

Clinical Characteristics and Genetic Variants of a Large Cohort of Patients with Retinitis Pigmentosa Using Multimodal Imaging and Next Generation Sequencing
 , ,
, , 
Abstract
:1. Introduction
2. Results
2.1. Demographic Information
2.2. Symptoms
2.3. Visual Acuity Results
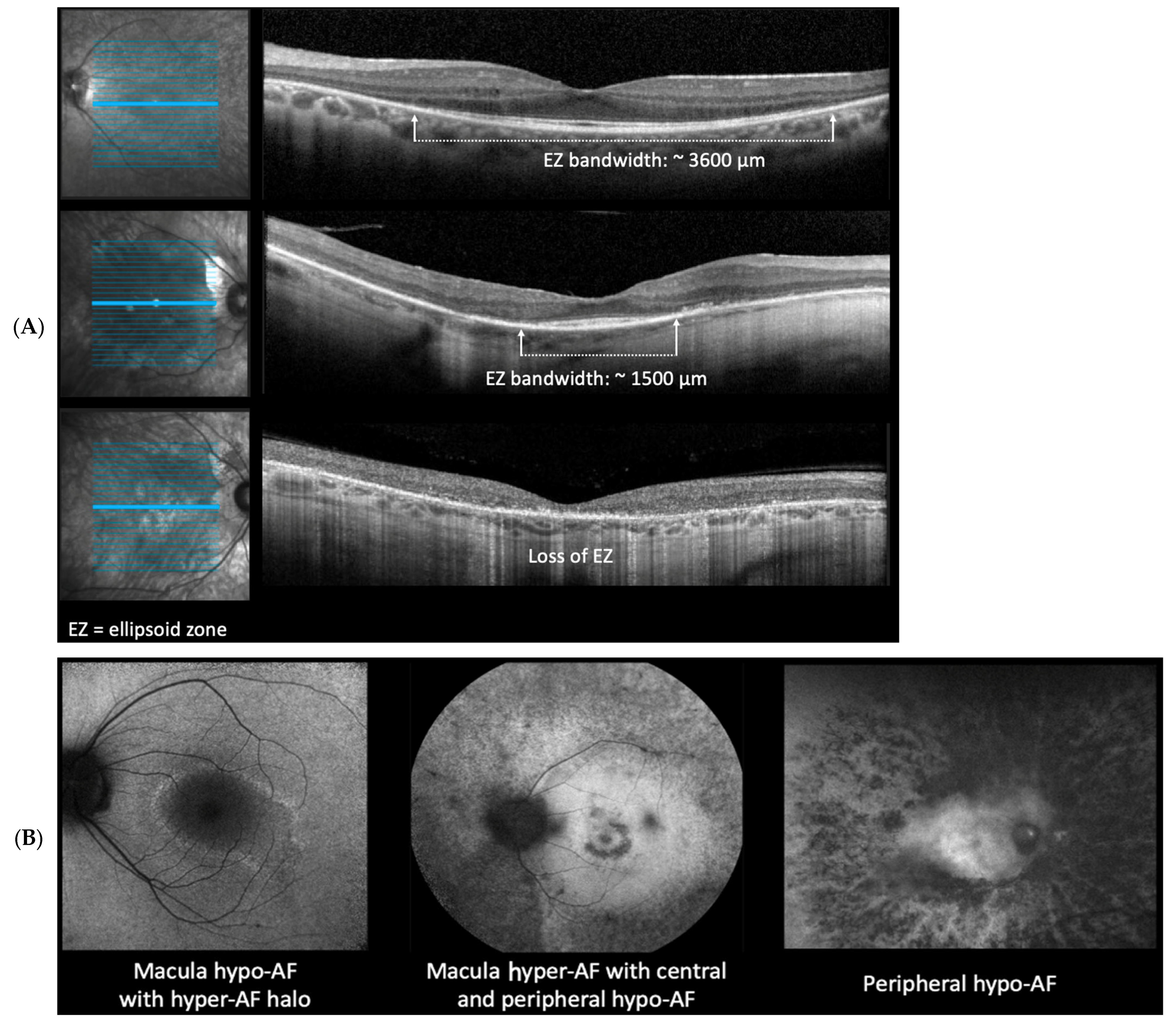
2.4. Ellipsoid Zone (EZ) Measurements
2.5. Fundus Autofluorescent (FAF) Patterns
2.6. Genetic Testing Reports
2.7. The Inheritance Pattern Distribution
2.8. Supplemental Data
3. Methods
3.1. Database
3.2. Genetic Testing
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sorrentino, F.S.; Gallenga, C.E.; Bonifazzi, C.; Perri, P. A challenge to the striking genotypic heterogeneity of retinitis pigmentosa: A better understanding of the pathophysiology using the newest genetic strategies. Eye 2016, 30, 1542–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiger, S.P.; Bowne, S.J.; Sullivan, L.S. Genes and Mutations Causing Autosomal Dominant Retinitis Pigmentosa. Cold Spring Harb. Perspect. Med. 2014, 5, a017129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, S.; Di Iorio, E.; Barbaro, V.; Ponzin, D.; Sorrentino, F.S.; Parmeggiani, F. Retinitis pigmentosa: Genes and disease mechanisms. Curr. Genom. 2011, 12, 238–249. [Google Scholar] [CrossRef]
- Arai, Y.; Maeda, A.; Hirami, Y.; Ishigami, C.; Kosugi, S.; Mandai, M.; Kurimoto, Y.; Takahashi, M. Retinitis Pigmentosa with EYS Mutations Is the Most Prevalent Inherited Retinal Dystrophy in Japanese Populations. J. Ophthalmol. 2015, 2015, 819760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Littink, K.W.; van den Born, L.I.; Koenekoop, R.K.; Collin, R.W.; Zonneveld, M.N.; Blokland, E.A.; Khan, H.; Theelen, T.; Hoyng, C.B.; Cremers, F.P.; et al. Mutations in the EYS Gene Account for Approximately 5% of Autosomal Recessive Retinitis Pigmentosa and Cause a Fairly Homogeneous Phenotype. Ophthalmology 2010, 117, 2026–2033.e7. [Google Scholar] [CrossRef]
- Boughman, J.; Vernon, M.; Shaver, K. Usher syndrome: Definition and estimate of prevalence from two high-risk populations. J. Chronic Dis. 1983, 36, 595–603. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar] [CrossRef]
- Radhakrishnan, R.; Dronamraju, V.R.; Leung, M.; Gruesen, A.; Solanki, A.K.; Walterhouse, S.; Roehrich, H.; Song, G.; da Costa Monsanto, R.; Cureoglu, S.; et al. The role of motor proteins in photoreceptor protein transport and visual function. Ophthalmic. Genet. 2022, 43, 285–300. [Google Scholar] [CrossRef]
- Özdek, Ş.; Onaran, Z.; Gürelik, G.; Bilgihan, K.; Acar, C.; Hasanreisoǧlu, B. Stargardt’s disease and retinitis pigmentosa: Different phenotypic presentations in the same family. Eye 2005, 19, 1222–1225. [Google Scholar] [CrossRef] [Green Version]
- Klevering, B.J.; Maugeri, A.; Wagner, A.; Go, S.L.; Vink, C.; Cremers, F.P.; Hoyng, C.B. Three families displaying the combination of Stargardt’s disease with cone–rod dystrophy or retinitis pigmentosa. Ophthalmology 2004, 111, 546–553. [Google Scholar] [CrossRef]
- Conley, S.M.; Naash, M.I. Gene Therapy for PRPH2-associated ocular disease: Challenges and prospects. Cold Spring Harb. Perspect. Med. 2014, 4, a017376. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.E.; Heitkotter, H.; Carroll, J. Challenges Associated with Ellipsoid Zone Intensity Measurements Using Optical Coherence Tomography. Transl. Vis. Sci. Technol. 2021, 10, 27. [Google Scholar] [CrossRef]
- Hariri, A.H.; Zhang, H.Y.; Ho, A.; Francis, P.; Weleber, R.G.; Birch, D.G.; Ferris, F.L.; Sadda, S.R.; for the Trial of Oral Valproic Acid for Retinitis Pigmentosa Group. Quantification of Ellipsoid Zone Changes in Retinitis Pigmentosa Using en Face Spectral Domain-Optical Coherence Tomography. JAMA Ophthalmol. 2016, 134, 628–635. [Google Scholar] [CrossRef] [Green Version]
- Birch, D.G.; Locke, K.G.; Wen, Y.; Locke, K.I.; Hoffman, D.R.; Hood, D.C. Spectral-domain optical coherence tomography measures of outer segment layer progression in patients with x-linked retinitis pigmentosa. JAMA Ophthalmol. 2013, 131, 1143–1150. [Google Scholar] [CrossRef] [Green Version]
- Bittner, A.K.; Iftikhar, M.H.; Dagnelie, G. Test-retest, within-visit variability of goldmann visual fields in retinitis pigmentosa. Investig. Opthalmol. Vis. Sci. 2011, 52, 8042–8046. [Google Scholar] [CrossRef] [Green Version]
- Trichonas, G.; Traboulsi, E.I.; Ehlers, J.P. Correlation of ultra-widefield fundus autofluorescence patterns with the underlying genotype in retinal dystrophies and retinitis pigmentosa. Ophthalmic. Genet. 2017, 38, 320–324. [Google Scholar] [CrossRef]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W.J. Molecular Therapies for Inherited Retinal Diseases—Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef] [Green Version]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.-F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65 -mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Sun, S.; Montezuma, S.R. Gene Therapy for Inherited Retinopathies: Update on Development Progress. J. Vitr. Dis. 2018, 2, 219–226. [Google Scholar] [CrossRef]
- Sather, R., III; Ihinger, J.; Khundkar, T.; Montezuma, S.R. Pathogenic gene variants identified in patients with retinitis pigmentosa at the referral center clinic of the University of Minnesota (UMN). Ophthalmic Genet. 2022, 1–3. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neveling, K.; Collin, R.W.; Gilissen, C.; van Huet, R.A.; Visser, L.; Kwint, M.P.; Gijsen, S.J.; Zonneveld, M.N.; Wieskamp, N.; de Ligt, J.; et al. Next-generation genetic testing for retinitis pigmentosa. Hum. Mutat. 2012, 33, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Martin-Merida, I.; Avila-Fernandez, A.; Del Pozo-Valero Valero, M.; Blanco-Kelly, F.; Zurita, O.; Perez-Carro, R.; Aguilera-Garcia, D.; Riveiro-Alvarez, R.; Arteche, A.; Trujillo-Tiebas, M.J.; et al. Genomic Landscape of Sporadic Retinitis Pigmentosa: Findings from 877 Spanish Cases. Ophthalmology 2019, 126, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Perea-Romero, I.; Gordo, G.; Iancu, I.F.; Del Pozo-Valero, M.; Almoguera, B.; Blanco-Kelly, F.; Carreño, E.; Jimenez-Rolando, B.; Lopez-Rodriguez, R.; Lorda-Sanchez, I.; et al. Genetic landscape of 6089 inherited retinal dystrophies affected cases in Spain and their therapeutic and extended epidemiological implications. Sci. Rep. 2021, 11, 1526. [Google Scholar] [CrossRef]
- Birtel, J.; Gliem, M.; Mangold, E.; Müller, P.L.; Holz, F.G.; Neuhaus, C.; Lenzner, S.; Zahnleiter, D.; Betz, C.; Eisenberger, T.; et al. Next-generation sequencing identifies unexpected genotype-phenotype correlations in patients with retinitis pigmentosa. PLoS ONE 2018, 13, e0207958. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Xu, M.; Verriotto, J.D.; Li, Y.; Wang, H.; Gan, L.; Lam, B.L.; Chen, R. Next-generation sequencing-based molecular diagnosis of 35 Hispanic retinitis pigmentosa probands. Sci. Rep. 2016, 6, 32792. [Google Scholar] [CrossRef] [Green Version]
- Karali, M.; Testa, F.; Brunetti-Pierri, R.; Di Iorio, V.; Pizzo, M.; Melillo, P.; Barillari, M.R.; Torella, A.; Musacchia, F.; D’angelo, L.; et al. Clinical and Genetic Analysis of a European Cohort with Pericentral Retinitis Pigmentosa. Int. J. Mol. Sci. 2019, 21, 86. [Google Scholar] [CrossRef] [Green Version]
- Haim, M. The epidemiology of retinitis pigmentosa in Denmark. Acta Ophthalmol. Scand. Suppl. 2002, 80, 1–34. [Google Scholar] [CrossRef]
- Ayuso, C.; Garcia-Sandoval, B.; Najera, C.; Valverde, D.; Carballo, M.; Antiñolo, G.; Spanish Multicentric and Multidisciplinary Group for Research into Retinitis Pigmentosa. Retinitis pigmentosa in Spain. Clin. Genet. 1995, 48, 120–122. [Google Scholar] [CrossRef]
- Fishman, G.A. Retinitis Pigmentosa: Genetic Percentages. Arch. Ophthalmol. 1978, 96, 822–826. [Google Scholar] [CrossRef]
- Bunker, C.H.; Berson, E.L.; Bromley, W.C.; Hayes, R.P.; Roderick, T.H. Prevalence of Retinitis Pigmentosa in Maine. Am. J. Ophthalmol. 1984, 97, 357–365. [Google Scholar] [CrossRef]
- Sullivan, L.S.; Bowne, S.J.; Birch, D.G.; Hughbanks-Wheaton, D.; Heckenlively, J.R.; Lewis, R.A.; Garcia, C.A.; Ruiz, R.S.; Blanton, S.H.; Northrup, H.; et al. Prevalence of Disease-Causing Mutations in Families with Autosomal Dominant Retinitis Pigmentosa: A Screen of Known Genes in 200 Families. Investig. Opthalmol. Vis. Sci. 2006, 47, 3052–3064. [Google Scholar] [CrossRef]
- Hu, D.-N. Genetic aspects of retinitis pigmentosa in China. Am. J. Med. Genet. 1982, 12, 51–56. [Google Scholar] [CrossRef]
- Donato, L.; Scimone, C.; Alibrandi, S.; Scalinci, S.Z.; Rinaldi, C.; D’angelo, R.; Sidoti, A. Epitranscriptome Analysis of Oxidative Stressed Retinal Epithelial Cells Depicted a Possible RNA Editing Landscape of Retinal Degeneration. Antioxidants 2022, 11, 1967. [Google Scholar] [CrossRef]
- Yépez, V.A.; Gusic, M.; Kopajtich, R.; Mertes, C.; Smith, N.H.; Alston, C.L.; Ban, R.; Beblo, S.; Berutti, R.; Blessing, H.; et al. Clinical implementation of RNA sequencing for Mendelian disease diagnostics. Genome Med. 2022, 14, 38. [Google Scholar] [CrossRef]
- Belkadi, A.; Bolze, A.; Itan, Y.; Cobat, A.; Vincent, Q.B.; Antipenko, A.; Shang, L.; Boisson, B.; Casanova, J.-L.; Abel, L. Whole-genome sequencing is more powerful than whole-exome sequencing for detecting exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 5473–5478. [Google Scholar] [CrossRef] [Green Version]
- González-Del Pozo, M.; Fernández-Suárez, E.; Martín-Sánchez, M.; Bravo-Gil, N.; Méndez-Vidal, C.; la Rúa, E.R.-D.; Borrego, S.; Antiñolo, G. Unmasking Retinitis Pigmentosa complex cases by a whole genome sequencing algorithm based on open-access tools: Hidden recessive inheritance and potential oligogenic variants. J. Transl. Med. 2020, 18, 73. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Age at Which the Patient Reported RP Eye Symptoms | Total Cases | |||
|---|---|---|---|---|
| Before the age of 10 | 61 | |||
| Between the ages of 10–19 | 32 | |||
| Between the ages of 20–40 | 34 | |||
| After the age of 40 | 17 | |||
| Unknown | 55 | |||
| Visual Acuity | ||||
| Snellen | ≥20/40 | <20/40–≥20/80 | <20/80 | |
| (LogMAR) | (≤0.3) | (>0.3–≤0.6) | (>0.6) | |
| Right eye | 93 | 31 | 74 | |
| Left eye | 99 | 19 | 80 | |
| EZ width | <1500 μm | 1500–3500 μm | 3501–6000 μm | |
| Right eye | 134 | 9 | 39 | |
| Left eye | 134 | 9 | 39 | |
| FAF Findings | Normal | Macula ring of hypo-AF | Peripheral hypo-AF | Macula ring of hyper-AF |
| Right eye | 4 | 69 | 181 | 91 |
| Left eye | 2 | 68 | 182 | 92 |
| Gene Panel * | Nº Patients Utilized | Nº Diagnostic Result | Diagnostic Yield Rate (%) |
|---|---|---|---|
| Invitae Laboratory | 71 | 31 | 43.7 |
| UMN NGS | 22 | 16 | 72.7 |
| PreventionGenetics | 12 | 6 | 50 |
| Blueprint Genetics | 9 | 8 | 88.9 |
| Other | 13 | 6 | 46.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sather, R., III; Ihinger, J.; Simmons, M.; Khundkar, T.; Lobo, G.P.; Montezuma, S.R. Clinical Characteristics and Genetic Variants of a Large Cohort of Patients with Retinitis Pigmentosa Using Multimodal Imaging and Next Generation Sequencing. Int. J. Mol. Sci. 2023, 24, 10895. https://doi.org/10.3390/ijms241310895
Sather R III, Ihinger J, Simmons M, Khundkar T, Lobo GP, Montezuma SR. Clinical Characteristics and Genetic Variants of a Large Cohort of Patients with Retinitis Pigmentosa Using Multimodal Imaging and Next Generation Sequencing. International Journal of Molecular Sciences. 2023; 24(13):10895. https://doi.org/10.3390/ijms241310895
Chicago/Turabian StyleSather, Richard, III, Jacie Ihinger, Michael Simmons, Tahsin Khundkar, Glenn P. Lobo, and Sandra R. Montezuma. 2023. "Clinical Characteristics and Genetic Variants of a Large Cohort of Patients with Retinitis Pigmentosa Using Multimodal Imaging and Next Generation Sequencing" International Journal of Molecular Sciences 24, no. 13: 10895. https://doi.org/10.3390/ijms241310895