
NIH/3T3 Fibroblasts Selectively Activate T Cells Specific for Posttranslationally Modified Collagen Type II

 ,
,  ,
, 

Abstract
:1. Introduction
2. Results
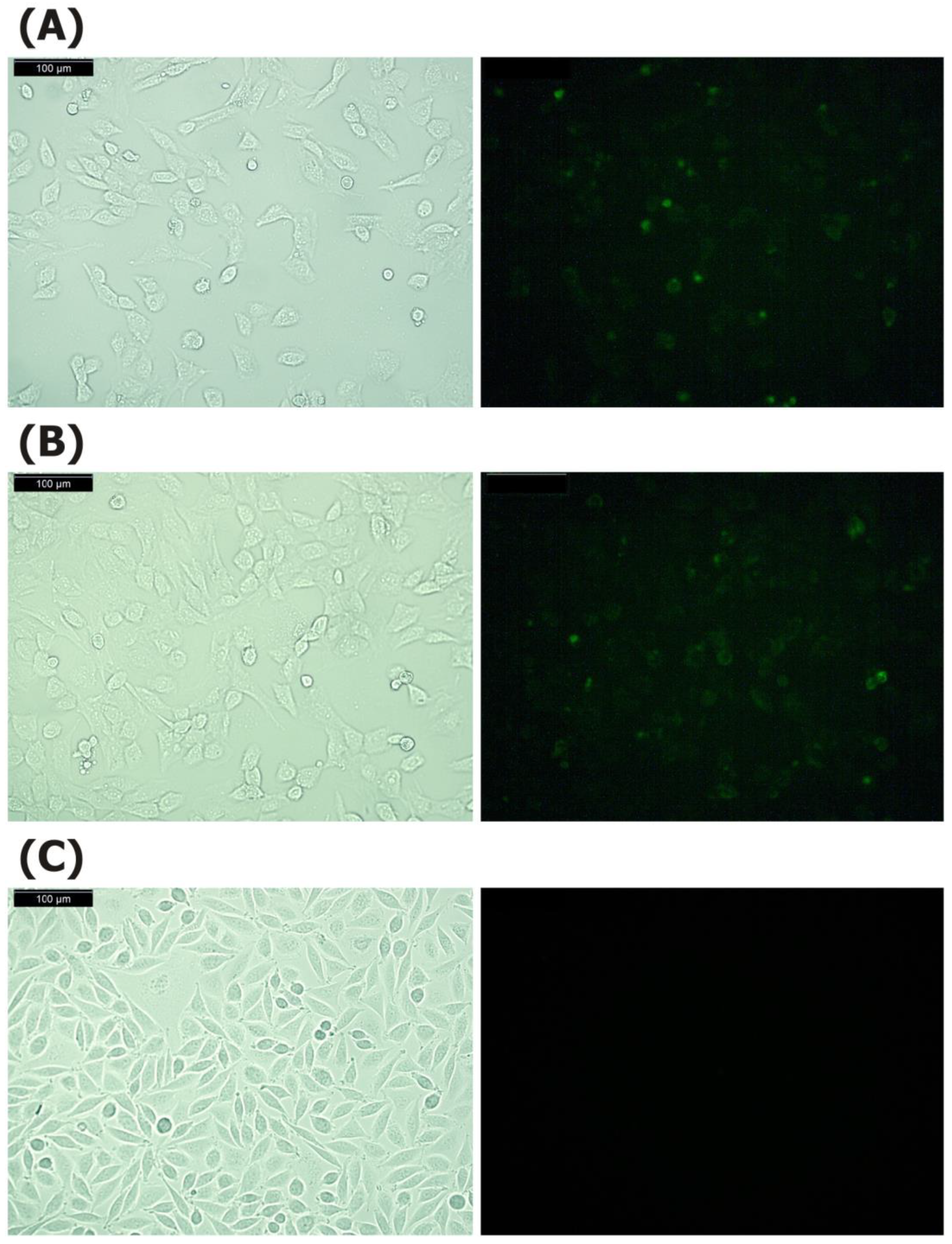
2.1. Destructive LS48 Fibroblasts, Synovial/Dermal and Thymic Fibroblasts Cannot Process and Present COL2 to T-Cells
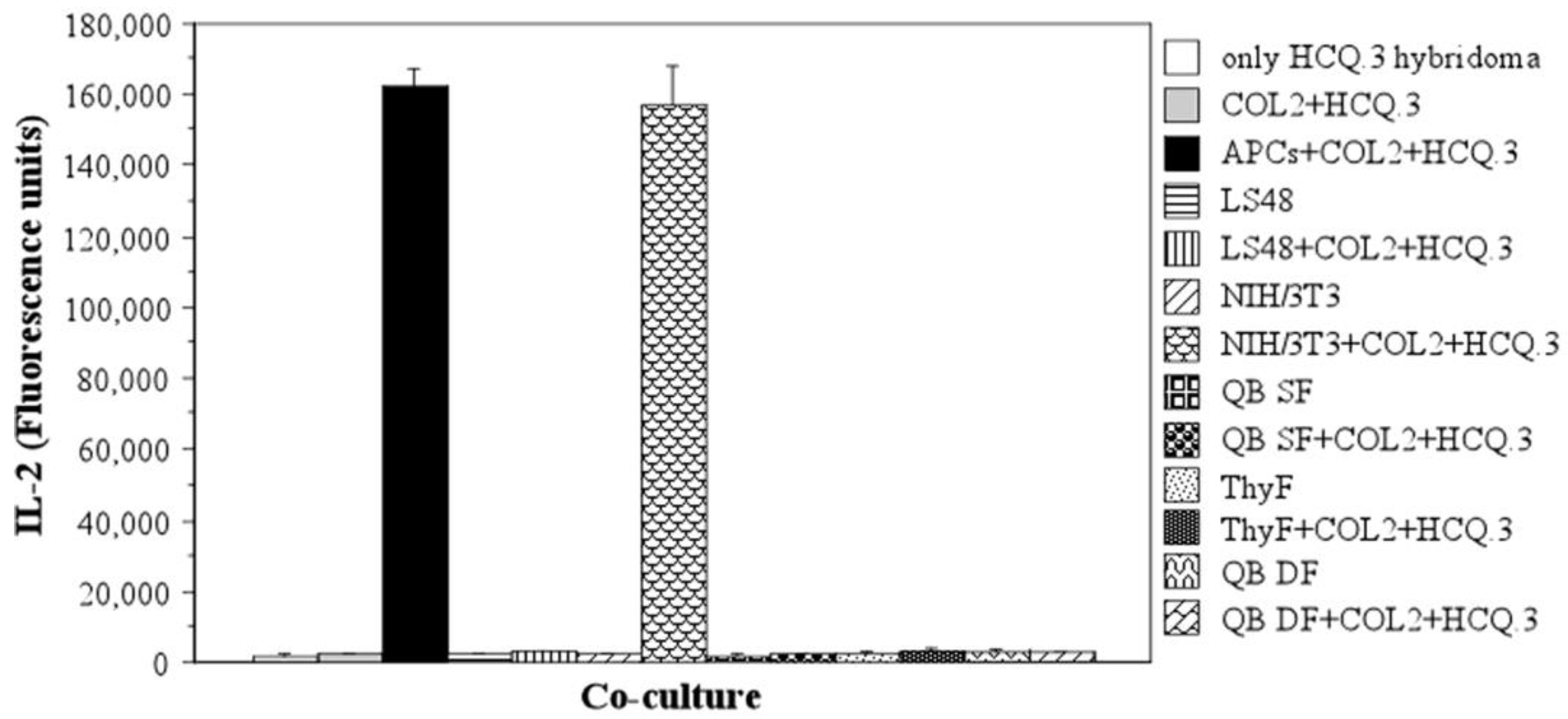
2.2. NIH/3T3 Cells Are Able to Stimulate the COL2-Specific HCQ.3 Hybridoma Even in the Absence of Antigen
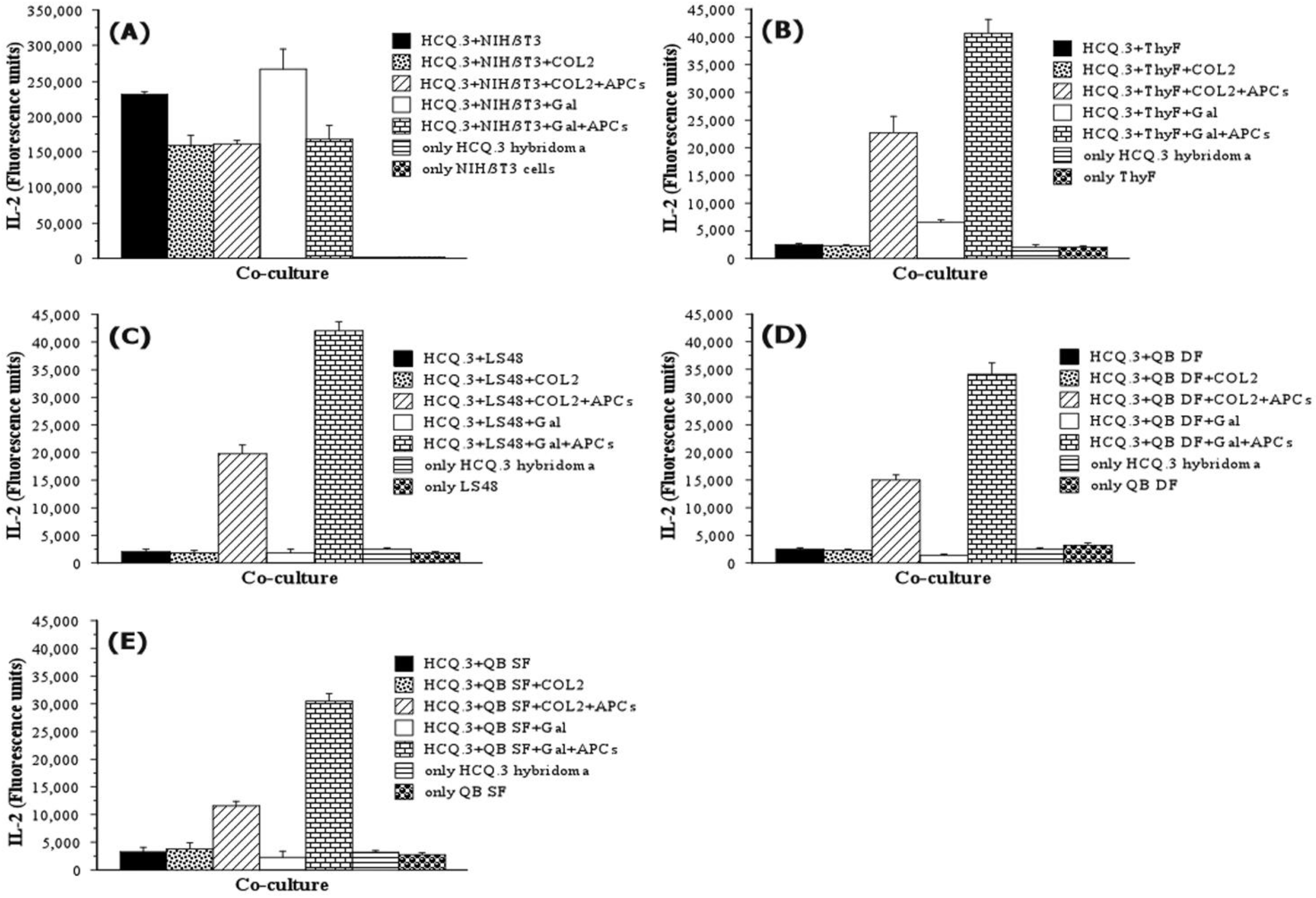
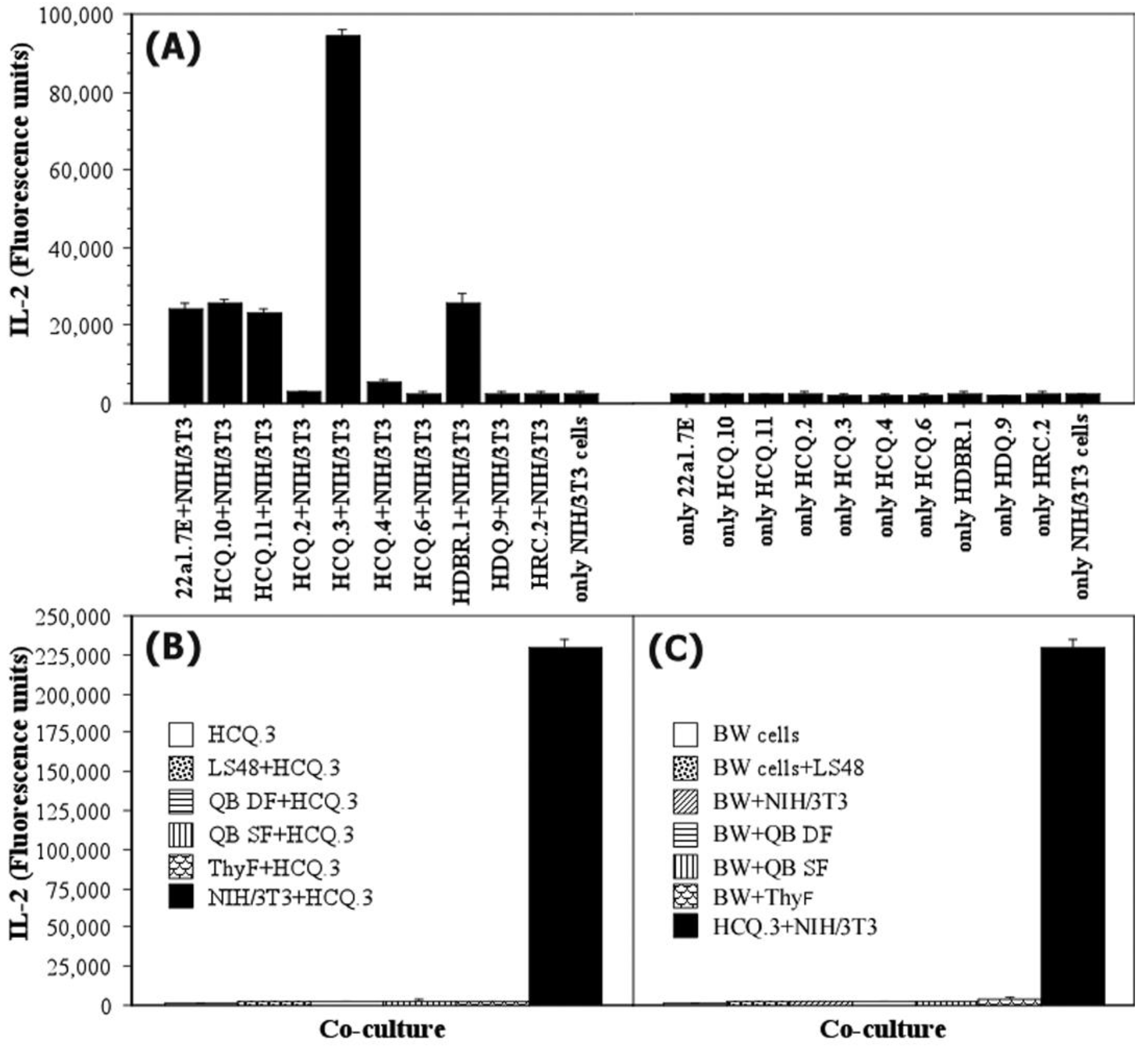
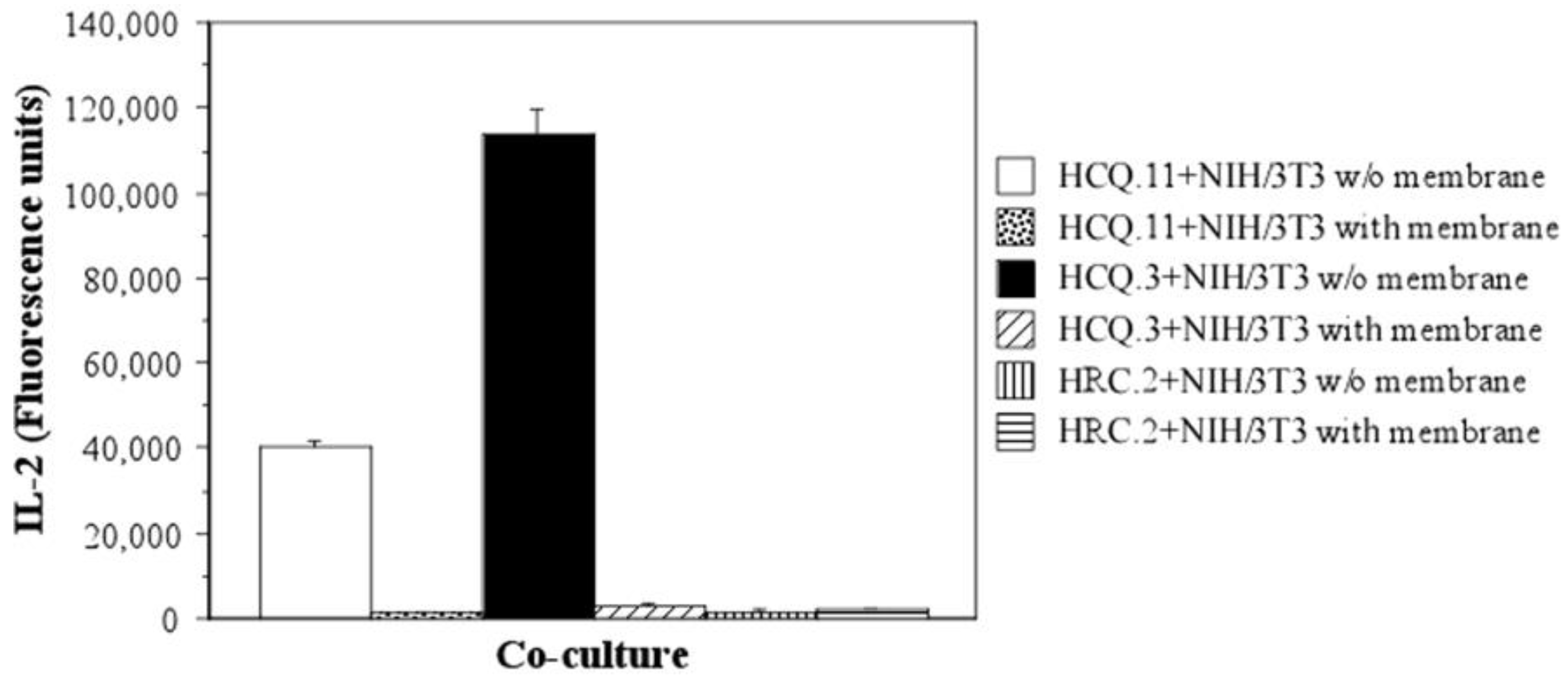
2.3. NIH/3T3 Fibroblasts Selectively Activate the COL2-Specific T-Cell Hybridoma HCQ.3
2.4. The Mechanism of T-Cell Hybridoma Activation by NIH/3T3 Fibroblasts Is Contact-Dependent
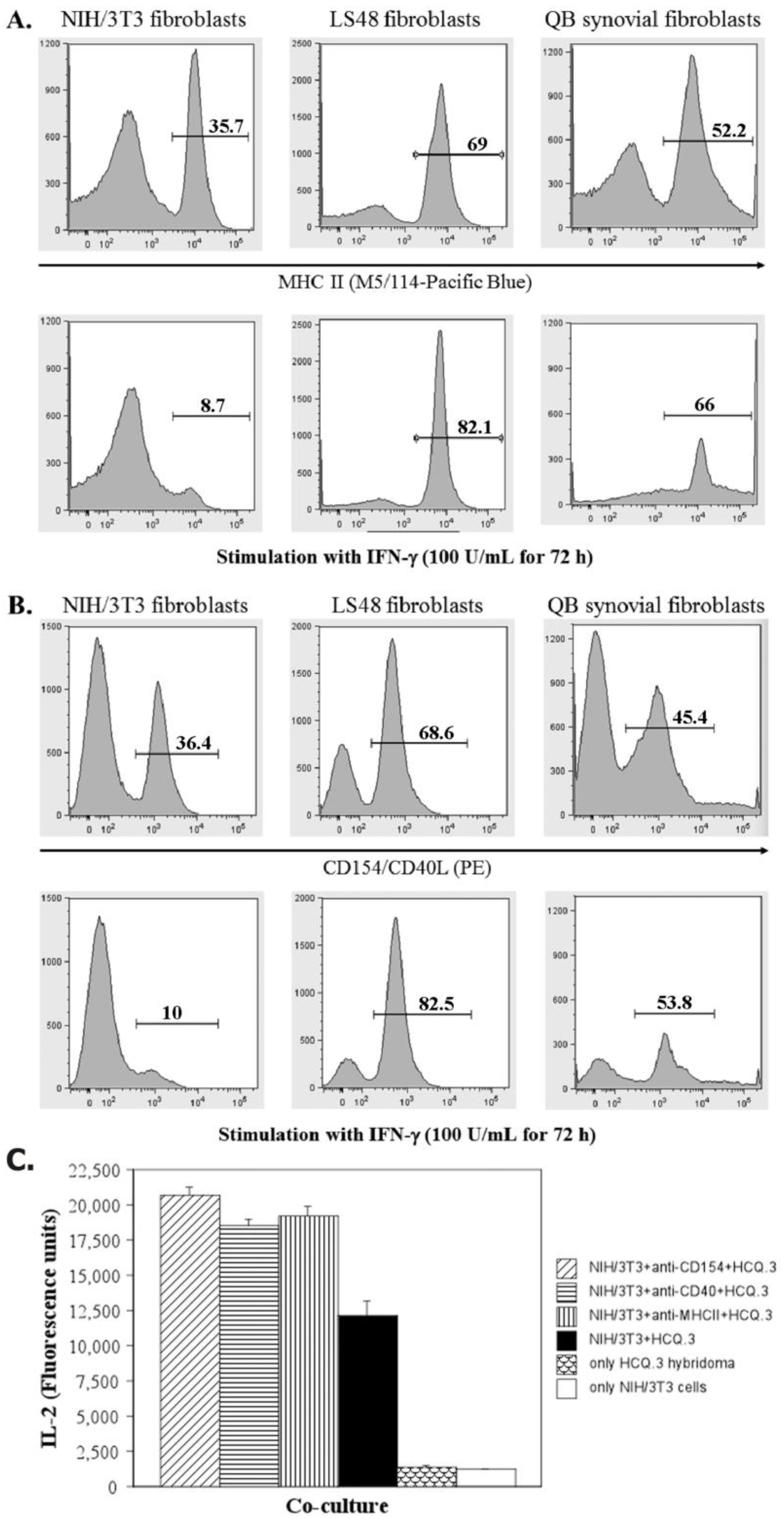
2.5. IFN-Gamma Down-Regulates the Expression of MHCII and CD154 on NIH/3T3 Fibroblasts, but Up-Regulates the Expression of These Markers on LS48 Destructive Fibroblasts and QB Synovial Fibroblasts
2.6. The Interaction between NIH/3T3 Fibroblasts and HCQ.3 Cells Involves the TCR Complex
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.1.1. T-Cell Hybridomas and Fine Specificities
4.1.2. LS48 Cell Line
4.1.3. NIH/3T3 Cell Line
4.2. Fluorescence Microscopy
4.3. Primary Murine Fibroblasts
4.4. Cell Culture Conditions
4.5. Co-Culture Experiments
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
4.7. Blocking of Cell Surface Molecules
4.8. Flow Cytometry
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guidelli, G.M.; Barskova, T.; Brizi, M.G.; Lepri, G.; Parma, A.; Talarico, R.; Cantarini, L.; Frediani, B. One year in review: Novelties in the treatment of rheumatoid arthritis. Clin. Exp. Rheumatol. 2015, 33, 102–108. [Google Scholar]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Bartok, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fan, B.; He, Z.; Yu, X.; Wang, J. Identification of HBEGF+ fibroblasts in the remission of rheumatoid arthritis by integrating single-cell RNA sequencing datasets and bulk RNA sequencing datasets. Arthritis Res. Ther. 2022, 24, 215. [Google Scholar] [CrossRef]
- Luo, P.; Wang, P.; Xu, J.; Hou, W.; Xu, P.; Xu, K.; Liu, L. Immunomodulatory role of T helper cells in rheumatoid arthritis: A comprehensive research review. Bone Jt. Res. 2022, 11, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Schonfeldova, B.; Zec, K.; Udalova, I.A. Synovial single-cell heterogeneity, zonation and interactions: A patchwork of effectors in arthritis. Rheumatology 2022, 61, 913–925. [Google Scholar] [CrossRef]
- Firestein, G.S. Etiology and pathogenesis in rheumatoid arthritis. In Kelly’s Textbook of Rheumatology, 8th ed.; Saunders Elsevier: Philadelphia, PA, USA, 2009; pp. 1035–1086. [Google Scholar]
- Smith, T.J. Insights into the role of fibroblasts in human autoimmune diseases. Clin. Exp. Immunol. 2005, 141, 388–397. [Google Scholar] [CrossRef]
- Ritchlin, C. Fibroblast biology. Effector signals released by the synovial fibroblast in arthritis. Arthritis Res. 2000, 2, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Pap, T.; Muller-Ladner, U.; Gay, R.E.; Gay, S. Fibroblast biology. Role of synovial fibroblasts in the pathogenesis of rheumatoid arthritis. Arthritis Res. 2000, 2, 361–367. [Google Scholar] [CrossRef] [Green Version]
- Yoshitomi, H. Regulation of Immune Responses and Chronic Inflammation by Fibroblast-Like Synoviocytes. Front. Immunol. 2019, 10, 1395. [Google Scholar] [CrossRef] [Green Version]
- Hu, F.; Liu, H.; Xu, L.; Li, Y.; Liu, X.; Shi, L.; Su, Y.; Qiu, X.; Zhang, X.; Yang, Y.; et al. Hypoxia-inducible factor-1alpha perpetuates synovial fibroblast interactions with T cells and B cells in rheumatoid arthritis. Eur. J. Immunol. 2016, 46, 742–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.X.; Wu, Y.J.; Zhang, J.; Wei, W. T-cells interact with B cells, dendritic cells, and fibroblast-like synoviocytes as hub-like key cells in rheumatoid arthritis. Int. Immunopharmacol. 2019, 70, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Naruishi, K. Biological Roles of Fibroblasts in Periodontal Diseases. Cells 2022, 11, 3345. [Google Scholar] [CrossRef]
- Wielento, A.; Lagosz-Cwik, K.B.; Potempa, J.; Grabiec, A.M. The Role of Gingival Fibroblasts in the Pathogenesis of Periodontitis. J. Dent. Res. 2023, 102, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Huber, L.C.; Distler, O.; Tarner, I.; Gay, R.E.; Gay, S.; Pap, T. Synovial fibroblasts: Key players in rheumatoid arthritis. Rheumatology 2006, 45, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckley, C.D.; Filer, A.; Haworth, O.; Parsonage, G.; Salmon, M. Defining a role for fibroblasts in the persistence of chronic inflammatory joint disease. Ann. Rheum. Dis. 2004, 63 (Suppl. 2), ii92–ii95. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Wang, B.; Sun, X.; Li, H.; Ouyang, X.; Wei, J.; Dai, B.; Zhang, Y.; Li, X. Rheumatoid arthritis fibroblast-like synoviocytes co-cultured with PBMC increased peripheral CD4(+) CXCR5(+) ICOS(+) T cell numbers. Clin. Exp. Immunol. 2017, 190, 384–393. [Google Scholar] [CrossRef] [Green Version]
- Tu, J.; Huang, W.; Zhang, W.; Mei, J.; Zhu, C. Two Main Cellular Components in Rheumatoid Arthritis: Communication Between T Cells and Fibroblast-Like Synoviocytes in the Joint Synovium. Front. Immunol. 2022, 13, 922111. [Google Scholar] [CrossRef]
- Smith, T.J. The putative role of fibroblasts in the pathogenesis of Graves’ disease: Evidence for the involvement of the insulin-like growth factor-1 receptor in fibroblast activation. Autoimmunity 2003, 36, 409–415. [Google Scholar] [CrossRef]
- Bahn, R.S. Clinical review 157: Pathophysiology of Graves’ ophthalmopathy: The cycle of disease. J. Clin. Endocrinol. Metab. 2003, 88, 1939–1946. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.S.; Smith, T.J.; Blieden, T.M.; Phipps, R.P. Fibroblasts as sentinel cells. Synthesis of chemokines and regulation of inflammation. Am. J. Pathol. 1997, 151, 317–322. [Google Scholar] [PubMed]
- Aldridge, J.; Ekwall, A.-K.H.; Mark, L.; Bergström, B.; Andersson, K.; Gjertsson, I.; Lundell, A.-C.; Rudin, A. T helper cells in synovial fluid of patients with rheumatoid arthritis primarily have a Th1 and a CXCR3+Th2 phenotype. Arthritis Res. Ther. 2020, 22, 245. [Google Scholar] [CrossRef]
- Bottini, N.; Firestein, G.S. Duality of fibroblast-like synoviocytes in RA: Passive responders and imprinted aggressors. Nat. Rev. Rheumatol. 2013, 9, 24–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, A.P.; Campos, J.; Jansen, K.; Turner, J.D.; Marshall, J.; Attar, M.; Savary, L.; Wehmeyer, C.; Naylor, A.J.; Kemble, S.; et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 2019, 570, 246–251. [Google Scholar] [CrossRef]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally distinct disease-associated fibroblast subsets in rheumatoid arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [Green Version]
- Lefevre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnaker, E.M.; Tarner, I.H.; Robbins, P.D.; et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nygaard, G.; Firestein, G.S. Restoring synovial homeostasis in rheumatoid arthritis by targeting fibroblast-like synoviocytes. Nat. Rev. Rheumatol. 2020, 16, 316–333. [Google Scholar] [CrossRef] [PubMed]
- Machado, C.R.L.; Dias, F.F.; Resende, G.G.; Oliveira, P.G.; Xavier, R.M.; Andrade, M.V.M.; Kakehasi, A.M. Morphofunctional analysis of fibroblast-like synoviocytes in human rheumatoid arthritis and mouse collagen-induced arthritis. Adv. Rheumatol. 2023, 63, 1. [Google Scholar] [CrossRef]
- Eyre, D.R. The collagens of articular cartilage. Semin. Arthritis Rheum. 1991, 21, 2–11. [Google Scholar] [CrossRef]
- Armiento, A.R.; Alini, M.; Stoddart, M.J. Articular fibrocartilage—Why does hyaline cartilage fail to repair? Adv. Drug. Deliv. Rev. 2019, 146, 289–305. [Google Scholar] [CrossRef]
- Alcaide-Ruggiero, L.; Molina-Hernández, V.; Granados, M.M.; Domínguez, J.M. Main and Minor Types of Collagens in the Articular Cartilage: The Role of Collagens in Repair Tissue Evaluation in Chondral Defects. Int. J. Mol. Sci. 2021, 22, 13329. [Google Scholar] [CrossRef]
- Responte, D.J.; Natoli, R.M.; Athanasiou, K.A. Collagens of Articular Cartilage: Structure, Function, and Importance in Tissue Engineering. Crit. Rev. ™ Biomed. Eng. 2007, 35, 363–411. [Google Scholar] [CrossRef]
- Bruckner, P.; van der Rest, M. Structure and function of cartilage collagens. Microsc. Res. Tech. 1994, 28, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Gelse, K. Collagens—Structure, function, and biosynthesis. Adv. Drug. Deliv. Rev. 2003, 55, 1531–1546. [Google Scholar] [CrossRef] [Green Version]
- Sophia Fox, A.J.; Bedi, A.; Rodeo, S.A. The Basic Science of Articular Cartilage: Structure, Composition, and Function. Sport. Health Multidiscip. Approach 2009, 1, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.-U.; Cho, M.-L.; Jung, Y.O.; Min, S.-Y.; Park, S.-W.; Min, D.-J.; Yoon, J.-H.; Kim, H.-Y. Type II Collagen Autoimmunity in Rheumatoid Arthritis. Am. J. Med. Sci. 2004, 327, 202–211. [Google Scholar] [CrossRef]
- Cremer, M.A.; Rosloniec, E.F.; Kang, A.H. The cartilage collagens: A review of their structure, organization, and role in the pathogenesis of experimental arthritis in animals and in human rheumatic disease. J. Mol. Med. 1998, 76, 275–288. [Google Scholar] [CrossRef]
- Brand, D.D.; Kang, A.H.; Rosloniec, E.F. Immunopathogenesis of Collagen Arthritis. Springer Semin. Immunopathol. 2003, 25, 3–18. [Google Scholar] [CrossRef]
- Holmdahl, R.; Bockermann, R.; Backlund, J.; Yamada, H. The molecular pathogenesis of collagen-induced arthritis in mice—A model for rheumatoid arthritis. Ageing Res. Rev. 2002, 1, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Staines, N.A.; Wooley, P.H. Collagen Arthritis—What Can It Teach Us? Rheumatology 1994, 33, 798–807. [Google Scholar] [CrossRef] [PubMed]
- Bäcklund, J.; Carlsen, S.; Höger, T.; Holm, B.; Fugger, L.; Kihlberg, J.; Burkhardt, H.; Holmdahl, R. Predominant selection of T cells specific for the glycosylated collagen type II epitope (263–270) in humanized transgenic mice and in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 2002, 99, 9960–9965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-Y.; Kim, W.-U.; Cho, M.-L.; Lee, S.K.; Youn, J.; Kim, S.-I.; Yoo, W.-H.; Park, J.-H.; Min, J.-K.; Lee, S.-H.; et al. Enhanced T cell proliferative response to type II collagen and synthetic peptide CII (255-274) in patients with rheumatoid arthritis. Arthritis Rheum. 1999, 42, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Snir, O.; Bäcklund, J.; Boström, J.; Andersson, I.; Kihlberg, J.; Buckner, J.H.; Klareskog, L.; Holmdahl, R.; Malmström, V. Multifunctional T cell reactivity with native and glycosylated type II collagen in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2482–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullazehi, M.; Mathsson, L.; Lampa, J.; Ronnelid, J. High anti-collagen type-II antibody levels and induction of proinflammatory cytokines by anti-collagen antibody-containing immune complexes in vitro characterise a distinct rheumatoid arthritis phenotype associated with acute inflammation at the time of disease onset. Ann. Rheum. Dis. 2006, 66, 537–541. [Google Scholar] [CrossRef] [Green Version]
- Terato, K.; DeArmey, D.A.; Ye, X.J.; Griffiths, M.M.; Cremer, M.A. The Mechanism of Autoantibody Formation to Cartilage in Rheumatoid Arthritis: Possible Cross-Reaction of Antibodies to Dietary Collagens with Autologous Type II Collagen. Clin. Immunol. Immunopathol. 1996, 79, 142–154. [Google Scholar] [CrossRef]
- Wooley, P.H.; Luthra, H.S.; O’Duffy, J.D.; Bunch, T.W.; Moore, S.B.; Stuart, J.M. Anti-type II collagen antibodies in rheumatoid arthritis. Tissue Antigens 2008, 23, 263–269. [Google Scholar] [CrossRef]
- Manivel, V.; Sohrabian, A.; Wick, M.C.; Mullazehi, M.; Håkansson, L.; Rönnelid, J. Anti-type II collagen immune complex-induced granulocyte reactivity is associated with joint erosions in RA patients with anti-collagen antibodies. Arthritis Res. Ther. 2015, 17, 8. [Google Scholar] [CrossRef] [Green Version]
- Wooley, P.H.; Luthra, H.S.; Stuart, J.M.; David, C.S. Type II collagen-induced arthritis in mice. I. Major histocompatibility complex (I region) linkage and antibody correlates. J. Exp. Med. 1981, 154, 688–700. [Google Scholar] [CrossRef]
- Myers, L.K.; Miyahara, H.; Terato, K.; Seyer, J.M.; Stuart, J.M.; Kang, A.H. Collagen-induced arthritis in B10.RIII mice (H-2r): Identification of an arthritogenic T-cell determinant. Immunology 1995, 84, 509–513. [Google Scholar]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of Rheumatoid Arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Weisse, S.; Xu, B.; Dobritzsch, D.; Viljanen, J.; Kihlberg, J.; Do, N.-N.; Schneider, N.; Lanig, H.; Holmdahl, R.; et al. Key interactions in the trimolecular complex consisting of the rheumatoid arthritis-associated DRB1*04:01 molecule, the major glycosylated collagen II peptide and the T-cell receptor. Ann. Rheum. Dis. 2022, 81, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Rosloniec, E.F.; Whittington, K.B.; Brand, D.D.; Myers, L.K.; Stuart, J.M. Identification of MHC Class II and TCR Binding Residues in the Type II Collagen Immunodominant Determinant Mediating Collagen-Induced Arthritis. Cell. Immunol. 1996, 172, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Kjellén, P.; Brunsberg, U.; Broddefalk, J.; Hansen, B.; Vestberg, M.; Ivarsson, I.; Engström, Å.; Svejgaard, A.; Kihlberg, J.; Fugger, L.; et al. The structural basis of MHC control of collagen-induced arthritis; binding of the immunodominant type II collagen 256 – 270 glycopeptide to H-2Aq and H-2Ap molecules. Eur. J. Immunol. 1998, 28, 755–766. [Google Scholar] [CrossRef]
- Lomholt, S.; Pedersen, M.J.; Glerup, M.; Kragstrup, T.W. Synovial fibroblasts in juvenile idiopathic arthritis: A scoping review. Semin. Arthritis Rheum. 2023, 58, 152159. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.N.; Davis, M.J.; Tesmer, L.A.; Endres, J.L.; Motyl, C.D.; Smuda, C.; Somers, E.C.; Chung, K.C.; Urquhart, A.G.; Lundy, S.K.; et al. Presentation of arthritogenic peptide to antigen-specific T cells by fibroblast-like synoviocytes. Arthritis Rheum. 2007, 56, 1497–1506. [Google Scholar] [CrossRef] [Green Version]
- Boots, A.M.; Wimmers-Bertens, A.J.; Rijnders, A.W. Antigen-presenting capacity of rheumatoid synovial fibroblasts. Immunology 1994, 82, 268–274. [Google Scholar]
- Geppert, T.D.; Lipsky, P.E. Dissection of defective antigen presentation by interferon-gamma-treated fibroblasts. J. Immunol. 1987, 138, 385–392. [Google Scholar] [CrossRef]
- Ohyama, H.; Nishimura, F.; Meguro, M.; Takashiba, S.; Murayama, Y.; Matsushita, S. Counter-antigen presentation: Fibroblasts produce cytokines by signalling through HLA class II molecules without inducing T-cell proliferation. Cytokine 2002, 17, 175–181. [Google Scholar] [CrossRef]
- Corrigall, V.M.; Panayi, G.S. Autoantigens and immune pathways in rheumatoid arthritis. Crit. Rev. Immunol. 2002, 22, 281–293. [Google Scholar]
- Sack, U.; Sehm, B.; Kahlenberg, F.; Murr, A.; Lehmann, J.; Tannapfel, A.; Uberla, K.; Moessner, A.; Dietrich, A.; Emmrich, F.; et al. Investigation of arthritic joint destruction by a novel fibroblast-based model. Ann. N. Y. Acad. Sci. 2005, 1051, 291–298. [Google Scholar] [CrossRef]
- Sack, U.; Hirth, A.; Funke, B.; Wiedemeyer, K.; Lange, F.; Troltzsch, M.; Tannapfel, A.; Gebhardt, R.; Emmrich, F.; Lehmann, J. A novel model of fibroblast-mediated cartilage destruction. Scand. J. Immunol. 2005, 61, 18–28. [Google Scholar] [CrossRef]
- Todaro, G.J.; Green, H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J. Cell Biol. 1963, 17, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Corthay, A.; Backlund, J.; Broddefalk, J.; Michaelsson, E.; Goldschmidt, T.J.; Kihlberg, J.; Holmdahl, R. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur. J. Immunol. 1998, 28, 2580–2590. [Google Scholar] [CrossRef]
- Pober, J.S.; Collins, T.; Gimbrone, M.A., Jr.; Cotran, R.S.; Gitlin, J.D.; Fiers, W.; Clayberger, C.; Krensky, A.M.; Burakoff, S.J.; Reiss, C.S. Lymphocytes recognize human vascular endothelial and dermal fibroblast Ia antigens induced by recombinant immune interferon. Nature 1983, 305, 726–729. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.; Sime, P.J.; Phipps, R.P. Expression of CD154 (CD40 ligand) by human lung fibroblasts: Differential regulation by IFN-gamma and IL-13, and implications for fibrosis. J. Immunol. 2004, 172, 1862–1871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, C.S.; Cho, M.L.; Min, S.Y.; Kim, W.U.; Min, D.J.; Lee, S.S.; Park, S.H.; Choe, J.; Kim, H.Y. CD40 engagement on synovial fibroblast up-regulates production of vascular endothelial growth factor. J. Immunol. 2000, 164, 5055–5061. [Google Scholar] [CrossRef] [Green Version]
- Yellin, M.J.; Winikoff, S.; Fortune, S.M.; Baum, D.; Crow, M.K.; Lederman, S.; Chess, L. Ligation of CD40 on fibroblasts induces CD54 (ICAM-1) and CD106 (VCAM-1) up-regulation and IL-6 production and proliferation. J. Leukoc. Biol. 1995, 58, 209–216. [Google Scholar] [CrossRef]
- Schneider, E.L.; Mitsui, Y.; Au, K.S.; Shorr, S.S. Tissue-specific differences in cultured human diploid fibroblasts. Exp. Cell Res. 1977, 108, 1–6. [Google Scholar] [CrossRef]
- Korsunsky, I.; Wei, K.; Pohin, M.; Kim, E.Y.; Barone, F.; Major, T.; Taylor, E.; Ravindran, R.; Kemble, S.; Watts, G.F.M.; et al. Cross-tissue, single-cell stromal atlas identifies shared pathological fibroblast phenotypes in four chronic inflammatory diseases. Med 2022, 3, 481–518.e14. [Google Scholar] [CrossRef]
- Shanaj, S.; Donlin, L.T. Synovial Tissue: Cellular and Molecular Phenotyping. Curr. Rheumatol. Rep. 2019, 21, 52. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The inflammatory speech of fibroblasts. Immunol. Rev. 2021, 302, 126–146. [Google Scholar] [CrossRef] [PubMed]
- Kundig, T.M.; Bachmann, M.F.; DiPaolo, C.; Simard, J.J.; Battegay, M.; Lother, H.; Gessner, A.; Kuhlcke, K.; Ohashi, P.S.; Hengartner, H.; et al. Fibroblasts as efficient antigen-presenting cells in lymphoid organs. Science 1995, 268, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, A.; Snijders, A.; Abraham-Inpijn, L.; Kapsenberg, M.L.; Kievits, F. Antigen-presenting properties of gingival fibroblasts in chronic adult periodontitis. Clin. Exp. Immunol. 1997, 110, 277–284. [Google Scholar] [CrossRef]
- Martu, M.-A.; Luchian, I.; Mares, M.; Solomon, S.; Ciurcanu, O.; Danila, V.; Rezus, E.; Foia, L. The Effectiveness of Laser Applications and Photodynamic Therapy on Relevant Periodontal Pathogens (Aggregatibacter actinomycetemcomitans) Associated with Immunomodulating Anti-rheumatic Drugs. Bioengineering 2023, 10, 61. [Google Scholar] [CrossRef]
- Zhao, S.; Grieshaber-Bouyer, R.; Rao, D.A.; Kolb, P.; Chen, H.; Andreeva, I.; Tretter, T.; Lorenz, H.M.; Watzl, C.; Wabnitz, G.; et al. Effect of JAK Inhibition on the Induction of Proinflammatory HLA-DR+CD90+ Rheumatoid Arthritis Synovial Fibroblasts by Interferon-gamma. Arthritis Rheumatol. 2022, 74, 441–452. [Google Scholar] [CrossRef]
- Mori, M.; Hashimoto, M.; Matsuo, T.; Fujii, T.; Furu, M.; Ito, H.; Yoshitomi, H.; Hirose, J.; Ito, Y.; Akizuki, S.; et al. Cell-contact-dependent activation of CD4(+) T cells by adhesion molecules on synovial fibroblasts. Mod. Rheumatol. 2017, 27, 448–456. [Google Scholar] [CrossRef] [Green Version]
- Gjertsson, I.; Laurie, K.L.; Devitt, J.; Howe, S.J.; Thrasher, A.J.; Holmdahl, R.; Gustafsson, K. Tolerance induction using lentiviral gene delivery delays onset and severity of collagen II arthritis. Mol. Ther. 2009, 17, 632–640. [Google Scholar] [CrossRef] [Green Version]
- Treese, C.; Mittag, A.; Lange, F.; Tarnok, A.; Loesche, A.; Emmrich, F.; Lehmann, J.; Sack, U. Characterization of fibroblasts responsible for cartilage destruction in arthritis. Cytometry A 2008, 73, 351–360. [Google Scholar] [CrossRef]
- Rissoan, M.C.; Van Kooten, C.; Chomarat, P.; Galibert, L.; Durand, I.; Thivolet-Bejui, F.; Miossec, P.; Banchereau, J. The functional CD40 antigen of fibroblasts may contribute to the proliferation of rheumatoid synovium. Clin. Exp. Immunol. 1996, 106, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Yellin, M.J.; Sippel, K.; Inghirami, G.; Covey, L.R.; Lee, J.J.; Sinning, J.; Clark, E.A.; Chess, L.; Lederman, S. CD40 molecules induce down-modulation and endocytosis of T cell surface T cell-B cell activating molecule/CD40-L. Potential role in regulating helper effector function. J. Immunol. 1994, 152, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Ruth, J.H.; Rottman, J.B.; Kingsbury, G.A.; Coyle, A.J.; Haines, G.K., 3rd; Pope, R.M.; Koch, A.E. ICOS and B7 costimulatory molecule expression identifies activated cellular subsets in rheumatoid arthritis. Cytometry A 2007, 71, 317–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saalbach, A.; Kraft, R.; Herrmann, K.; Haustein, U.F.; Anderegg, U. The monoclonal antibody AS02 recognizes a protein on human fibroblasts being highly homologous to Thy-1. Arch. Dermatol. Res. 1998, 290, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Holm, L.; Bockermann, R.; Wellner, E.; Backlund, J.; Holmdahl, R.; Kihlberg, J. Side-chain and backbone amide bond requirements for glycopeptide stimulation of T-cells obtained in a mouse model for rheumatoid arthritis. Bioorg Med. Chem. 2006, 14, 5921–5932. [Google Scholar] [CrossRef]
- White, J.; Blackman, M.; Bill, J.; Kappler, J.; Marrack, P.; Gold, D.P.; Born, W. Two better cell lines for making hybridomas expressing specific T cell receptors. J. Immunol. 1989, 143, 1822–1825. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hybridoma | COL2 Protein | Non-Modified COL2259–273 Peptide | COL2259–273 Hyl-264 Peptide | COL2259–273 Gal-Hyl-264 Peptide | COL2259–273 Gal-Hyl-264/270 Peptide |
|---|---|---|---|---|---|
| HRC.2 | + | + | – | – | – |
| HDQ.9 | + | + | – | – | – |
| HCQ.4 | + | + | – | – | – |
| HDBR.1 | + | – | + | – | – |
| HCQ.2 | + | – | – | + | – |
| HCQ.3 | + | – | – | + | – |
| HCQ.6 | + | – | – | + | – |
| HCQ.10 | + | – | – | + | – |
| 22a1.7E | + | – | – | + | – |
| HCQ.11 | + | – | – | – | + |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dzhambazov, B.; Batsalova, T.; Merky, P.; Lange, F.; Holmdahl, R. NIH/3T3 Fibroblasts Selectively Activate T Cells Specific for Posttranslationally Modified Collagen Type II. Int. J. Mol. Sci. 2023, 24, 10811. https://doi.org/10.3390/ijms241310811
Dzhambazov B, Batsalova T, Merky P, Lange F, Holmdahl R. NIH/3T3 Fibroblasts Selectively Activate T Cells Specific for Posttranslationally Modified Collagen Type II. International Journal of Molecular Sciences. 2023; 24(13):10811. https://doi.org/10.3390/ijms241310811
Chicago/Turabian StyleDzhambazov, Balik, Tsvetelina Batsalova, Patrick Merky, Franziska Lange, and Rikard Holmdahl. 2023. "NIH/3T3 Fibroblasts Selectively Activate T Cells Specific for Posttranslationally Modified Collagen Type II" International Journal of Molecular Sciences 24, no. 13: 10811. https://doi.org/10.3390/ijms241310811