
Comparative Transcriptome Profiling of CMS-D2 and CMS-D8 Systems Characterizes Fertility Restoration Genes Network in Upland Cotton

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
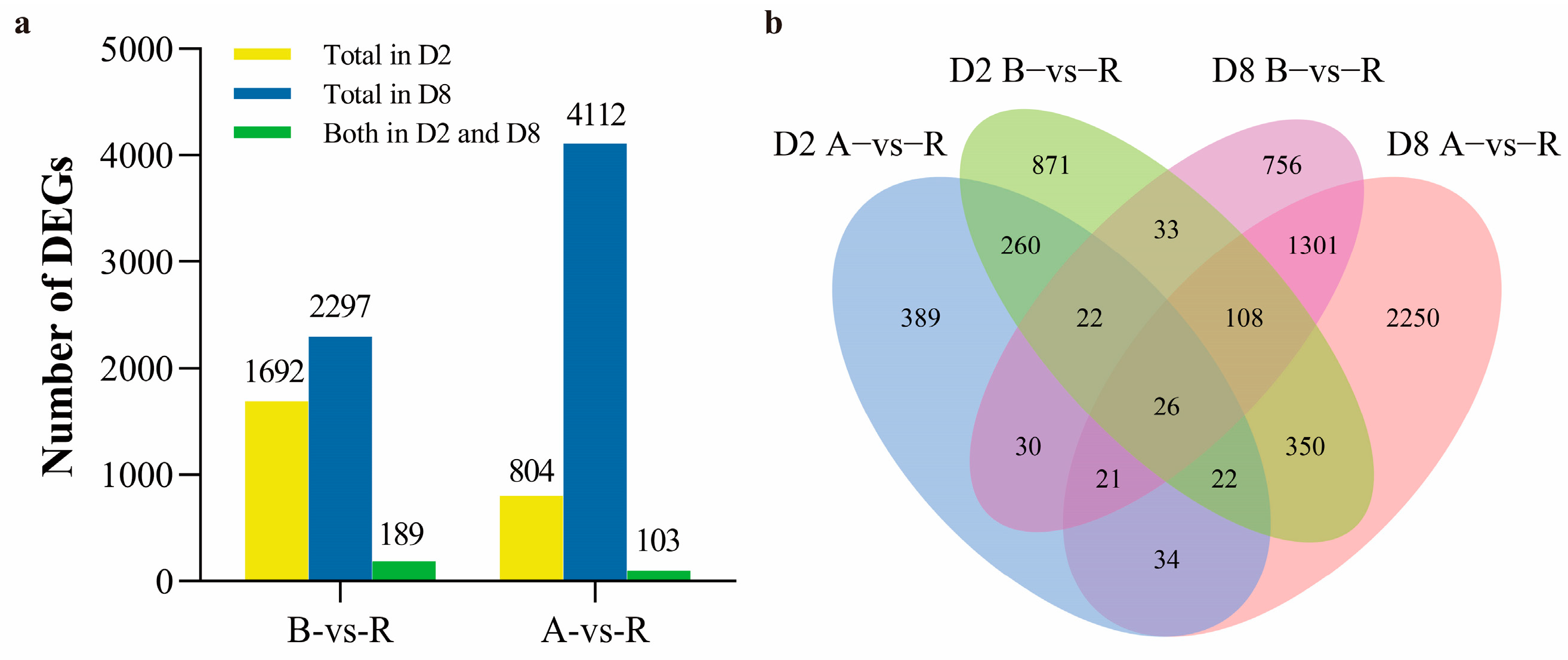
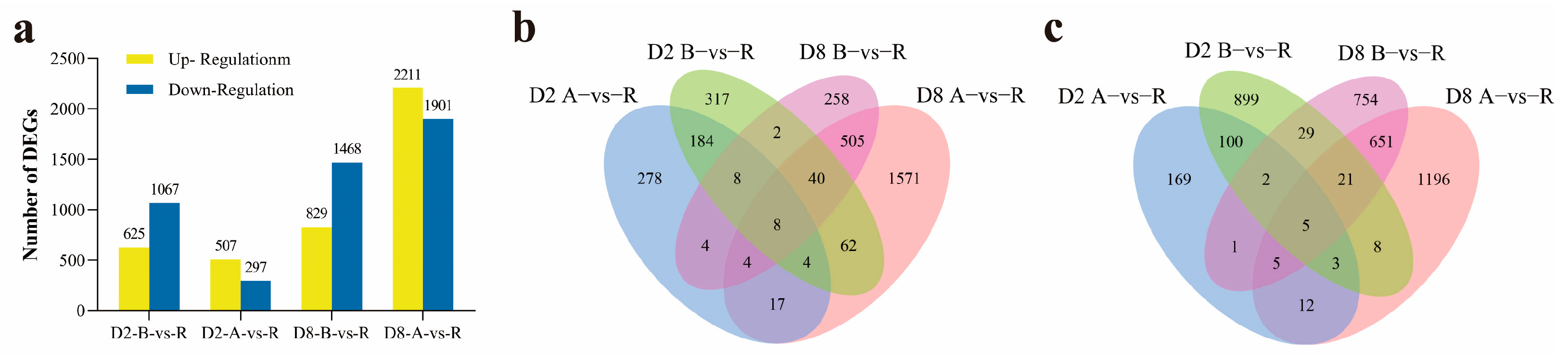
2.1. Differentially Expressed Genes (DEGs) Analysis in Both CMS-D2 and CMS-D8 Systems
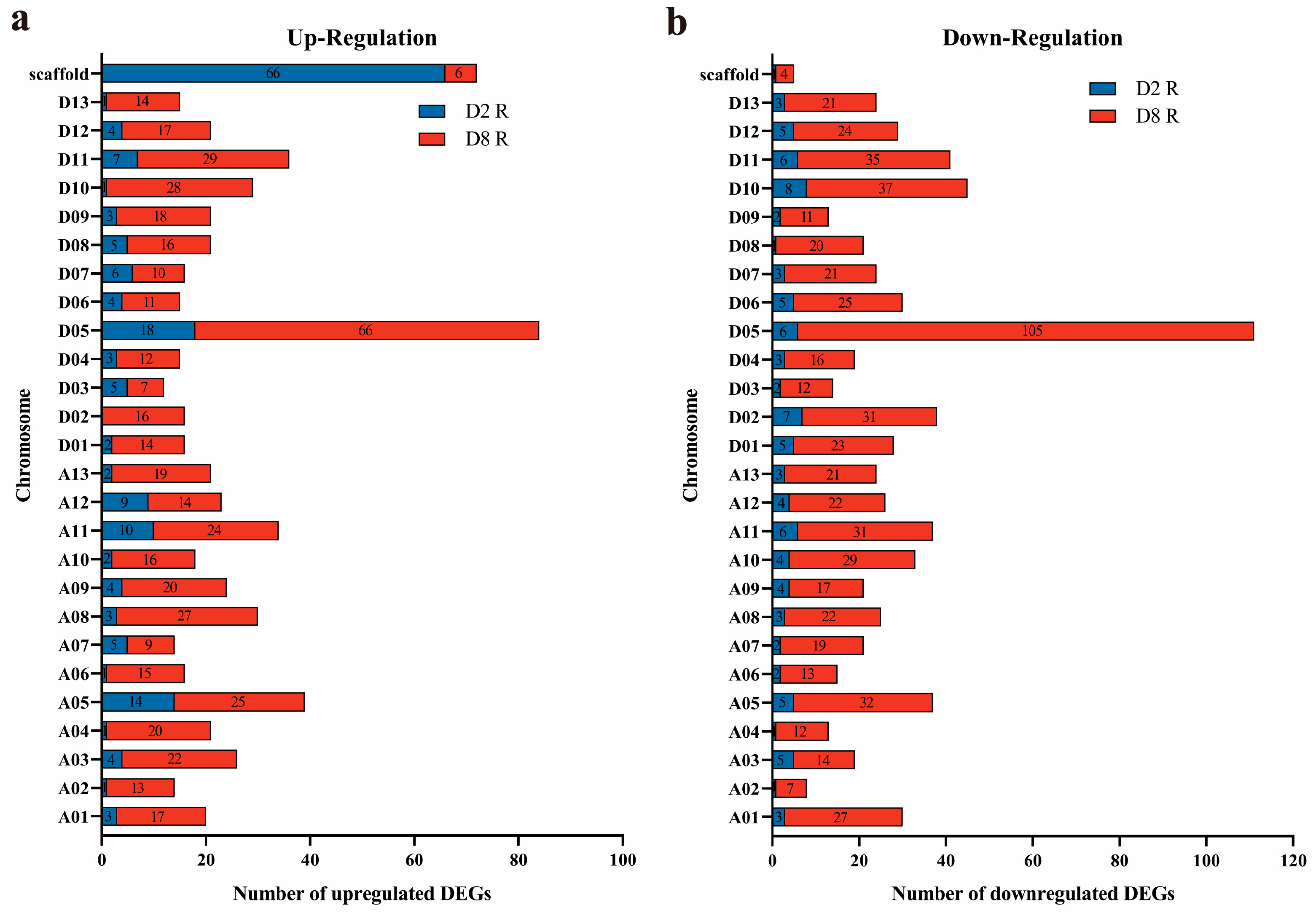
2.2. Chromosome Co-Location Analysis of Specifical DEGs of Both CMS-D2 and CMS-D8 R Lines
2.3. Gene Ontology (GO) Function Enrichment Analysis of Specific DEGs in the R Line
2.4. KEGG Pathway Enrichment Analysis of Specific DEGs in Restorer Line
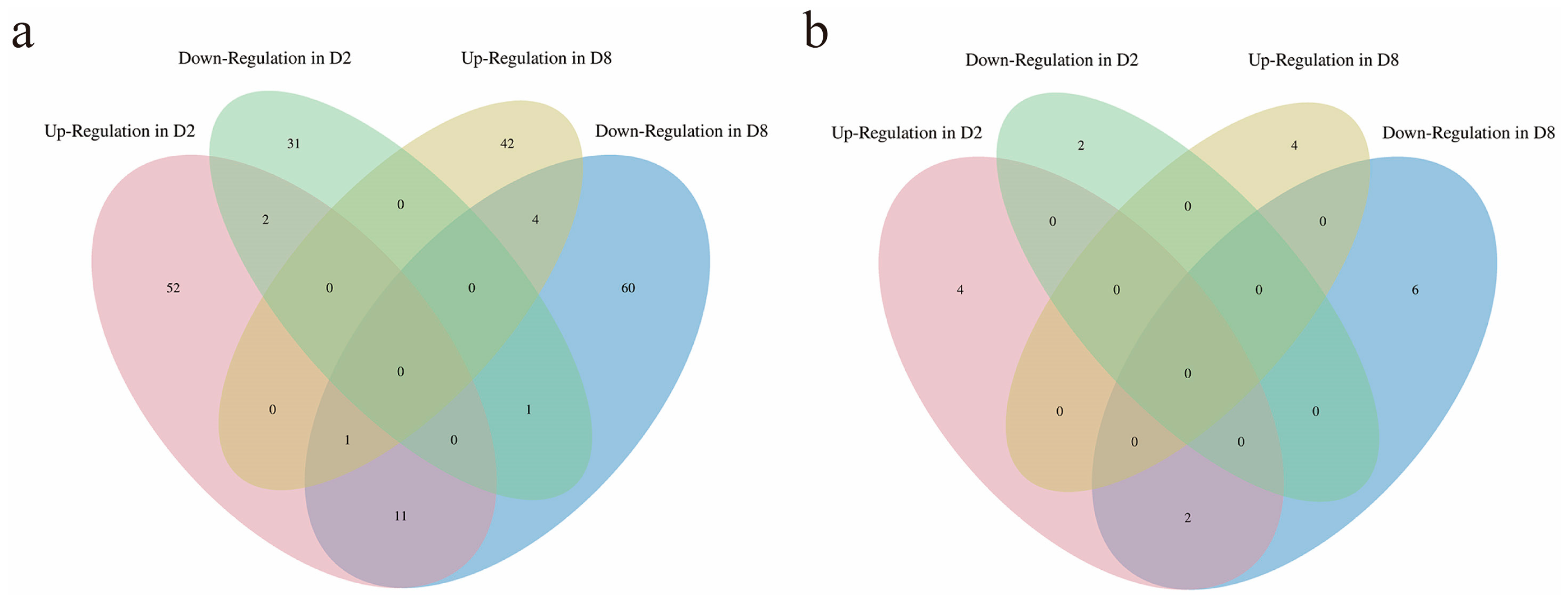
2.5. Co-Localization Analysis of the Up-Regulated DEGs in Two R Lines Located on Chr_D05 and Identification of Candidate Restoration Genes
3. Discussion
3.1. Overview of Differential Transcriptome Dynamics in Two Cotton CMS Systems
3.2. Differential Gene Regulation Network Probably Influences the Retrieval of Pollen Fertility for CMS-D2 and CMS-D8 Cotton
3.3. Discovery of Gene Regulatory Network Located in the Target Region of Fertility Restorer Genes in CMS Cotton
4. Materials and Methods
4.1. Plant Materials
4.2. RNA Extraction, Sequencing Library Construction, and Data Analysis
4.3. Differential Expression and GO and KEGG (Kyoto Encyclopedia of Genes and Genomes) Enrichment Analysis
4.4. Chromosome Location of DEGs on D05 in the Target Region of Restorer Genes
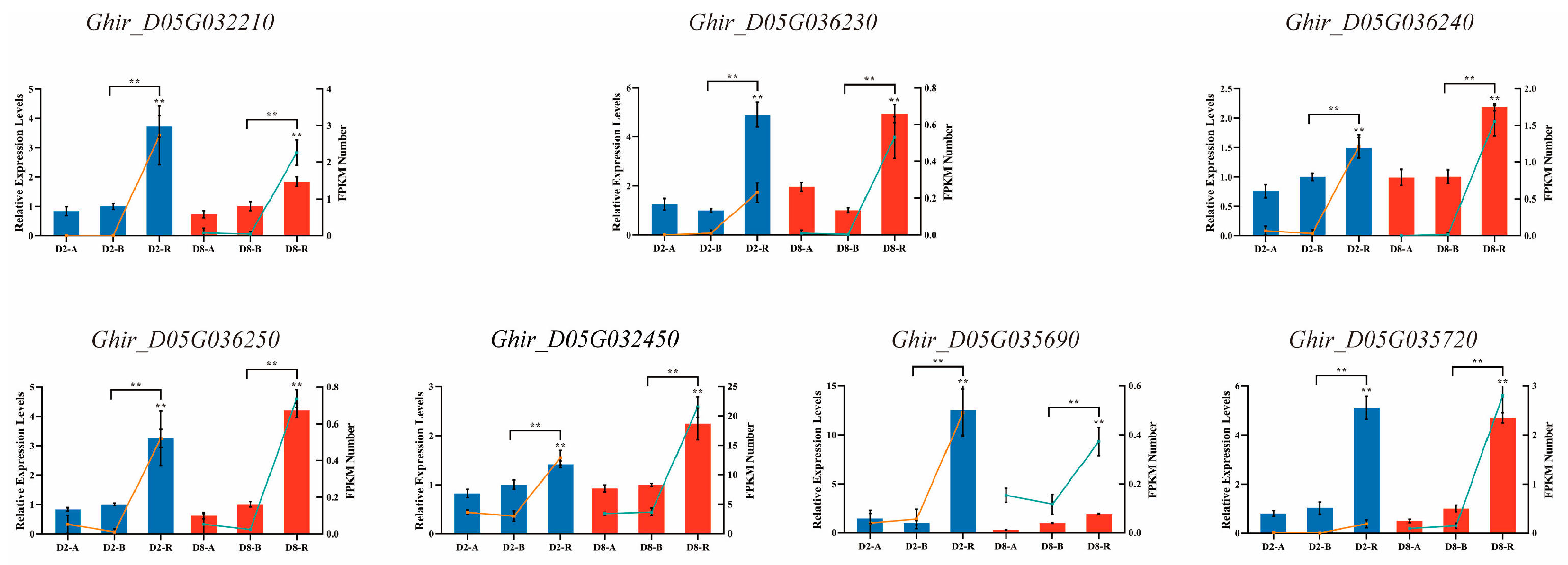
4.5. qRT-PCR Validation for DEGs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Shahzad, K.; Zhang, X.; Guo, L.; Qi, T.; Bao, L.; Zhang, M.; Zhang, B.; Wang, H.; Tang, H.; Qiao, X.; et al. Comparative Transcriptome Analysis between Inbred and Hybrids Reveals Molecular Insights into Yield Heterosis of Upland Cotton. BMC Plant Biol. 2020, 20, 239. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Xiao, M.; Hayward, A.; Jiang, G.; Zhu, L.; Zhou, Q.; Li, J.; Zhang, M. What is Crop Heterosis: New Insights into an Old Topic. J. Appl. Genet. 2015, 56, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Fan, S.; Wang, H.; Wei, H.; Pang, C. Progresses in Research on Cotton High Yield Breeding in China. Sci. Agric. Sin. 2016, 49, 3465–3476. [Google Scholar]
- Xing, C.; Guo, L.; Li, W.; Wu, J.; Yang, D.; Qi, T.; Ma, X.; Zhang, X. Ten-year Achievements and Future Development of Cotton Heterosis Utilization. Cotton Sci. 2017, 29, 28–36. [Google Scholar]
- Kubo, T.; Kitazaki, K.; Matsunaga, M.; Kagami, H.; Mikami, T.J.C.R. Male Sterility-Inducing Mitochondrial Genomes: How Do They Differ? Crit. Rev. Plant Sci. 2011, 30, 378–400. [Google Scholar] [CrossRef]
- Horn, R.; Gupta, K.J.; Colombo, N. Mitochondrion Role in Molecular Basis of Cytoplasmic Male Sterility. Mitochondrion 2014, 19 Pt B, 198–205. [Google Scholar] [CrossRef]
- Budar, F.; Pelletier, G. Male Sterility in Plants: Occurrence, Determinism, Significance And Use. Comptes Rendus De L’academie Sciences. Ser. III Sci. La Vie 2001, 324, 543–550. [Google Scholar] [CrossRef]
- Zheng, H.; Wang, R.; Jiang, Q.; Zhang, D.; Mu, R.; Xu, Y.; Nnaemeka, V.E.; Mei, J.; Zhao, Y.; Cai, F.; et al. Identification and Functional Analysis of a Pollen Fertility-Associated Gene GhGLP4 of Gossypium hirsutum L. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2021, 134, 3237–3247. [Google Scholar] [CrossRef]
- Han, Z.; Wang, J.; Shen, G.; Zhao, F.; Wang, Z.; Li, R. Research and Application of Cytoplasmic-nulear Male Sterility and Fertility Restoration in Cotton. J. China Agric. Univ. 2011, 16, 36–41. [Google Scholar]
- Meyer, V.G. Male Sterility from Gossypium harknessii. J. Hered. 1975, 66, 23–27. [Google Scholar] [CrossRef]
- Weaver, D.B., Jr. Inheritance of Pollen Fertility Restoration in Cytoplasmic Male-Sterile Upland Cotton. Crop Sci. 1977, 17, 497–499. [Google Scholar] [CrossRef]
- Stewart, J. A New Cytoplasmic Male Sterile and Restorer for Cotton. In Proceedings of the Beltwide Cotton Conferences; National Cotton Council: Memphis, TN, USA, 1992; p. 610. [Google Scholar]
- Stewart, J. Observations on Fertility Reatoration in D8 CMS Cotton. In Proceedings of the Beltwide Cotton Conference, San Antonio, TX, USA, 4–7 January 1995; p. 507. [Google Scholar]
- Black, C.S.J. Morphological Description of D8 CMS Cotton. In Proceedings of the Beltwide Cotton Conference, San Antonio, TX, USA, 4–7 January 1995; p. 507. [Google Scholar]
- Stewart, J.; Zhang, J.; Dugger, C.; Richter, D. Cytoplasmic Influence on the Inheritance of the D8 Restorer Gene. In Proceedings of the Beltwide Cotton Conference, Nashville, TN, USA, 9–12 January 1996; pp. 622–623. [Google Scholar]
- Zhang, J.; Stewart, J.M. CMS-D8 Restoration in Cotton is Conditioned by one Dominant Gene. Crop Sci. 2001, 41, 283–288. [Google Scholar] [CrossRef]
- Zhang, J.; Stewart, J.M. Inheritance and Genetic Relationships of the D8 and D2-2 Restorer Genes for Cotton Cytoplasmic Male Sterility. Theor. Appl. Genet. 2001, 41, 289–294. [Google Scholar] [CrossRef]
- Mutz, K.-O.; Heilkenbrinker, A.; Lönne, M.; Walter, J.-G.; Stahl, F. Transcriptome Analysis Using Next-Generation Sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef]
- Wei, C.; Li, M.; Qin, J.; Xu, Y.; Zhang, Y.; Wang, H. Transcriptome Analysis Reveals the Effects of Grafting on Sweetpotato Scions during the Full Blooming Stages. Genes Genom. 2019, 41, 895–907. [Google Scholar] [CrossRef]
- Thirugnanasambandam, P.P.; Mason, P.J.; Hoang, N.V.; Furtado, A.; Botha, F.C.; Henry, R.J. Analysis of the Diversity and Tissue Specificity of Sucrose Synthase Genes in the Long Read Rranscriptome of Sugarcane. BMC Plant Biol. 2019, 19, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiao, D.; Yang, C.; Chen, J.; Guo, Y.; Li, Y.; Niu, S.; Cao, K.; Chen, Z. Comprehensive Identification of the Full-Length Transcripts and Alternative Splicing Related to the Secondary Metabolism Pathways in the Tea Plant (Camellia sinensis). Sci. Rep. 2019, 9, 2709. [Google Scholar] [CrossRef] [PubMed]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D. RNA-seq Analysis of Short Fiber Mutants Ligon-lintless-1 (Li 1) and -2 (Li 2) Revealed Important Role of Aquaporins in Cotton (Gossypium hirsutum L.) Fiber Elongation. BMC Plant Biol. 2015, 15, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.S.; Fang, D.D.; Thyssen, G.N.; Delhom, C.D.; Liu, Y.; Kim, H.J. Comparative Fiber Property and Transcriptome Analyses Reveal Key Genes Potentially Related to High Fiber Strength in Cotton (Gossypium hirsutum L.) Line MD52ne. BMC Plant Biol. 2016, 16, 36. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Zhu, L.; Tu, L.; Liu, L.; Yuan, D.; Jin, L.; Long, L.; Zhang, X. Lignin Metabolism Has a Central Role in the Resistance of Cotton to the Wilt Fungus Verticillium Dahliae as Revealed by RNA-Seq-dependent Rranscriptional Analysis and Histochemistry. J. Exp. Bot. 2011, 62, 5607–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.; Pang, C.; Fan, S.; Song, M.; Wei, H.; Yu, S. Global Analysis of the Gossypium hirsutum L. Transcriptome during Leaf Senescence by RNA-Seq. BMC Plant Biol. 2015, 15, 43. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, H.; Rodriguez-Uribe, L.; Xu, J.; Zhang, J. Transcriptome Analysis of Cytoplasmic Male Sterility and Restoration in CMS-D8 Cotton. Plant Cell Rep. 2013, 32, 1531–1542. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, M.; Zhang, B.; Zhang, X.; Guo, L.; Qi, T.; Wang, H.; Zhang, J.; Xing, C. Genome-wide Comparative Transcriptome Analysis of CMS-D2 and its Maintainer and Restorer Lines in Upland Cotton. BMC Genom. 2017, 18, 454. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Zhang, X.; Liu, G.; Guo, L.; Qi, T.; Zhang, M.; Li, X.; Wang, H.; Tang, H.; Qiao, X.; et al. A Combined Small RNA and Transcriptome Sequencing Analysis Reveal Regulatory Roles of miRNAs during Anther Development of Upland Cotton Carrying Cytoplasmic Male Sterile Gossypium harknessii (D2) Cytoplasm. BMC Plant Biol. 2018, 18, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Wu, Y.; Zhang, M.; Zhang, J.; Stewart, J.M.; Xing, C.; Wu, J.; Jin, S. Transcriptome, Cytological and Biochemical Analysis of Cytoplasmic Male Sterility and Maintainer Line in CMS-D8 Cotton. Plant Mol. Biol. 2018, 97, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Guo, W.; Zhu, X.; Zhang, T. Inheritance and Fine Mapping of Fertility Restoration for Cytoplasmic Male Sterility in Gossypium hirsutum L. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2003, 106, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.D.; Stewart, J.M.; Zhang, J.F. STS Markers Linked to the Rf1 Fertility Restorer Gene of Cotton. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2005, 110, 237–243. [Google Scholar] [CrossRef]
- Yin, J.; Guo, W.; Yang, L.; Liu, L.; Zhang, T. Physical Mapping of the Rf1 Fertility-Restoring Gene to a 100 kb Region in Cotton. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2006, 112, 1318–1325. [Google Scholar] [CrossRef]
- Yang, L. Map-Based Cloning of Fertility Restoring Gene of CMS and Analysis of PPR Gene Family in Cotton; Nanjing Agricultural University: Nanjing, China, 2009. [Google Scholar]
- Wu, J.; Cao, X.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Zhang, J.; Xing, C.J.M.B. Development of a Candidate Gene Marker for Rf1 Based on a PPR Gene in Cytoplasmic Male Sterile CMS-D2 Upland Cotton. Mol. Breed. 2014, 34, 231–240. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, M.; Zhang, X.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Zhang, J.; Xing, C.J.E. Development of InDel Markers for the Restorer Gene Rf1 and Assessment of their Utility for Marker-Assisted Selection in Cotton. Euphytica 2017, 213, 1–8. [Google Scholar] [CrossRef]
- Zhang, J.; Stewart, J.M. Identification of molecular markers linked to the fertility restorer genes for CMS-D8 in Cotton. Crop Sci. 2004, 44, 1209–1217. [Google Scholar] [CrossRef]
- Wang, F.; Stewart, J.M.; Zhang, J. Molecular Markers Linked to the Rf2 Fertility Restorer Gene in cotton. Genome 2007, 50, 818–824. [Google Scholar] [CrossRef]
- Feng, J.; Zhu, H.; Zhang, M.; Zhang, X.; Guo, L.; Qi, T.; Tang, H.; Wang, H.; Qiao, X.; Zhang, B.; et al. Development and Utilization of an InDel Marker Linked to the Fertility Restorer Genes of CMS-D8 and CMS-D2 in Cotton. Mol. Biol. Rep. 2020, 47, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Zhang, X.; Zhang, M.; Guo, L.; Qi, T.; Tang, H.; Zhu, H.; Wang, H.; Qiao, X.; Xing, C.; et al. Physical Mapping and InDel Marker Development for the Restorer Gene Rf(2) in Cytoplasmic Male Sterile CMS-D8 Cotton. BMC Genom. 2021, 22, 24. [Google Scholar] [CrossRef]
- Li, T.; Zhang, X.; Guo, L.; Qi, T.; Tang, H.; Wang, H.; Qiao, X.; Zhang, M.; Zhang, B.; Feng, J.; et al. Single-Molecule Real-Time Transcript Sequencing of Developing Cotton Anthers Facilitates Genome Annotation and Fertility Restoration Candidate Gene Discovery. Genomics 2021, 113, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Li, Y.; Zhang, J.; Zhang, M.; Zhang, X.; Shahzad, K.; Guo, L.; Qi, T.; Tang, H.; Wang, H.; et al. Transcript Complexity and New Insights of Restorer Line in CMS-D8 Cotton Through Full-Length Transcriptomic Analysis. Front. Plant Sci. 2022, 13, 930131. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; He, M.; Wu, H. Differential Mantle Transcriptomics and Characterization of Growth-Related Genes in the Diploid and Triploid Pearl Oyster Pinctada Fucata. Mar. Genom. 2017, 33, 31–38. [Google Scholar] [CrossRef]
- Hamid, R.; Marashi, H.; Tomar, R.S.; Malekzadeh Shafaroudi, S.; Sabara, P.H. Transcriptome Analysis Identified Aberrant Gene Expression in Pollen Developmental Pathways Leading to CGMS in Cotton (Gossypium hirsutum L.). PLoS ONE 2019, 14, e0218381. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, X.; Guo, L.; Qi, T.; Liu, G.; Feng, J.; Shahzad, K.; Zhang, B.; Li, X.; Wang, H.; et al. Single-base Resolution Methylome of Cotton Cytoplasmic Male Sterility System Reveals Epigenomic Changes in Response to High-Temperature Stress during Anther Development. J. Exp. Bot. 2020, 71, 951–969. [Google Scholar]
- Yang, C.; Vizcay-Barrena, G.; Conner, K.; Wilson, Z.A. MALE STERILITY1 is Required for Tapetal Development and Pollen Wall Biosynthesis. Plant Cell 2007, 19, 3530–3548. [Google Scholar] [CrossRef] [Green Version]
- He, M.; Ding, N.Z. Plant Unsaturated Fatty Acids: Multiple Roles in Stress Response. Front. Plant Sci. 2020, 11, 562785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, M.; Guo, L.; Qi, T.; Tang, H.; Li, Y.; Zuo, Z.; Shahzad, K.; Feng, J.; Zang, R.; et al. Integrated Analysis of Metabolome and Transcriptome Reveals the Cytoplasmic Effects of CMS-D2 on Pollen Fertility Resulting from Disrupted Lipid Metabolism. Front. Plant Sci. 2022, 13, 998203. [Google Scholar] [CrossRef]
- Alvarez-Buylla, E.R.; Benítez, M.; Corvera-Poiré, A.; Cador, Á.C.; de Folter, S.; de Buen, A.G.; Garay-Arroyo, A.; García-Ponce, B.; Jaimes-Miranda, F.; Pérez-Ruiz, R.V. Flower Development. Arab. Book/Am. Soc. Plant Biol. 2010, 8, e0127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, X.; Wu, S.; Li, Z.; An, X.; Tian, Y. Lipid Metabolism: Critical Roles in Male Fertility and Other Aspects of Reproductive Development in Plants. Mol. Plant 2020, 13, 955–983. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Lam, P.Y.; Lui, A.C.W.; Zhu, F.Y.; Chen, M.X.; Liu, H.; Zhang, J.; Lo, C. Flavonoids are Indispensable for Complete Male Fertility in Rice. J. Exp. Bot. 2020, 71, 4715–4728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, X.; Guo, L.; Qi, T.; Wang, H.; Tang, H.; Qiao, X.; Shahzad, K.; Xing, C.; Wu, J. Genome-wide Analysis of Rf-PPR-like (RFL) Genes and a New InDel Marker Development for Rf1 Gene in Cytoplasmic Male Sterile CMS-D2 Upland Cotton. J. Cotton Res. 2018, 1, 1–11. [Google Scholar]
- Zhao, C.; Zhao, G.; Geng, Z.; Wang, Z.; Wang, K.; Liu, S.; Zhang, H.; Guo, B.; Geng, J. Physical Mapping and Candidate Gene Prediction of Fertility Restorer Gene of Cytoplasmic Male Sterility in Cotton. BMC Genom. 2018, 19, 6. [Google Scholar] [CrossRef]
- Wang, F.; Yue, B.; Hu, J.; Stewart, J.M.; Zhang, J. A Target Region Amplified Polymorphism Marker for Fertility Restorer Gene Rf1 and Chromosomal Localization of Rf1 and Rf2 in Cotton. Crop Sci. 2009, 49, 1602–1608. [Google Scholar] [CrossRef]
- Wang, W. Gene Cloning of ADP-Ribosylation Factor from Gosspium hirsutum L.; Southwest University: Chongqing, China, 2002. [Google Scholar]
- Ren, M.Z.; Chen, Q.J.; Zhang, R.; Guo, S.D. The Structural Characteristics, Alternative Splicing and Genetic Experession Analysis of ADP-Ribosylation-Factor 1 (arf1) in Cotton. Yi Chuan Xue Bao = Acta Genet. Sin. 2004, 31, 850–857. [Google Scholar]
- Chen, Q. Molecular Cloning of ADP-Ribosylation Factor 1 Gene (arf1) from Gossypium hirsutum and Function Analysis of Promoter; Xinjiang Agricultural University: Ürümqi, China, 2004. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps And QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing Real-time PCR Data by the Comparative C(T) Method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Rychlik, W. OLIGO 7 Primer Analysis Software. Methods Mol. Biol. 2007, 402, 35–60. [Google Scholar] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Zhang, M.; Shahzad, K.; Zhang, X.; Guo, L.; Qi, T.; Tang, H.; Wang, H.; Qiao, X.; Feng, J.; et al. Comparative Transcriptome Profiling of CMS-D2 and CMS-D8 Systems Characterizes Fertility Restoration Genes Network in Upland Cotton. Int. J. Mol. Sci. 2023, 24, 10759. https://doi.org/10.3390/ijms241310759
Song X, Zhang M, Shahzad K, Zhang X, Guo L, Qi T, Tang H, Wang H, Qiao X, Feng J, et al. Comparative Transcriptome Profiling of CMS-D2 and CMS-D8 Systems Characterizes Fertility Restoration Genes Network in Upland Cotton. International Journal of Molecular Sciences. 2023; 24(13):10759. https://doi.org/10.3390/ijms241310759
Chicago/Turabian StyleSong, Xiatong, Meng Zhang, Kashif Shahzad, Xuexian Zhang, Liping Guo, Tingxiang Qi, Huini Tang, Hailin Wang, Xiuqin Qiao, Juanjuan Feng, and et al. 2023. "Comparative Transcriptome Profiling of CMS-D2 and CMS-D8 Systems Characterizes Fertility Restoration Genes Network in Upland Cotton" International Journal of Molecular Sciences 24, no. 13: 10759. https://doi.org/10.3390/ijms241310759
APA StyleSong, X., Zhang, M., Shahzad, K., Zhang, X., Guo, L., Qi, T., Tang, H., Wang, H., Qiao, X., Feng, J., Han, Y., Xing, C., & Wu, J. (2023). Comparative Transcriptome Profiling of CMS-D2 and CMS-D8 Systems Characterizes Fertility Restoration Genes Network in Upland Cotton. International Journal of Molecular Sciences, 24(13), 10759. https://doi.org/10.3390/ijms241310759