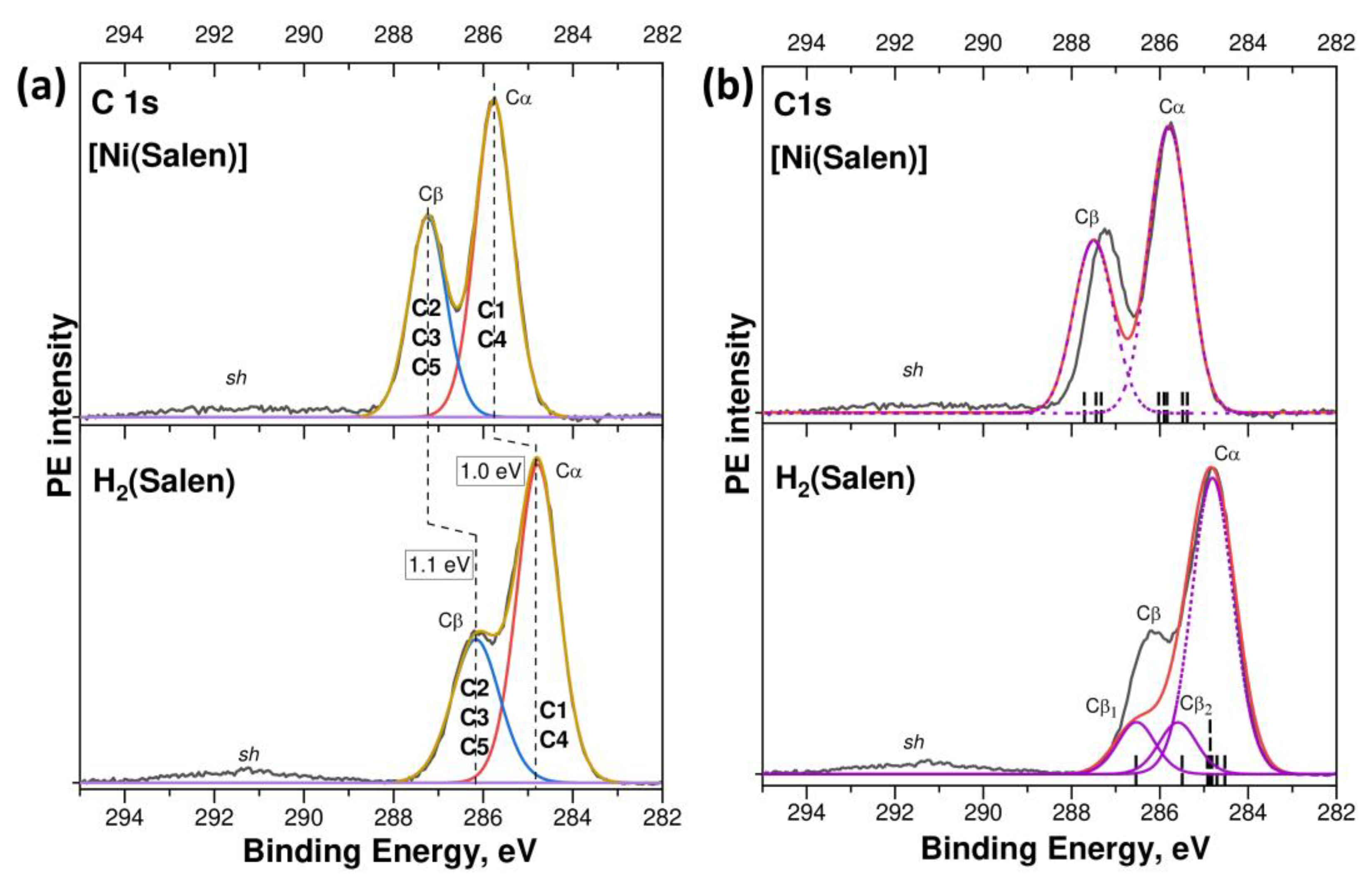
Figure 2a presents the experimental C 1s PE spectra of the H
2(Salen) and [Ni(Salen)] on the electron BE scale, relative to the Fermi level, which are compared with those calculated using the DFT method in
Figure 2b. The experimental C 1s PE spectra are similar in their spectral shape and are characterized by two resolved low- and high-energy components, Cα and Cβ, at the E
bind’s of 284.8 eV and 286.2 eV (H
2(Salen)), and 285.75 eV and 287.25 eV ([Ni(Salen)]), which are separated from each other by a close energy distance Δ, equal to 1.4 eV and 1.5 eV, respectively. In addition, the spectra show a low-intensity broad band
sh over the E
bind range of 290–292 eV. It should also be noted that these results for the complex agree in terms of the spectral shape and energy positions of the PE bands with the data that were previously obtained using synchrotron radiation [
33].
At the same time, the compared PE spectra reveal significant differences in the energy positions of individual components Cα and Cβ, their relative intensities, and their spectral resolution. Thus, when passing from the H2(Salen) spectrum to that of [Ni(Salen)], close high-energy shifts of 1.0 eV and 1.1 eV are observed for the Cα and Cβ PE components, respectively. The high-energy shift of the C 1s PE spectrum of [Ni(Salen)] as a whole, by about 1 eV relative to the H2(Salen) spectrum, unambiguously indicates a change in the chemical states of all the carbon atoms and an increase in their effective charges in the complex compared to the ligand molecule.
These energy shifts will be discussed below, simultaneously with consideration of the N 1s and O 1s PE spectra of H2(Salen) and [Ni(Salen)].
Along with this, there is a noticeable increase in the peak intensity of the Cβ band relative to the Cα one, and these bands also become better resolved. The fitting parameters for the compared spectra are presented, in detail, in
Table S1. Obviously, the spectral shape of the C 1s PE spectra of H
2(Salen) and [Ni(Salen)], consisting of two components, is due to the presence of two groups of carbon atoms in different chemical states in the atomic structure of the molecule and the complex (
Figure 1). In a previous work [
33], it was suggested that the carbon atoms linked to the oxygen and nitrogen atoms in [Ni(Salen)] are in close chemical states, which differ from those of the carbon atoms linked only to other carbon atoms. As a result, the sixteen carbon atoms of the [Ni(Salen)] complex were divided into two groups, Cα and Cβ: the first one consists of the C1 and C4 atoms of the phenyl rings of C
6H
5 (10 atoms in total) and the second one consists of the C2, C3, and C5 atoms (bonded to N or O atoms in addition to carbon atoms; 6 atoms in total). Such a division of the number of carbon atoms
n(Cα):
n(Cβ) = 1.0: 0.6 coincides practically with the peak intensity ratio of 1.0: 0.63 for the corresponding PE components in the [Ni(Salen)] spectrum (
Figure 2a,
Table S1).
In the H
2(Salen) spectrum, the peak intensity of the Cβ band, with respect to that of the Cα band, is only 0.47 (
Figure 1a), which is noticeably lower than the above-mentioned relative intensity of the Cβ band (0.63) in the [Ni(Salen)] spectrum. A comparison of the total intensities of the Cα and Cβ components, based on fitting the PE spectrum with two individual bands, only slightly increases the relative intensity of the Cβ component to a value of 0.52 (
Table S1). Based on the relative intensity of the PE band Cβ, it can be assumed that the number of carbon atoms of Cβ and Cα in the H
2(Salen) molecule are 5 and 11, respectively, since, in this case, the relative intensity of the Cβ component in the C 1s PE spectrum of H
2(Salen) is expected to be 0.45, which is close to the observed intensity. It should be noted that this is a rather unexpected result, since the carbon atoms of Cβ in H
2(Salen) (
Figure 1) interact in pairs with oxygen atoms (two C5 atoms) and nitrogen atoms (two C3 atoms and two C2 atoms) and they should have the same chemical states in these pairs.
A significant redistribution of the charge (chemical) state of the carbon atoms in the salen ligand, in going from a molecule to a complex, is also evidenced by a comparison of the FWHM of the PE bands in the compared spectra. Indeed, the C 1s spectra of H
2(Salen) and [Ni(Salen)] were measured under the same experimental conditions; nevertheless, the individual Cα and Cβ bands obtained as a result of fitting exhibit noticeably larger FWHM (1.09 and 1.25 eV,
Table S1) in the spectrum of the molecule in comparison to the full widths of 0.97 eV for both bands in the spectrum of the complex. The Cα and Cβ bands in the PE spectrum of the H
2(Salen) molecule are formed by PE signals from the 1s electron shells of the C1, C4 and C2, C3, C5 carbon atoms, respectively. Each type of carbon atom C1 … C5 is characterized by its own E
bind for the 1s electron shell and these energies differ little from each other within the groups of the Cα and Cβ atoms. However, this small spread in the binding energies of the C 1s electrons in a group leads to an additional broadening of the corresponding PE band beyond the instrumental FWHM. As can be seen in
Figure 2a and
Table S1, this broadening in the C 1s PE spectrum of the molecule is noticeably larger than in that of the complex.
It is now interesting to compare the binding energies of the C 1s electrons in the H
2(Salen) molecule with the corresponding energies for the phenol and ethylenediamine fragments of this molecule. The energy position of the low-energy Cα component (284.80 eV) in the H
2(Salen) spectrum agrees well with the E
bind of the 1s electrons of the carbon atoms in the phenol C
6H
5OH, which are not bound to the oxygen atom (284.5 eV [
37]). In turn, the high-energy Cβ component is located at the E
bind of 286.2 eV, which coincides with the C 1s binding energy in the phenol or ethylenediamine C
2H
4(NH
2)
2 molecules for the carbon atoms that are bonded to oxygen or nitrogen atoms (286.1 eV [
37] and 286.0 eV [
38], respectively). Thus, the observed correlation between the energy positions of the Cα and Cβ components in the C 1s PE spectrum of the H
2(Salen) molecule and the C 1s PE lines in the spectra of the phenol and ethylenediamine molecules confirms the correct assignment of the carbon atoms C1, C4 and C5, C2 to the two groups, Cα and Cβ, of the carbon atoms, which do not directly interact and interact with the oxygen and nitrogen atoms in H
2(Salen). In this case, the decrease in the relative intensity of the Cβ band and the broadening of the Cα and Cβ bands in the H
2(Salen) spectrum can be logically associated with the location of the C 1s PE line of the C3 atoms in the region of the binding energies between the Cβ and Cα bands.
Let us now compare the experimental C 1s PE spectra of the H
2(Salen) molecule and the [Ni(Salen)] complex with their calculated spectra (
Figure 2b). The calculated E
bind of the C 1s electrons in the different carbon atoms of the molecule and complex (
Table S2) are shown as vertical black bars in
Figure 2. Then, to facilitate this comparison with the experimental spectra, the obtained spectra (red curves) were aligned with the intensity of the Cα bands and shifted to the high-energy side by 5.64 eV (H
2(Salen)) and 6.86 eV ([Ni(Salen)]), so that the Cα band coincided in its energy position with the corresponding bands in the experimental spectra. The main individual components (Cα, Cβ, Cβ
1, and Cβ
2) of the calculated spectra are shown by purple dashed lines.
As can be seen, the calculated spectra reproduce well the two-band shape of both spectra, the energy distance Δ between the Cα and Cβ bands, and the increase in the relative intensity of the Cβ band upon passing from the spectrum of the molecule to that of the complex. It is also important to note that the calculation confirms the assumption made above about a significant change in the 1s binding energies of the carbon atoms assigned to the Cβ group upon passing from the spectrum of [Ni(Salen)] to the spectrum of H2(Salen). Indeed, the 1s binding energies of the carbon atoms in H2(Salen) are divided into three groups (Cα, Cβ2, and Cβ1), while in the spectrum of [Ni(Salen)], a clear division into only two groups is observed.
The main difference between the compared spectra is that the relative intensity of the Cβ band in the calculated spectrum of H
2(Salen) (0.33) is significantly less than the intensity in the experimental one (0.52), while these intensities practically coincide in the spectra of [Ni(Salen)] (
Figure 2b,
Table S1). Despite this discrepancy, we can state that the experimental and calculated results indicate a similar change in the chemical state of the carbon atoms in the salen ligand upon passing from a molecule to a complex. Indeed, the compared C1s PE spectra demonstrate a high-energy shift of about 1 eV, which indicates a decrease in the density of the valence electrons of the carbon atoms in the [Ni(Salen)] complex compared to those in H
2(Salen). Obviously, this is a consequence of the chemical bonding between the complexing nickel atom and the oxygen and nitrogen atoms of the salen ligand.
In this regard, it is interesting to understand how the chemical states of oxygen and nitrogen atoms, which directly interact with the nickel atom during the formation of the complex, change. To do this, let us compare the measured N 1s and O 1s PE spectra of the salen ligand H
2(Salen) and [Ni(Salen)] complex, which are presented in
Figure 3 on the BE scale, relative to the Fermi level. The N 1s and O 1s PE spectra are represented by single lines at binding energies of 398.65 eV and 532.5 eV (H
2(Salen)), and 400.55 eV and 532.10 eV ([Ni(Salen)]). The binding energies of the N 1s and O 1s electrons obtained for [Ni(Salen)] are in good agreement with the data (400.3 eV and 532.0 eV) of this work [
33], in which these spectra were excited by monochromatic synchrotron radiation with photon energies of 500 eV and 615 eV, respectively.
The simple shape of the 1s PE spectra of the N and O atoms, in contrast to the 1s PE spectrum of the C atoms (
Figure 2), implies that the chemical states of both nitrogen atoms (oxygen atoms) are the same in the ligand molecule and the complex. When comparing the binding energies of the N 1s and O 1s electrons in the molecule and complex, a significant high-energy shift of 1.9 eV is observed in the spectra of the nitrogen atoms and a small low-energy shift of –0.4 eV in the spectra of the oxygen atoms. Taking into account the positive charge of the Ni
2+ cation and the high-energy shift found above for all the carbon atoms in the complex, it can be argued that the formation of the [Ni(Salen)] complex is accompanied by a significant redistribution of the electron density for all the atoms in the salen ligand, due to its transfer to the region of the oxygen atoms. In other words, this means that the transfer of electron density to the O atoms in [Ni(Salen)] occurs not only from the Ni atom, but also from the N and C atoms. An important role in this process is apparently played by the delocalized conjugated pi system of the 2p electron states of carbon atoms.
Finally, all the C, N, and O 1s PE spectra of the ligand molecule and complex contain low-intensity satellites
sh at higher E
bind’s (
Figure 2a and
Figure 3), which usually accompany the photoionization of the 1s electron shells of light atoms and arise due to the shake-up processes of valence electrons [
39,
40]. In the C 1s spectra of H
2(Salen) and [Ni(Salen)], these satellites are very low-intensity and broad due to the more complex shape of the parent C 1s PE line compared to that of the N 1s and O 1s PE line, and their energy position is difficult to characterize correctly. The energy positions of the shake-up satellites in the N 1s and O 1s PE spectra are 402.5 eV and 404.9 eV, and 538.8 eV and 538.4 eV, for H
2(Salen) and [Ni(Salen)], respectively. The energy distances between the main lines and their satellites are equal to 4.4 eV (N 1s PE spectra) and 6.3 eV (O 1s PE spectra). Since these satellites are due to the additional excitation of the valence electrons from upper MOs localized on the nitrogen (oxygen) atom to the lower, unoccupied MOs [
19], these values can be used to estimate the energy separation between the MOs participating in the shake-up process. They can also be regarded as upper bounds for the energy separation between the highest occupied MO (HOMO) and the lowest unoccupied MO (LUMO) in H
2(Salen) and [Ni(Salen)]. The differences in the corresponding energy separations in the N 1s and O 1s PE spectra can be explained by the effect of the core hole on the excitation process of valence electrons.
2.2. UV PES
Let us now consider the UV PE spectra of the H
2(Salen) molecule and [Ni(Salen)] complex, which reflect the energy distribution and properties of the occupied electronic states in the valence band below the Fermi level. These spectra, measured at photon energies
hν of 21.2 eV, are shown in
Figure 4. They are normalized to the intensity of the PE band
d and plotted on the scale of the valence electron BE’s relative to the Fermi level (
EF). A preliminary discussion of these spectra was carried out in the framework of a study devoted to a consideration of the structural evolution of the VB PE spectra of the [Ni(Salen)] complex with a change in the energy of its exciting photons in the range of 21.2–848.0 eV [
34]. Here, the qualitative results of this discussion are supplemented with a detailed comparative analysis of the UV PE spectra of H
2(Salen) and [Ni(Salen)], using the results of the DFT calculations of their valence bands.
The H
2(Salen) spectrum (
Figure 4, purple line) is quite noisy due to sample charging; therefore, its structural elements are revealed only by a direct comparison with the [Ni(Salen)] spectrum (blue line). Both spectra are characterized by a similar spectral distribution of the PE intensities, with the most intense PE signals in the high E
bind range from 6 eV to 16 eV. This suggests that the energy distributions of the occupied electronic states in this energy region of the H
2(Salen) and [Ni(Salen)] valence bands are very close to each other and are essentially determined by the corresponding states of the salen ligand. It should be noted here that this feature of the compared PE spectra is due to the dominance of the C 2p AOs of the phenol and ethylenediamine fragments in the formation of the electronic structure of the valence bands of the molecule and complex. Indeed, the number of carbon atoms in both compounds (16) is more than three times greater than the total number of oxygen, nitrogen, and nickel atoms (5). In addition, to characterize the PE spectrum, it is important that the photoionization cross-sections of the valence 2p AOs of the C, N, and O atoms and 3d AOs of the Ni atom are approximately equal (10–5 Mb) to an energy of the absorbed photons of 21.2 eV [
41]. Therefore, the designations of the PE bands
c–
f in the H
2(Salen) spectrum are made, taking into account their correlation with the corresponding PE bands in the [Ni(Salen)] spectrum and the results of their subsequent identification based on the DFT calculations.
The low-energy region of 2–6 eV in the [Ni(Salen)] spectrum is represented by the clear PE bands
a′,
a, and
b, while it can be confidently assumed that the H
2(Salen) spectrum contains only a weak
a′ band at a binding energy of ~2.5 eV. At the same time, in the spectrum of H
2(Salen), a structureless rise in the PE intensity is observed instead of the PE bands
a and
b in the [Ni(Salen)] spectrum. The PE bands
a and
b, located in the [Ni(Salen)] spectrum at the binding energies of 3.6 and 5.2 eV, were previously assigned to the photoionization of the valence MOs, with significant contributions from the valence 3d orbitals of the Ni atom [
33]. Therefore, it can be assumed that the disappearance of bands
a and
b in the UV PE spectrum of the H
2(Salen) molecule is associated with the absence of the Ni 3d-derived MOs in its valence band. In this case, it is logical to attribute the
a′ band, as in the [Ni(Salen)] spectrum [
33], to the photoionization of HOMO, formed by the π C2p MOs of the phenol fragments of the H
2(Salen) molecule.
In order to more fully understand and describe the changes in the electronic structure of the salen ligand upon passing from the H
2(Salen) molecule to the [Ni(Salen)] complex, DFT calculations of the valence bands for both compounds were performed. In
Figure 5, the calculated spectra of the total and partial densities of the occupied valence states for [Ni(Salen)] (
a) and H
2(Salen) (
b) are compared with the experimental UV PE spectra in the binding energy range from −1 to 18 eV. The corresponding spectra are aligned at the energy position of the
a′ band. The higher occupied MOs of the H
2(Salen) molecule and [Ni(Salen)] complex are shown in
Figure S2.
First of all, it should be emphasized that the total DOS calculated for [Ni(Salen)] in this work does not practically differ from the data of the previous study [
33]. The small differences in the intensities of the individual bands of the total and partial DOS of the occupied electronic states are apparently due to the use of a more powerful all-electron aug-cc-pVTZ basis in this work. When comparing the DOS spectra calculated for [Ni(Salen)] and H
2(Salen), first of all, we note a significant difference in their total DOS spectra in the E
bind region below 4 eV, namely: a single band
a′ in the spectrum of the H
2(Salen) molecule is structured in the form of three bands
a′,
a, and
b in the spectrum of the [Ni(Salen)] complex. In addition, the
d′ and
d bands in the total DOS spectrum for H
2(Salen) merge into one band upon passing to the DOS spectrum of the complex. The partial DOSs of the ligand atoms are also characterized by significant changes in their spectral distributions upon passing from the molecule to the complex. Thus, in the case of [Ni(Salen)], in comparison with H
2(Salen), the center of gravity of the partial N 2p and C 2p DOS is shifted to the region of a higher E
bind, while that of the O 2p DOS is shifted in the opposite direction. This finding unambiguously indicates an increase in the effective charges of the carbon and nitrogen atoms, as well as a decrease in the effective charge of the oxygen atoms in the ligand during the formation of the [Ni(Salen)] complex. It should be emphasized that these directions of charge transfer are in perfect agreement with those established above when comparing the chemical shifts of the 1s PE spectra of the C, N, and O atoms for H
2(Salen) and [Ni(Salen)].
As can be clearly seen from
Figure 5, the spectra of the partial DOS for all the atoms strongly overlap in the entire valence band of the molecule and the complex, which reflects the strong mixing of the valence AOs due to the low symmetry of the compounds. This factor complicates the unambiguous assignment of the bands in the UV PE spectra to the valence MOs, with contributions from specific AOs. However, a careful examination of the partial DOS spectra allows us to assume that the PE band
a′ in the UV PE spectra of both compounds is due to the photoionization of HOMO, formed mainly by the atomic 2pπ orbitals of the carbon atoms (C1 and C4) of the phenolic fragments. In turn, the atomic 2pσ orbitals of the C1 and C4 atoms are apparently responsible for the PE bands
c and
d in the spectra of the complex and the molecule. It is also logical to assert that the atomic N 2p and 2p orbitals of the C2 and C3 carbon atoms of the ethylenediamine fragment significantly contribute to the MOs responsible for the PE bands
c–
e in the [Ni(Salen)] spectrum. Finally, the results of the performed calculation confirm the important role of the Ni 3d states in the formation of the
a–
d bands of the occupied electronic states in the valence band of the [Ni(Salen)] complex, which was first pointed out in [
33]. A more detailed analysis of the electronic structures of the valence bands for the H
2(Salen) molecule and the [Ni(Salen)] complex requires additional calculations, taking into account the site symmetry for individual atoms.
2.3. NEXAFS
To obtain information about low-lying unoccupied electronic states in a polyatomic system, one should consider the NEXAFS spectra of the atoms forming this system. It is conventional to describe these spectra using the multiple (resonant) scattering of photoelectrons ejected from the core of an absorbing atom on its nearest neighbor atoms [
17,
18]. At certain photoelectron energies, a quasi-molecular fragment formed by neighborhood atoms can temporarily trap a photoelectron, resulting in the formation of metastable states (shape resonances). These resonances, depending on their lifetime, are observed in the spectra as narrow lines or broad absorption bands. Resonance localization in the quasi-molecule field allows us to consider them as a result of the dipole-allowed transitions of core electrons to the unoccupied MOs of this quasi-molecular fragment.
Let us begin our comparative analysis of the obtained X-ray absorption spectra of the H
2(Salen) molecule and [Ni(Salen)] molecular complex by considering the 1s NEXAFS spectra of the nitrogen and oxygen atoms. These atoms directly interact with the nickel cation in the complex; therefore, their spectra are expected to most fully reflect the changes in the local electronic structure of the salen ligand during the formation of the Ni complex. The N 1s NEXAFS spectra of the H
2(Salen) ligand and [Ni(Salen)] complex are shown in
Figure 6 as a dependence of the total electron yield (TEY) of the photoemission on the energy
hν of the absorbed photons. The designations of the absorption structures in both spectra correspond to those of the structures for the [Ni(Salen)] complex spectrum in our previous work [
33].
It is clearly seen that the compared spectra are characterized by a strong similarity in their overall spectral distribution in the photon energy range of 395–420 eV. In both spectra, the low-energy region is dominated by an intense absorption resonance
B at the photon energies of 398.85 eV and 399.20 eV for H
2(Salen) and [Ni(Salen)], respectively. In the H
2(Salen) spectrum, this resonance is accompanied by a low-intensity band
B2, an extended structureless rise
C, and broad absorption bands
D–
E at the higher photon energies. These structures completely characterize the N 1s NEXAFS spectrum of H
2(Salen). The main differences in the [Ni(Salen)] spectrum are a high-energy shift of the main absorption resonance
B by 0.35 eV and a noticeable increase in its relative intensity, as well as the appearance of an additional low-intensity
B1 band. These changes are observed in the low-energy region of the [Ni(Salen)] spectrum, in which absorption structures are associated with the transitions of the N 1s electrons to empty π*N 2p MOs [
33]. All the other absorption bands,
C–
E, in the [Ni(Salen)] spectrum do not practically change their energy positions and relative intensities in comparison with those in the ligand spectrum.
It is well known that the 2p electronic states of the light atoms in planar molecules are split by the molecular field into the π2p
z- and σ2p
x,y-states, which characterize the occupied and vacant MOs of molecules oriented perpendicular and parallel to the molecular plane. Unoccupied electronic states are described by antibonding π* and σ* MOs, which appear in 1s NEXAFS spectra as π* and σ* absorption resonances due to dipole-allowed 1s→2p electronic transitions. The energy distance ΔE(π*–σ*) between them in the spectrum depends inversely on the distance between the absorbing atom and the atoms of its nearest surroundings [
42]. In view of the above, it is logical to attribute the absorption resonances
D and
E in the N 1s NEXAFS spectrum of H
2(Salen) to the N 1s electron transitions to the σ* MOs oriented to the carbon atoms C2 and C3, respectively, since the interatomic distances are
R(N–C2) = 1.448 Å and
R(N–C3) = 1.279 Å [
35].
These absorption resonances,
D and
E, do not change their energy positions upon transitioning to the spectrum of [Ni(Salen)], which implies that the interatomic distances
R(N–C2) and
R(N–C3) in the complex remain unchanged. This conclusion agrees with the data for [Ni(Salen)] obtained via gas-phase electron diffraction [
36].
The observed similarity of the N 1s NEXAFS spectra of the ligand molecule and the complex indicates weak changes in the local electronic and atomic structure of the salen ligand in the region of the nitrogen atom upon passing from H2(Salen) to [Ni(Salen)]. In other words, during such a transition, the spectrum of the vacant electronic states and the interatomic distances between the nitrogen atom and its nearest neighbors remain practically unchanged. In H2(Salen), the nitrogen atom is part of the imine (–C=N–) group linked with the phenolic and ethylene groups. In the [Ni(Salen)] complex, the nitrogen atoms are additionally coordinated by the complexing nickel atom, but this interaction has very little effect on the structure of the N 1s absorption spectrum, since the bonding is realized due to the lone pair of valence electrons of the nitrogen atom. Thus, this experimental finding for the N 1s NEXAFS spectra reflects the fact that the fine structure of the compared N 1s spectra is mainly determined by the electronic structure of the ethylenediamine bridge, while the interaction of the nitrogen and nickel atoms in the [Ni(Salen)] complex only leads to a change in the spectral parameters of the B resonance and the appearance of an additional low-intensity B1 structure.
As is known, the resonance
B is due to the N 1s electron transitions to the unoccupied π*-states of the [Ni(Salen)] complex, which, in turn, can be described using the empty anti-bonding π*-MO of the NiN
2O
2 coordination center [
33]. The existence of a similar absorption band in the Ni 2p
3/2 and O 1s NEXAFS spectra of the coordination center indicates the presence of π bonding between the complexing Ni atom and the nitrogen and oxygen atoms of the ligand in [Ni(Salen)]. Hence, it is clear that the vacant π*-MO, which is responsible for the resonance
B, is characterized by significant contributions from the valence AO atoms of the NiN
2O
2 quasimolecule (Ni 3d, N 2p, and O 2p). The formation of such a π-type MO in planar complexes is a signature of a π-back-donation between the nickel and ligand atoms [
43]. It should be noted that the low-intensity absorption band
B1 is apparently also associated with the transitions of the N 1s electrons to the vacant π*-MO, which reflects the presence of a weak π-conjugation between the nitrogen atom and the carbon atoms of the benzene ring (through the C3 carbon atom).
The high-energy shift of the band B in the N 1s NEXAFS spectrum of the complex is indicative of a higher positive effective charge on the N atom in [Ni(Salen)] compared to the salen ligand, which is consistent with the above results for the analysis of the N 1s PE spectra of the molecule and complex. Taking into account the π-back-donation effect, this means that the N 2p electron density shifts from the N atoms to the 3d AO of Ni and then to the 2p AO of the oxygen atoms. Since the N 2p population of the occupied valence π-MO decreases, the N 2p contribution to the unoccupied π*-MO increases. Thus, the transfer of the electron density (of the lone pair) of the nitrogen atom to the complexing nickel cation also explains the increase in the intensity of the absorption B resonance upon passing from the N 1s spectrum of the free ligand to the that of the complex.
Let us now consider the O 1s NEXAFS spectra of H
2(Salen) and [Ni(Salen)], which are shown in
Figure 7 as a dependence of the TEY of the photoemission on the energy
hν of the absorbed photons in the energy range from 525 eV to 560 eV. The designations of the absorption structures in both spectra correspond to those of the structures for the [Ni(Salen)] complex spectrum in our previous work [
33]. The fine structure of the H
2(Salen) spectrum consists of three broad absorption bands,
B–
D, and the last band
D has an extended tail
E on the high-energy side. These absorption structures correspond to the dipole-allowed transitions of the O 1s electrons to the unoccupied MOs localized in the region of the C–O–H molecular fragment, with significant contributions from the O 2p valence states. It is clearly seen that the fine structure of the O 1s absorption spectrum, in contrast to the N 1s spectrum, undergoes significant changes in going from H
2(Salen) to [Ni(Salen)]. Thus, in the low-energy part of the spectrum of the complex, the absorption band
B at a photon energy of 531.7 eV is split into two narrow absorption lines,
B and
B1 (531.8 eV and 533.4 eV), the relative intensities of the
C and
D bands are reversed, a distinct shoulder
D1 appears near the
D band. It is clearly seen that the fine structure of the O 1s absorption spectrum, in contrast to the N 1s spectrum, undergoes significant changes in going from H
2(Salen) to [Ni(Salen)]. Thus, in the low-energy part of the spectrum of the complex, the absorption band
B at a photon energy of 531.7 eV is split into two narrow absorption lines,
B and
B1 (531.8 eV and 533.4 eV), the relative intensities of the
C and
D bands are reversed, a distinct shoulder
D1 appears near the
D band, and finally, the high-energy extended tail
E transforms into a broad band. The absence of narrow π-like absorption bands in the low-energy part of the H
2(Salen) spectrum unambiguously indicates that the total molecular field in the region of the C–O–H fragment is very different from the flat one, due to its different orientations in the molecule. It is quite possible that this finding is partly due to the presence in the sample of several isomeric structures, in which these fragments have different orientations in the H
2(Salen) molecule. As a consequence, the absorption transitions of the O 1s electrons to a vacant low-energy MO with O 2p contributions have slightly different energies, which explains the large width of the low-energy absorption band
B in the H
2(Salen) spectrum.
In moving from H
2(Salen) to [Ni(Salen)], the salen ligand loses its hydrogen atoms and its oxygen atoms are linked to the complexing Ni atom. In the [Ni(Salen)] complex, the oxygen atoms, together with the nitrogen atoms, are coordinated by the Ni atom and form a square-planar coordination center, NiO
2N
2 [
36]. This increase in the symmetry of the molecular field in the region of the oxygen atoms now makes it possible to consider the O 2p electronic states as split into 2p
π and 2pσ components. Thus, it is logical to attribute the narrow absorption resonances
B and
B1 in the [Ni(Salen)] spectrum to the transitions of the O 1s electrons to two vacant antibonding π* MOs, separated by 1.6 eV from each other. The first one reflects the presence of π-bonding with the Ni atom (π-back-donation) [
33], while the second π*-MO seems to indicate π-conjugation between the oxygen atoms and the benzene ring. It should be noted that a similar assignment of the absorption band
B1 was made above when discussing the N 1s NEXAFS spectra of H
2(Salen) and [Ni(Salen)]. It is important that the energy distance between the corresponding resonances
B and
B1 practically coincides in the spectra of the oxygen (1.6 eV) and nitrogen (1.5 eV) of the [Ni(Salen)] complex. The high relative intensity of the
B1 resonance in the oxygen spectrum, in comparison with the nitrogen one, apparently indicates a higher degree of π-conjugation between the oxygen atoms and carbon atoms of benzene rings in the complex.
Using an analogy with the N 1s spectrum, it is constructive to attribute the absorption structures
C–
E in the O 1s spectrum of H
2(Salen) to the transitions of the O 1s electrons to vacant antibonding σ*O 2p MOs, reflecting the σ-bonding of the O atom with the nearest hydrogen and carbon atoms. It can be seen from
Figure 7 that, upon transitioning to the spectrum of [Ni(Salen)], the
C band practically disappears, which makes it possible to associate it with the σ* MO, which is responsible for the σ bonding of the oxygen atom with the hydrogen atom. In this case, the
D band can be attributed to σ* MO, which characterizes the chemical bonding between the oxygen and carbon (C5) atoms (
Figure 1). It is important that this assignment is consistent with the presence of this absorption structure in the O 1s spectrum of [Ni(Salen)]. It is quite possible that the appearance of an additional absorption band
E in the spectrum of the complex, which is absent in the spectrum of H
2(Salen), is due to the σ bonding of the oxygen atom with the complexing Ni atom.
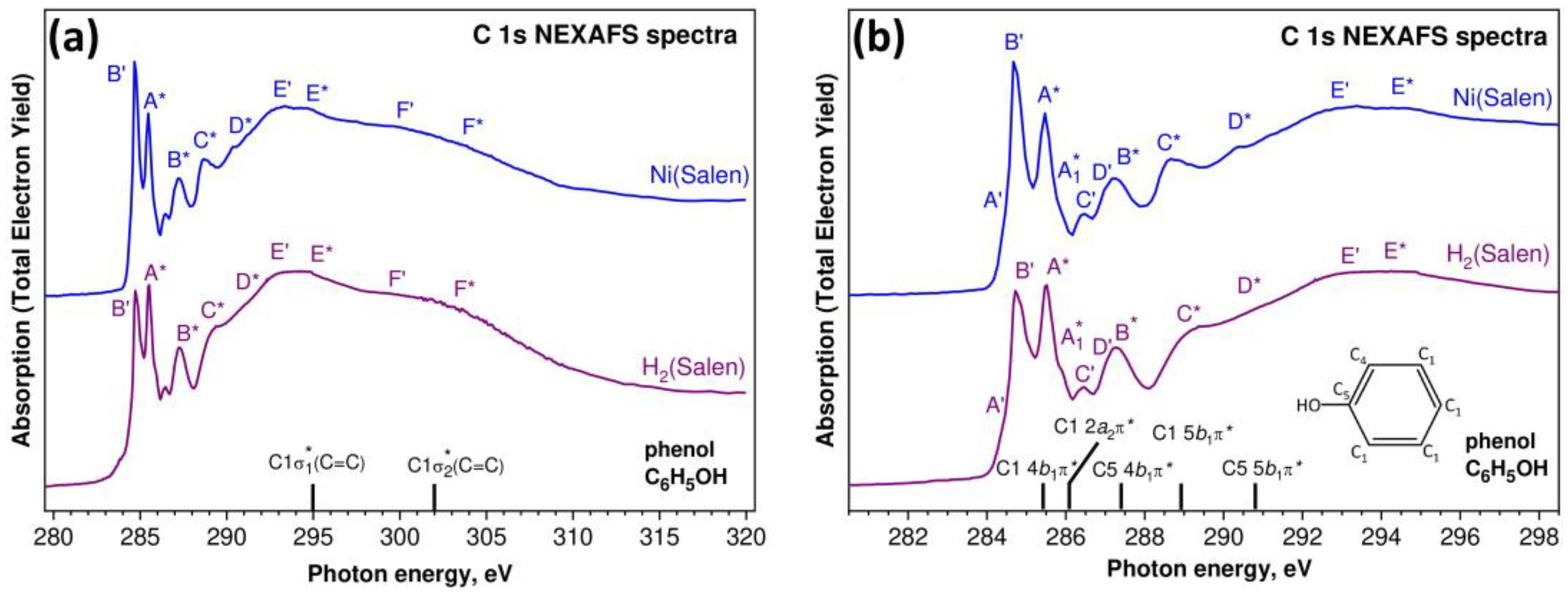
Let us now compare the C 1s NEXAFS spectra of the H
2(Salen) ligand and the [Ni(Salen)] complex (
Figure 8). Compared to the spectra of the nitrogen and oxygen atoms, the C 1s NEXAFS spectra show a richer fine structure, which is due to the presence of several chemically nonequivalent positions of the carbon atoms in the ligand (
Figure 1). An examination of the spectra (
Figure 8a) clearly shows that, as in the case of N 1s absorption spectra, the C 1s NEXAFS spectra of the free salen ligand and the [Ni(Salen)] complex are characterized by a strong similarity of their overall spectral distributions in the photon energy range of 280–320 eV and are consistent in the numbers of their main absorption bands and their energy positions. This finding is quite obvious, since the carbon atoms of the ligand do not directly interact with the complexing Ni atom. Nevertheless, when passing from a free ligand to a complex, small differences are still observed in the structure of the absorption spectrum of the [Ni(Salen)] complex, namely, (i) the relative intensity of the first absorption peak
B’ noticeably increases, and (ii) the shoulders
C* and
D* become more contrasted and turn into clear absorption bands
C* and
D*. It is clear that these spectral differences are a consequence of the indirect influence of the nickel atom through the nitrogen and oxygen atoms on the properties of the vacant MOs spatially localized in the region of the carbon atoms. It is also possible that these changes are partly associated with an increase in molecular symmetry upon moving from H
2(Salen) to [Ni(Salen)].
As noted above, carbon atoms are part of two polyatomic fragments (quasimolecules) of the ligand–phenolic and ethylenediamine groups. Let us begin a detailed discussion of the compared C 1s NEXAFS spectra by considering the contribution to the fine structure of the 1s excitations of the C atoms of the phenolic groups, since these C atoms predominate in the ligand. It is well known that the C 1s NEXAFS spectrum of the phenol molecule C
6H
5OH is very similar to that of the parent benzene molecule C
6H
6 [
37,
44]. At the same time, the addition of an electronegative oxygen atom to the benzene ring is accompanied by noticeable changes in the electronic structure of this benzene molecule, which are reflected in the C 1s NEXAFS spectrum of the phenol group.
Generally speaking, all the carbon atoms in a phenol molecule are in a different chemical (charge) states, since an electronegative oxygen atom attached to one of the C atoms of the benzene molecule C
6H
6 causes a redistribution of the electron density throughout the benzene ring. However, these differences are small for all the C atoms of the benzene ring, with the exception of the atom bonded to the oxygen atom, and do not lead to noticeable chemical shifts of the 1s levels of these carbon atoms. This allows us to consider the carbon atoms of the phenol group in two chemical states—the C1(C4) atoms bonded only to other carbon atoms and the C5 atom bonded also to an oxygen atom (insert in
Figure 8b). The correctness of this separation of the carbon atoms in the phenol group is confirmed by the results of the photoemission measurements of the core C 1s spectra for the phenol molecule [
45] and the [Ni(Salen)] complex, which demonstrate the two-component shape with close energy separations between these components—1.8 eV and 1.5 eV, respectively.
In addition to the splitting of the C 1s levels in the phenol molecule, the oxygen atom attached to the parent benzene molecule lowers the point symmetry of the molecule from the D
6h group (C
6H
6) to C
2v (C
6H
5OH), which, in turn, causes changes in the spectrum of the unoccupied MOs upon going from benzene to phenol. As a result, low-energy π* MOs of e
2u and b
2g symmetry types unoccupied in the benzene molecule [
46] are transformed in the phenol molecule into the 4b
1 + 2a
2 and 5b
1 π* MOs, respectively [
37,
44]. Thus, the splitting of the LUMO of π*e
2u symmetry into two 4b
1 and 2a
2 components and the presence of two types of chemically different carbon atoms are characteristic features of the electronic structure of the phenol molecule.
The C 1s NEXAFS spectrum of a gaseous phenol molecule C
6H
5OH (insert in
Figure 8b) is well known and quite fully interpreted [
37,
44]. The results of this interpretation are shown in
Figure 8b by vertical black bars with the C 1s absorption transition assignments. It is clearly seen that these C 1s → π* transitions are in good agreement in terms of their energy positions with the absorption bands marked by letters with asterisks in the spectra of H
2(Salen) and [Ni(Salen)]. It should also be noted that the
E* and
F* absorption bands in the spectra of H
2(Salen) and [Ni(Salen)] also agree well in terms of their energy positions with those of the σ
1* and σ
2* resonances at 295 eV and 302 eV in the spectrum of the phenol molecule (
Figure 8a). This implies that the association of two phenol molecules with the ethylenediamine group in the H
2(Salen) ligand, as well as the subsequent formation of the [Ni(Salen)] complex, have little effect on the energy spectrum of the unoccupied MOs of the phenol fragment in the compared polyatomic systems. The small changes observed in the C 1s NEXAFS spectrum of the complex are most likely associated with an increase in the symmetry of the polyatomic system in going from H
2(Salen) to [Ni(Salen)].
It follows from the above that the other bands marked with dashed letters are due to the absorption transitions of the 1s electrons of the carbon atoms in the ethylenediamine group. This fragment also contains carbon atoms in two chemical states, which is due to their nearest surroundings in the C
2H
4 ethylene molecule (C2 atoms) and the –N=C– imine functional group (C3 atoms). First of all, this approach makes it possible to relate the intense sharp peak
B’ at the photon energy of 284.75 eV in the spectra of H
2(Salen) and [Ni(Salen)] to the transitions of the C 1s electrons to the empty antibonding π* MO of the C
2H
4 molecule (284.7 eV) [
47]. Moreover, the broad bands
E’ and
F’ in the C 1s NEXAFS spectra of H
2(Salen) and [Ni(Salen)] (~293 eV and ~301 eV) should be considered to be as a result of the electronic transitions in the unoccupied σ
1* and σ
2* MOs of the C
2H
4 molecule (292.6 eV and 301 eV). Finally, in this case, it is logical to assume that the C 1s excitations of the carbon atoms in imine functional groups are responsible for the weak absorption bands
A’,
C’, and
D’ in the compared spectra. An increase in the intensity of the
B’ resonance in the [Ni(Salen)] spectrum indicates a certain effect of the complexing Ni atom on the atomic orbital composition of the unoccupied π* MO of the ethylene fragment, which manifests itself in an increase in the contribution of the valence C 2p states to this MO.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}