

Exploring the Mitogenomes of Mantodea: New Insights from Structural Diversity and Higher-Level Phylogenomic Analyses



Abstract
:1. Introduction
2. Results
2.1. Mantodean Mitogenome Structure
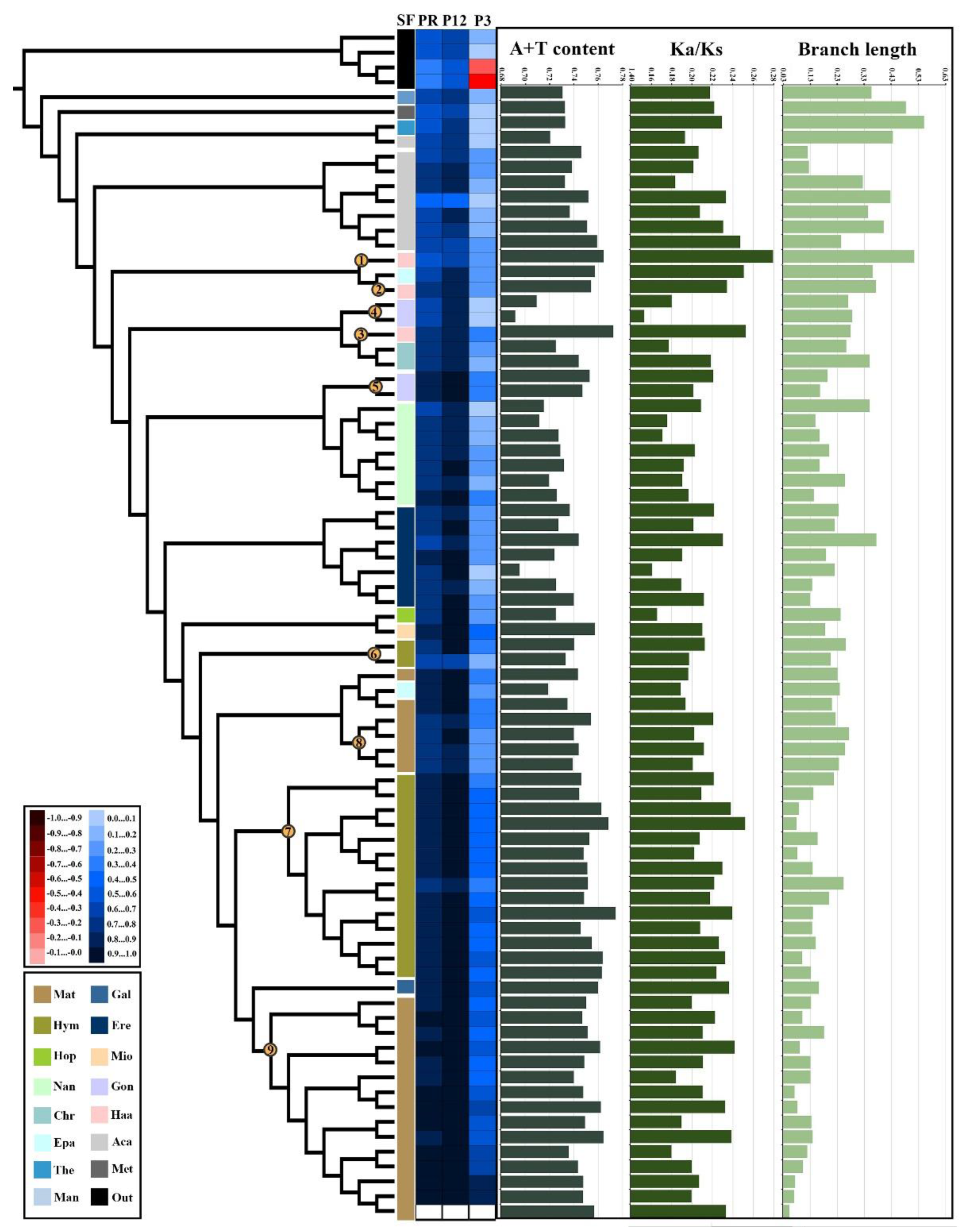
2.2. Nucleotide Composition and Evolutionary Rates of Mantodean Mitogenomes
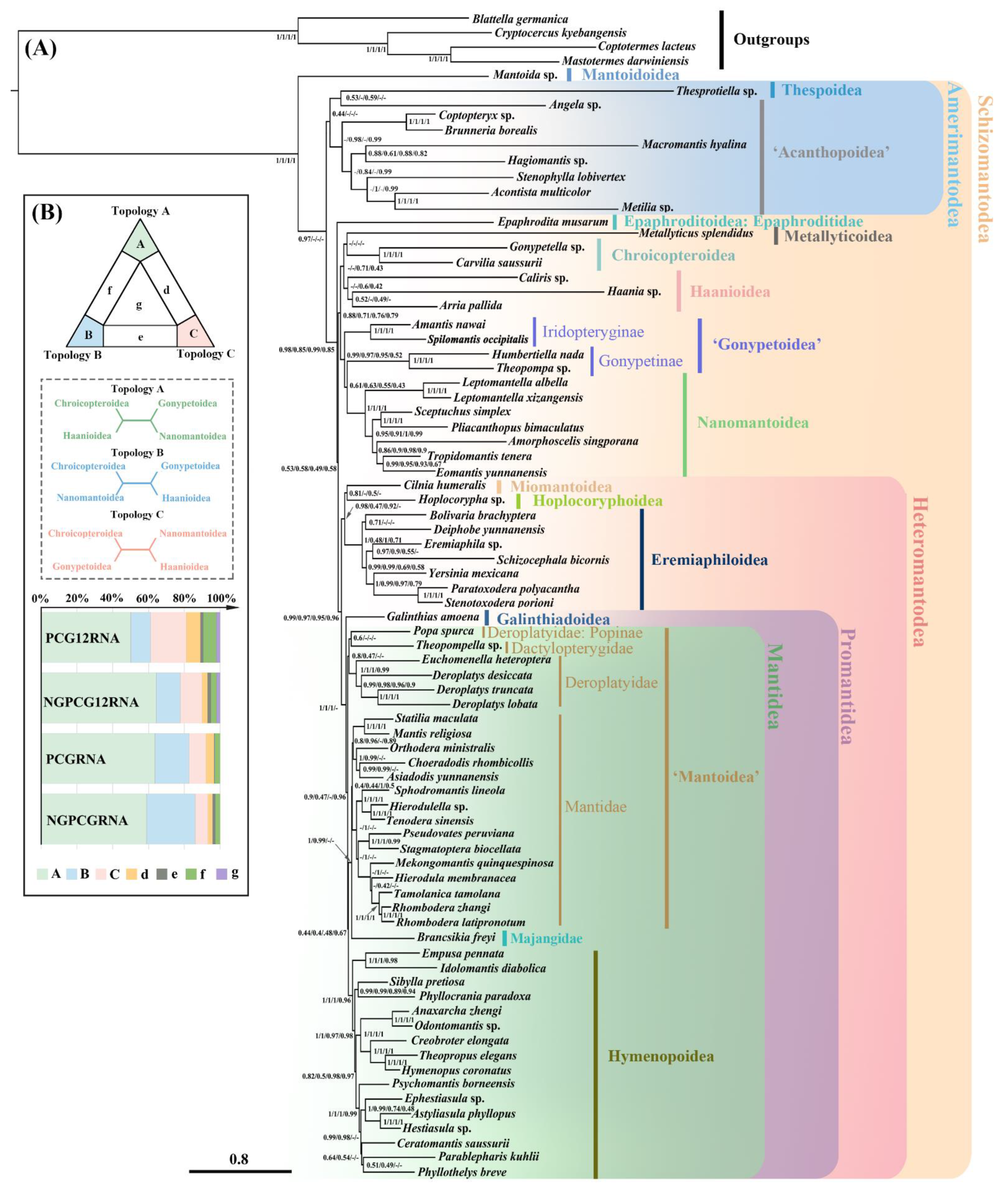
2.3. Phylogenetic Analyses under Site-Homogenous Models and Parsimony Methods
2.4. Systematic Errors under Site-Homogeneous Models
2.5. Phylogenetic Analyses under Site-Heterogeneous Model
2.6. Testing Alternative Hypotheses
3. Discussion
3.1. Gene Arrangements in Mantodean Mitogenomes
3.2. Phylogenetic Implication
4. Materials and Methods
4.1. Taxon Sampling and DNA Extraction
4.2. Molecular Marker Generation
4.3. Sequence Alignment, Concatenation, and Analyses
4.4. Phylogenetic Analyses
4.5. Phylogenetic Hypothesis Testing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wieland, F.; Svenson, G.J. Biodiversity of Mantodea. In Insect Biodiversity, Science and Society; Footit, R.G., Adler, P.H., Eds.; Wiley-Blackwell: Oxford, UK, 2018; Volume 2, pp. 389–416. [Google Scholar]
- Schwarz, C.J.; Roy, R. The systematics of Mantodea revisited: An updated classification incorporating multiple data sources (Insecta: Dictyoptera). Ann. Soc. Entomol. Fr. 2019, 55, 101–196. [Google Scholar] [CrossRef]
- Wieland, F. The phylogenetic system of Mantodea (Insecta: Dictyoptera). Species Phylogeny Evol. 2013, 3, 1–222. [Google Scholar]
- Hennig, W. Insect Phylogeny; John Wiley & Sons: Hoboken, NJ, USA, 1981. [Google Scholar]
- Klass, K.D.; Meier, R. A phylogenetic analysis of Dictyoptera (Insecta) based on morphological characters. Entomol. Abh. 2006, 63, 3–50. [Google Scholar]
- Misof, B.; Liu, S.; Meusemann, K.; Peters, R.S.; Donath, A.; Mayer, C.; Frandsen, P.B.; Ware, J.; Flouri, T.; Beutel, R.G.; et al. Phylogenomics resolves the timing and pattern of insect evolution. Science 2014, 346, 763–767. [Google Scholar] [CrossRef] [PubMed]
- Rivera, J.; Svenson, G.J. The Neotropical ‘polymorphic earless praying mantises’—Part I: Molecular phylogeny and revised higher-level systematics (Insecta: Mantodea, Acanthopoidea). Syst. Entomol. 2016, 41, 607–649. [Google Scholar] [CrossRef]
- Svenson, G.J.; Whiting, M.F. Phylogeny of Mantodea based on molecular data: Evolution of a charismatic predator. Syst. Entomol. 2004, 29, 359–370. [Google Scholar] [CrossRef]
- Svenson, G.J.; Whiting, M.F. Reconstructing the origins of praying mantises (Dictyoptera, Mantodea): The role of Gondwanan vicariance and morphological convergence. Cladistics 2009, 25, 468–514. [Google Scholar] [CrossRef]
- Grimaldi, D. A revision of Cretaceous mantises and their relationships, including new taxa (Insecta: Dictyoptera: Mantodea). Am. Mus. Novit. 2003, 3412, 1–47. [Google Scholar] [CrossRef]
- Giglio-Tos, E. Das Tierreich: Orthoptera Mantidae; Walter de Gruyter & Co: Berlin, Germany, 1927. [Google Scholar]
- Beier, M. Blattopteroidea, Mantodea. In Klassen und Ordnungen des Tierreichs. Fünfter Band: Arthropoda. III. Abteilung: Insecta, 6th ed.; Bronns, H.G., Ed.; Geest & Portig: Leipzig, Germany, 1964; pp. 849–970. [Google Scholar]
- Ehrmann, R. Mantodea: Gottesanbeterinnen der Welt; Natur und Tier: Munster, Germany, 2002. [Google Scholar]
- Svenson, G.J.; Rodrigues, H.M. A Cretaceous-aged Palaeotropical dispersal established an endemic lineage of Caribbean praying mantises. Proc. R. Soc. B 2017, 284, 20171280. [Google Scholar] [CrossRef] [Green Version]
- Djernæs, M.; Klass, K.D.; Eggleton, P. Identifying possible sister groups of Cryptocercidae + Isoptera: A combined molecular and morphological phylogeny of Dictyoptera. Mol. Phylogenet. Evol. 2015, 84, 284–303. [Google Scholar] [CrossRef] [PubMed]
- Yager, D.D.; Svenson, G.J. Patterns of praying mantis auditory system evolution based on morphological, molecular, neuro-physiological, and behavioral data. Biol. J. Linn. Soc. Lond. 2008, 94, 541–568. [Google Scholar] [CrossRef] [Green Version]
- Svenson, G.J.; Hardy, N.B.; Wightman, H.M.C.; Wieland, F. Of flowers and twigs: Phylogenetic revision of the plant mimicking praying mantises (Mantodea: Empusidae and Hymenopodidae) with a new suprageneric classification. Syst. Entomol. 2015, 40, 789–834. [Google Scholar] [CrossRef]
- Timmermans, M.J.T.N.; Barton, C.; Haran, J.; Ahrens, D.; Culverwell, C.L.; Ollikainen, A.; Dodsworth, S.; Foster, P.G.; Bocak, L.; Vogler, A.P. Family-level sampling of mitochondrial genomes in Coleoptera: Compositional heterogeneity and phylogenetics. Genome Biol. Evol. 2015, 8, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Shao, R.F.; Song, N.; Jiang, P.; Li, Z.H.; Cai, W.Z. Higher-level phylogeny of paraneopteran insects inferred from mitochondrial genome sequences. Sci. Rep. 2015, 5, 8527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, F.; Li, H.; Jiang, P.; Zhou, X.G.; Liu, J.P.; Sun, C.H.; Vogler, A.P.; Cai, W.Z. Capturing the phylogeny of Holometabola with mitochondrial genome data and Bayesian site-heterogeneous mixture models. Genome Biol. Evol. 2016, 8, 1411–1426. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leavengood, J.M.; Chapman, E.G.; Burkhardt, D.; Song, F.; Jiang, P.; Liu, J.P.; Zhou, X.G.; Cai, W.Z. Mitochondrial phylogenomics of Hemiptera reveals adaptive innovations driving the diversification of true bugs. Proc. R. Soc. B 2017, 284, 20171223. [Google Scholar] [CrossRef] [Green Version]
- Ye, F.; Lan, X.E.; Zhu, W.B.; You, P. Mitochondrial genomes of praying mantises (Dictyoptera, Mantodea): Rearrangement, duplication, and reassignment of tRNA genes. Sci. Rep. 2016, 6, 25634. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.L.; Ye, F. Comparative mitogenomic analyses of praying mantises (Dictyoptera, Mantodea): Origin and evolution of usual intergenic gaps. Int. J. Biol. Sci. 2017, 13, 367–382. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.P.; Yu, D.N.; Storey, K.B.; Cheng, H.Y.; Zhang, J.Y. Higher tRNA gene duplication in mitogenomes of praying mantises (Dictyoptera, Mantodea) and the phylogeny within Mantodea. Int. J. Biol. Macromol. 2018, 111, 787–795. [Google Scholar] [CrossRef]
- Zhang, L.P.; Cai, Y.Y.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. Gene characteristics of the complete mitochondrial genomes of Paratoxodera polyacantha and Toxodera hauseri (Mantodea: Toxoderidae). PeerJ 2018, 6, e4595. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Li, L.Y.; Liu, Q.P.; Ali, M.Y.; Yuan, Z.L.; Smagghe, G.; Liu, T.X. Complete mitochondrial genomes of four species of praying mantises (Dictyoptera, Mantidae) with ribosomal second structure, evolutionary and phylogenetic analyses. PLoS ONE 2021, 16, e0254914. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.D.; Guan, J.Y.; Zhang, Z.Y.; Cao, Y.R.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Novel tRNA Gene rearrangements in the mitochondrial genomes of praying mantises (Mantodea: Mantidae): Translocation, duplication and pseudogenization. Int. J. Biol. Macromol. 2021, 185, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.J.; Cai, L.N.; Zhao, Y.Y.; Cheng, H.Y.; Storey, K.B.; Yu, D.N.; Zhang, J.Y. Novel mitochondrial gene rearrangement and intergenic regions exist in the mitochondrial genomes from four newly established families of praying mantises (Insecta: Mantodea). Insects 2022, 13, 564. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, L.P.; Lin, Y.J.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. Phylogenetic relationships and divergence dating of Mantodea using mitochondrial phylogenomics. Syst. Entomol. 2023, 1–14. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Song, H.J.; Cameron, S.L.; Whiting, M.F. Nonstationary evolution and compositional heterogeneity in beetle mitochondrial phylogenomics. Syst. Biol. 2009, 58, 381–394. [Google Scholar] [CrossRef] [Green Version]
- Talavera, G.; Vila, R. What is the phylogenetic signal limit from mitogenomes? The reconciliation between mitochondrial and nuclear data in the Insecta Class phylogeny. BMC Evol. Biol. 2011, 11, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.Q.; Song, F.; Jiang, P.; Wilson, J.; Cai, W.Z.; Li, H. Compositional heterogeneity in true bug mitochondrial phylogenomics. Mol. Phylogenet. Evol. 2019, 118, 135–144. [Google Scholar] [CrossRef]
- Lartillot, N.; Rodrigue, N.; Stubbs, D.; Richer, J. PhyloBayes MPI: Phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Syst. Biol. 2013, 62, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Lartillot, N.; Brinkmann, H.; Philippe, H. Suppression of long-branch attraction artifacts in the animal phylogeny using a site-heterogeneous model. BMC Evol. Biol. 2007, 7, S4. [Google Scholar] [CrossRef] [Green Version]
- Kapli, P.; Flouri, T.; Telford, M.J. Systematic errors in phylogenetic trees. Curr. Biol. 2021, 31, R59–R64. [Google Scholar] [CrossRef]
- Cai, C.Y.; Tihelka, E.; Giacomelli, M.; Lawrence, J.F.; Slipinski, A.; Donoghue, P.C.J. Integrated phylogenomics and fossil data illuminate the evolution of beetles. R. Soc. Open Sci. 2022, 9, 211771. [Google Scholar] [CrossRef]
- Guan, J.Y.; Jia, Y.Y.; Zhang, Z.Y.; Cao, S.S.; Ma, J.L.; Zhang, J.Y.; Yu, D.N. The complete mitochondrial genome of Xanthomantis bimaculata (Mantodea: Iridopterygidae) and its Phylogeny. Mitochondrial DNA Part B 2020, 5, 3097–3099. [Google Scholar] [CrossRef] [PubMed]
- Nie, R.E.; Andújar, C.; Gómez-Rodríguez, C.; Bai, M.; Xue, H.J.; Tang, M.; Yang, C.T.; Tang, P.; Yang, X.K.; Vogler, A.P. The phylogeny of leaf beetles (Chrysomelidae) inferred from mitochondrial genomes. Syst. Entomol. 2020, 45, 188–204. [Google Scholar] [CrossRef]
- Strimmer, K.; Von Haeseler, A. Likelihood-Mapping: A simple method to visualize phylogenetic content of a sequence alignment. Proc. Natl. Acad. Sci. USA 1997, 9, 6815–6819. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.J.; Wang, H.; Huang, H.M.; Zhao, Y.Z.; Zhou, C.F. Mitochondrial genomes of 10 Mantidae species and their phylogenetic implications. Arch. Insect Biochem. Physiol. 2022, 111, e21874. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.P.; Ma, Y.; Yu, D.N.; Storey, K.B.; Zhang, J.Y. The mitochondrial genomes of Statilia maculate and S. nemoralis (Mantidae: Mantinae) with different duplications of trnR genes. Int. J. Biol. Macromol. 2019, 121, 839–845. [Google Scholar] [CrossRef]
- Svenson, G.J. The Origins, Evolution, and Phylogeny of the Praying Mantises (Dictyoptera, Mantodea). Ph.D. Dissertation, Brigham Young University, Provo, UT, USA, 2007. [Google Scholar]
- Legendre, F.; Nel, A.; Svenson, G.J.; Robillard, T.; Pellens, R.; Grandcolas, F. Phylogeny of Dictyoptera: Dating the origin of cockroaches, praying mantises and termites with molecular data and controlled fossil evidence. PLoS ONE 2015, 10, e0130127. [Google Scholar] [CrossRef] [Green Version]
- Wieland, F. The genus Metallyticus reviewed (Insecta: Mantodea). Species Phylogeny Evol. 2008, 1, 147–170. [Google Scholar]
- Demers-Potvin, A.V.; Larsson, H.C.E.; Cournoyer, M.; Bethoux, O. Wing morphology of a new cretaceous praying mantis solves the phylogenetic jigsaw of early-diverging extant lineages. Syst. Entomol. 2021, 46, 205–223. [Google Scholar] [CrossRef]
- Ware, J.L.; Litman, J.; Klass, K.D.; Spearman, L.A. Relationships among the major lineages of Dictyoptera: The effect of outgroup selection on dictyopteran tree topology. Syst. Entomol. 2008, 33, 429–450. [Google Scholar] [CrossRef]
- Djernæs, M.; Klass, K.D.; Picker, M.D.; Damgaard, J. Phylogeny of cockroaches (Insecta, Dictyoptera, Blattodea), with placement of aberrant taxa and exploration of out-group sampling. Syst. Entomol. 2012, 37, 65–83. [Google Scholar] [CrossRef]
- Rodrigues, H.M.; Svenson, G.J. Epaphroditidae sensu novo, an endemic caribbean family of morphologically divergent praying mantises (Insecta, Mantodea). Neotrop. Entomol. 2018, 47, 502–507. [Google Scholar] [CrossRef] [PubMed]
- Hughes-Schrader, S. Meiosis without chiasmata in diploid and tetraploid spermatocytes of the mantid Callimantis antillarum Saussure. J. Morphol. 1943, 73, 111–141. [Google Scholar] [CrossRef]
- Kirby, W.F. A Synonymic Catalogue of Orthoptera; Orthoptera Euplexoptera, Cursoria, et Gressoria; British Museum (Natural History): London, UK, 1904; Volume I. [Google Scholar]
- Beier, M. Mantodea (Fangheuschrecken). In Handbuch der Zoologie; Band/Volume IV Arthropoda 2:, Insecta; Helmcke, J.G., Starck, D., Wermuth, H., Eds.; Walter de Gruyter & Co: Berlin, Germany, 1968; pp. 1–47. [Google Scholar]
- Schwarz, C.J.; Helmkampf, M.E. A remarkable new species of Mythomantis Giglio-Tos, 1916 from northern Borneo, with notes on the systematics of Deroplatyinae Westwood, 1889 (Mantodea: Mantidae). Zootaxa 2014, 3797, 120–129. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Schütte, K. Mise au point sur le genre Brancsikia Saussure & Zehntner (Mantodea, Epaphroditidae). Bull. Soc. Entomol. Fr. 2016, 121, 269–282. [Google Scholar] [CrossRef]
- Schwarz, C.J. Update on Tagalomantis manillensis (Saussure), with description of the female and comments on its systematic placement and life history (Insecta: Mantodea: Deroplatyinae). Stuttg. Beitr. Naturkd. A 2017, 10, 19–39. [Google Scholar] [CrossRef] [Green Version]
- Losos, J.B. Lizards in an Evolutionary Tree: Ecology and Adaptive Radiation of Anoles; University of California Press: Berkeley, CA, USA, 2009. [Google Scholar]
- Mahler, D.L.; Ingram, T.; Revell, L.J.; Losos, J.B. Exceptional convergence on the macroevolutionary landscape in island lizard radiations. Science 2013, 341, 292–295. [Google Scholar] [CrossRef]
- Cui, Y.; Evangelista, D.A.; Béthoux, O. Prayers for fossil mantis unfulfilled: Prochaeradodis enigmaticus Piton, 1940 is a cockroach (Blattodea). Geodiversitas 2018, 40, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Schubnel, T.; Nel, A. New Paleogene mantises from the Oise amber and their evolutionary importance. Acta Palaeontol. Pol. 2019, 64, 779–786. [Google Scholar] [CrossRef]
- Ho, S.Y.W.; Phillips, M.J. Accounting for calibration uncertainty in phylogenetic estimation of evolutionary divergence times. Syst. Biol. 2009, 58, 367–380. [Google Scholar] [CrossRef] [Green Version]
- Claramunt, S.; Cracraft, J. A new time tree reveals earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 1, e1501005. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Meng, G.L.; Li, Y.Y.; Yang, C.T.; Liu, S.L. MitoZ: A toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [Green Version]
- Kück, P.; Meid, S.A.; Groß, C.; Wägele, J.W.; Misof, B. AliGROOVE–Visualization of heterogeneous sequence divergence within multiple sequence alignments and detection of inflated branch support. BMC Bioinform. 2014, 15, 294. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Trifinopoulos, J.; Nguyen, L.T.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast-online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goloboff, P.A.; Catalano, S.A. TNT Version 1.5, Including a full implementation of phylogenetic morphometrics. Cladistics 2016, 32, 221–238. [Google Scholar] [CrossRef]
- Kishino, H.; Hasegawa, M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in Hominoidea. J. Mol. Evol. 1989, 29, 170–179. [Google Scholar] [CrossRef]
- Shimodaira, H.; Hasegawa, M. Multiple comparisons of log-likelihoods with applications to phylogenetic inference. Mol. Biol. Evol. 1999, 16, 1114–1116. [Google Scholar] [CrossRef] [Green Version]
- Shimodaira, H. An approximately unbiased test of phylogenetic tree selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishino, H.; Miyata, T.; Hasegawa, M. Maximum likelihood inference of protein phylogeny and the origin of chloroplasts. J. Mol. Evol. 1990, 31, 151–160. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| BI | ML | MP | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Dataset | PR | P12R | NPR | NP12R | PR | P12R | NPR | NP12R | PR | P12R | NPR | NP12R | |||||||||
| Partition | N | G | C | N | G | C | N | G | C | N | G | C | |||||||||
| Schizomantodea | 0.98 | 0.97 | 0.97 | 0.93 | 0.92 | NS | 0.92 | 0.95 | 1 | 0.92 | 0.97 | NS | 0.99 | 0.99 | 1.00 | 0.98 | 400 | 333 | 457 | 381 | |
| Heteromantodea * | 0.97 | 0.96 | 0.99 | 0.95 | 1.00 | 0.99 | 1.00 | 0.98 | 0.91 | 1.00 | 0.99 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | NS | NS | NS | NS | |
| Amerimantodea | NS | NS | 0.44 | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | |
| Mantidea * | 0.47 | NS | 0.9 | 0.48 | NS | NS | NS | NS | NS | NS | NS | 0.49 | NS | NS | NS | NS | NS | NS | NS | NS | |
| Promantidea * | 1.00 | 0.96 | 1.00 | 1.00 | 0.99 | 0.99 | 0.99 | 0.98 | 0.91 | 0.96 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | NS | NS | NS | NS | |
| Haanioidea | NS | 0.42 | NS | 0.6 | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | NS | 0.58 | NS | NS | NS | NS | |
| Nanomantoidea | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 22 | 22 | 17 | 20 | |
| Eremiaphiloidea | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 37 | 25 | 42 | 24 | |
| Chroicopteroidea | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 61 | 32 | 66 | 39 | |
| Hymenopoidea | 1.00 | 1.00 | 1.00 | 1.00 | NS | 0.93 | NS | 0.91 | 0.85 | 0.84 | 0.50 | 0.91 | NS | 0.94 | 1.00 | 0.94 | 27 | NS | 28 | 14 | |
| MHEC | 0.97 | 0.96 | 0.98 | 0.92 | NS | 0.50 | NS | 0.62 | 0.94 | 0.89 | 0.72 | 0.85 | 0.57 | 0.66 | 0.61 | 0.90 | NS | NS | 16 | 7 | |
| MECE | 0.85 | 0.85 | 0.98 | 0.99 | NS | NS | NS | NS | NS | NS | NS | NS | NS | 0.78 | NS | NS | 34 | NS | 7 | 13 | |
| GNC | 0.97 | 0.52 | 0.99 | 0.95 | NS | NS | NS | NS | NS | NS | NS | 0.89 | NS | NS | NS | NS | NS | NS | NS | NS | |
| GMC * | 1.00 | NS | 1.00 | 1.00 | NS | NS | NS | NS | NS | NS | NS | 1.00 | NS | NS | NS | NS | NS | NS | NS | NS | |
| Dataset | Tree | logL | deltaL | bp-RELL | p-KH | p-SH | p-WKH | p-WSH | c-ELW | p-AU |
|---|---|---|---|---|---|---|---|---|---|---|
| NGPCG12RNA | PCGRNA | −230,935.7142 | 78.335 | 0.0007− | 0.0084− | 0.0318− | 0.0084− | 0.0222− | 0.000728− | 0.00427 |
| PCG12RNA | −230,888.8728 | 31.494 | 0.131+ | 0.211+ | 0.337+ | 0.211+ | 0.391+ | 0.131+ | 0.196+ | |
| NGPCGRNA | −230,857.3788 | 0 | 0.497+ | 0.561+ | 1+ | 0.561+ | 0.818+ | 0.497+ | 0.634+ | |
| NGPCG12RNA | −230,861.4544 | 4.0756 | 0.371+ | 0.439+ | 0.7+ | 0.439+ | 0.689+ | 0.371+ | 0.541+ | |
| NGPCGRNA | PCGRNA | −452,278.8796 | 104.43 | 0.0031− | 0.004− | 0.0171− | 0.004− | 0.0095− | 0.00312− | 0.00735− |
| PCG12RNA | −452,296.1574 | 121.71 | 0.0044− | 0.0077− | 0.0098− | 0.0077− | 0.0171− | 0.0046− | 0.00849− | |
| NGPCGRNA | −452,174.449 | 0 | 0.985+ | 0.993+ | 1+ | 0.993+ | 1+ | 0.985+ | 0.996+ | |
| NGPCG12RNA | −452,253.6602 | 79.211 | 0.0075− | 0.0071− | 0.0704+ | 0.0071− | 0.0168− | 0.00728− | 0.0114− | |
| PCG12RNA | PCGRNA | −204,317.2383 | 34.324 | 0.0839+ | 0.0858+ | 0.206+ | 0.0858+ | 0.19+ | 0.0845+ | 0.0893+ |
| PCG12RNA | −204,282.914 | 0 | 0.915+ | 0.914+ | 1+ | 0.914+ | 0.987+ | 0.915+ | 0.928+ | |
| NGPCGRNA | −204,369.6892 | 86.775 | 0.0005− | 0.0028− | 0.0029− | 0.0028− | 0.007− | 0.000449− | 0.0027− | |
| NGPCG12RNA | −204,361.0502 | 78.136 | 0.0005− | 0.0013− | 0.0068− | 0.0013− | 0.0027− | 0.000508− | 0.000323− | |
| PCGRNA | PCGRNA | −422,801.9202 | 0 | 0.755+ | 0.79+ | 1+ | 0.79+ | 0.945+ | 0.756+ | 0.844+ |
| PCG12RNA | −422,828.4959 | 26.576 | 0.211+ | 0.21+ | 0.384+ | 0.21+ | 0.386+ | 0.21+ | 0.243+ | |
| NGPCGRNA | −422,848.051 | 46.131 | 0.0343− | 0.0503+ | 0.162+ | 0.0503+ | 0.116+ | 0.0346− | 0.0621+ | |
| NGPCG12RNA | −422,920.0939 | 118.17 | 0− | 0.0003− | 0.0004− | 0.0003− | 0.0016− | 1.21 × 10−7− | 5.1 × 10−5− | |
| PCGRNA | Schwarz and Roy, 2019 1 | −417,699.9417 | 0 | 0.996+ | 0.996+ | 1+ | 0.996+ | 0.996+ | 0.996+ | 0.996+ |
| Svenson and Rodrigues, 2017 2 | −417,803.5686 | 103.63 | 0.0041− | 0.0043− | 0.0043− | 0.0043− | 0.0043− | 0.0041− | 0.00378− | |
| PCG12RNA | Schwarz and Roy, 2019 1 | −202,107.3782 | 0 | 0.994+ | 0.994+ | 1+ | 0.994+ | 0.994+ | 0.994+ | 0.995+ |
| Svenson and Rodrigues, 2017 2 | −202,190.6792 | 83.301 | 0.0063− | 0.0061− | 0.0061− | 0.0061− | 0.0061− | 0.00617− | 0.00514− | |
| NGPCGRNA | Schwarz and Roy, 2019 1 | −447,001.7467 | 0 | 0.893+ | 0.895+ | 1+ | 0.895+ | 0.895+ | 0.893+ | 0.889+ |
| Svenson and Rodrigues, 2017 2 | −447,056.1321 | 54.385 | 0.107+ | 0.105+ | 0.105+ | 0.105+ | 0.105+ | 0.107+ | 0.111+ | |
| NGPCG12RNA | Schwarz and Roy, 2019 1 | −228,551.765 | 0 | 0.825+ | 0.825+ | 1+ | 0.825+ | 0.825+ | 0.825+ | 0.827+ |
| Svenson and Rodrigues, 2017 2 | −228,585.8363 | 34.071 | 0.175+ | 0.175+ | 0.175+ | 0.175+ | 0.175+ | 0.175+ | 0.173+ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Liu, Y.; Liu, Q.; Tian, L.; Li, H.; Song, F.; Cai, W. Exploring the Mitogenomes of Mantodea: New Insights from Structural Diversity and Higher-Level Phylogenomic Analyses. Int. J. Mol. Sci. 2023, 24, 10570. https://doi.org/10.3390/ijms241310570
Liu Q, Liu Y, Liu Q, Tian L, Li H, Song F, Cai W. Exploring the Mitogenomes of Mantodea: New Insights from Structural Diversity and Higher-Level Phylogenomic Analyses. International Journal of Molecular Sciences. 2023; 24(13):10570. https://doi.org/10.3390/ijms241310570
Chicago/Turabian StyleLiu, Qinpeng, Yingqi Liu, Qiaoqiao Liu, Li Tian, Hu Li, Fan Song, and Wanzhi Cai. 2023. "Exploring the Mitogenomes of Mantodea: New Insights from Structural Diversity and Higher-Level Phylogenomic Analyses" International Journal of Molecular Sciences 24, no. 13: 10570. https://doi.org/10.3390/ijms241310570
APA StyleLiu, Q., Liu, Y., Liu, Q., Tian, L., Li, H., Song, F., & Cai, W. (2023). Exploring the Mitogenomes of Mantodea: New Insights from Structural Diversity and Higher-Level Phylogenomic Analyses. International Journal of Molecular Sciences, 24(13), 10570. https://doi.org/10.3390/ijms241310570