
Unmasking the Deceptive Nature of Cancer Stem Cells: The Role of CD133 in Revealing Their Secrets
 , ,
, ,  ,
,  and
and
Abstract
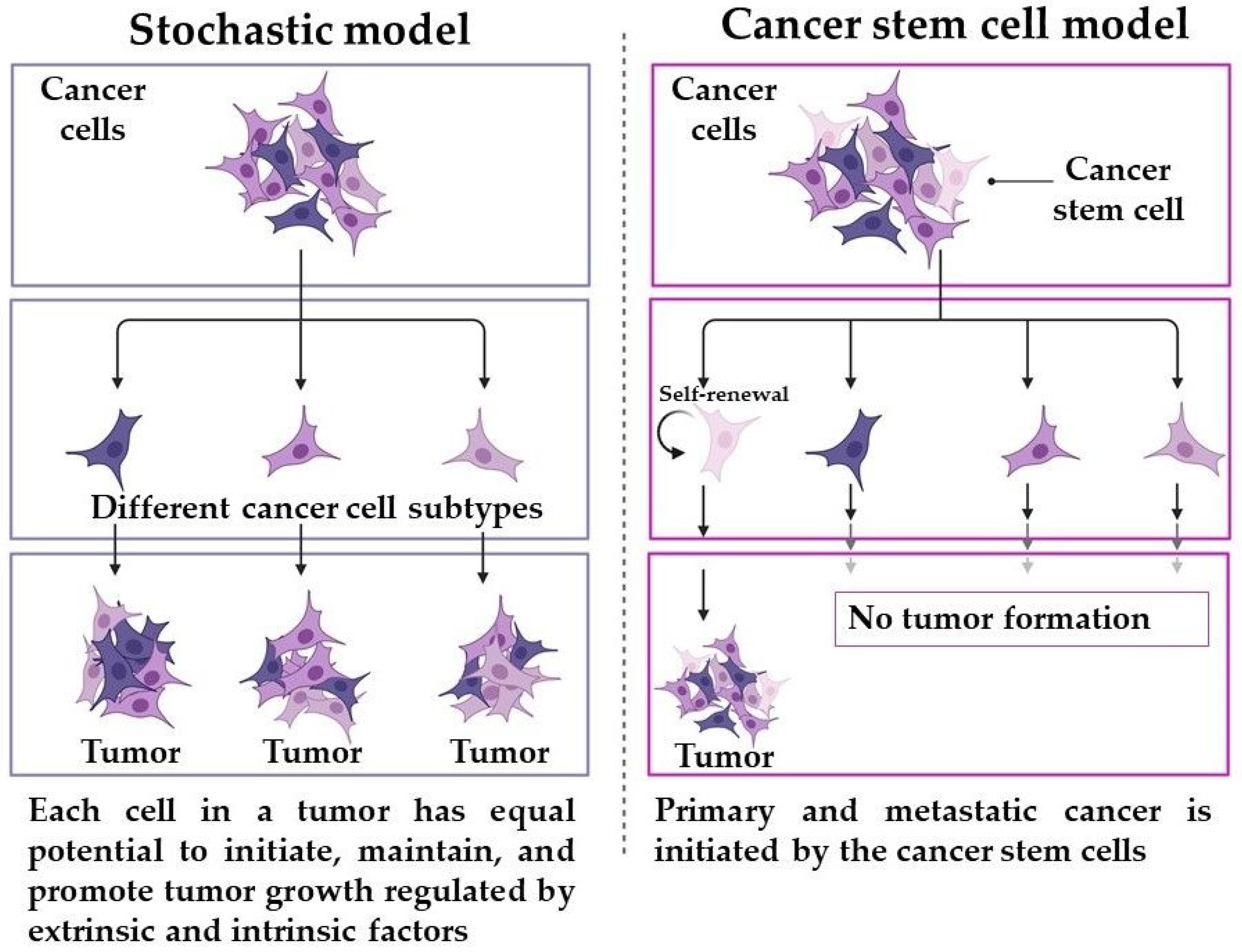
:1. Introduction
2. A Brief 26-Year History of CD133: From Discovery to Understanding the Role of Protein
2.1. The Discovery of CD133
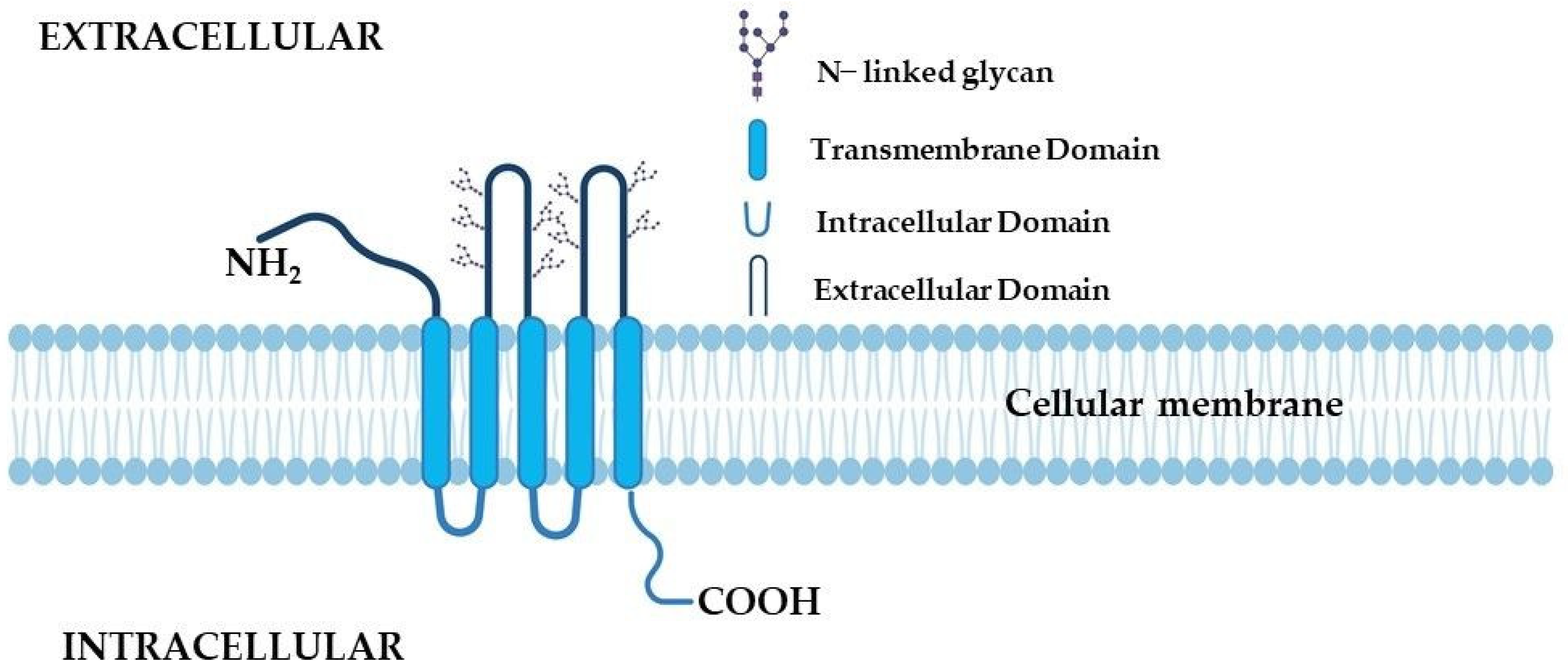
2.2. The Receptor Structure
2.3. CD133 Physiological Functions—Cell Differentiation, Proliferation, and Survival
2.4. The Role of CD133 Glycosylation
3. CD133 as a Target in Cancer Therapies
3.1. Strategy for Gene Therapy
3.2. The Relationship between CD133 and Chemotherapeutic Drugs
3.3. Possibility to Enhance Immunotherapy
3.4. Approach to Improve the Selectivity of Photodynamic Therapy
3.5. Dealing with Cancer Stemness by Suppression CD133
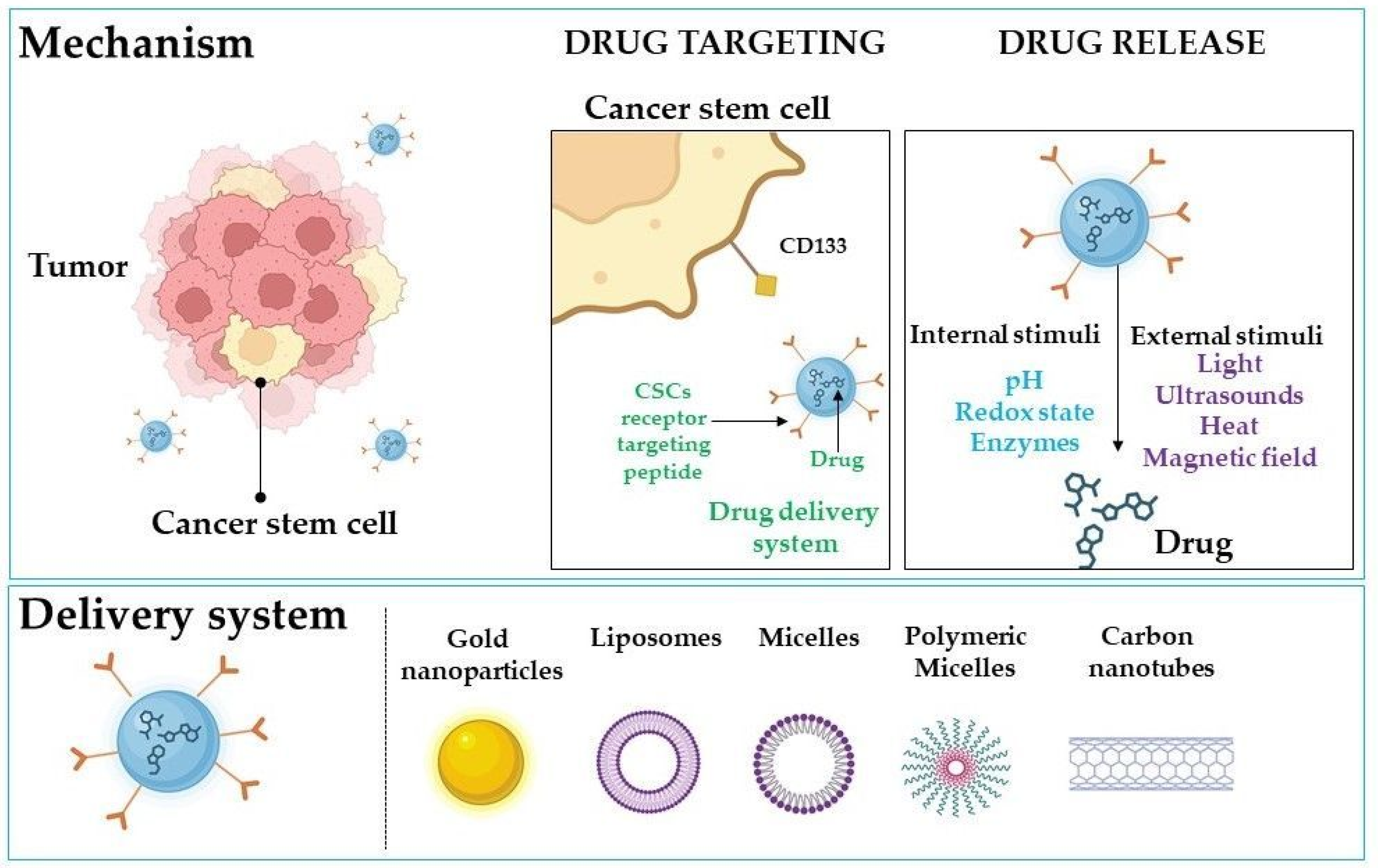
4. Drug Delivery Systems for CD133-Targeted Therapy Based on Nanotechnology
4.1. Gold Nanoparticles in CD133-Targeted Therapy
4.2. Nanoliposomes
4.3. Other Delivery Systems
5. Limitations and Advantages
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deo, S.V.S.; Sharma, J.; Kumar, S. GLOBOCAN 2020 Report on Global Cancer Burden: Challenges and Opportunities for Surgical Oncologists. Ann. Surg. Oncol. 2022, 29, 6497–6500. [Google Scholar] [CrossRef]
- Chhikara, B.S.; Parang, K. Global Cancer Statistics 2022: The Trends Projection Analysis. Chem. Biol. Lett. 2023, 10, 451. [Google Scholar]
- Rezayatmand, H.; Razmkhah, M.; Razeghian-Jahromi, I. Drug Resistance in Cancer Therapy: The Pandora’s Box of Cancer Stem Cells. Stem Cell. Res. Ther. 2022, 13, 181. [Google Scholar] [CrossRef]
- Yu, Z.; Pestell, T.G.; Lisanti, M.P.; Pestell, R.G. Cancer Stem Cells. Int. J. Biochem. Cell. Biol. 2012, 44, 2144–2151. [Google Scholar] [CrossRef] [Green Version]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Brown, G. Oncogenes, Proto-Oncogenes, and Lineage Restriction of Cancer Stem Cells. Int. J. Mol. Sci. 2021, 22, 9667. [Google Scholar] [CrossRef]
- Lee, E.C.Y.; Kok, J.S.T.; Teh, B.T.; Lim, K.S. Interplay between the DNA Damage Response and Immunotherapy Response in Cancer. Int. J. Mol. Sci. 2022, 23, 13356. [Google Scholar] [CrossRef] [PubMed]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-Suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Afify, S.; Seno, M. Conversion of Stem Cells to Cancer Stem Cells: Undercurrent of Cancer Initiation. Cancers 2019, 11, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, G.J.; Saya, H. Therapeutic Strategies Targeting Cancer Stem Cells. Cancer Sci. 2016, 107, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Raghav, P.K.; Mann, Z. Cancer Stem Cells Targets and Combined Therapies to Prevent Cancer Recurrence. Life Sci. 2021, 277, 119465. [Google Scholar] [CrossRef]
- Saga, I.; Shibao, S.; Okubo, J.; Osuka, S.; Kobayashi, Y.; Yamada, S.; Fujita, S.; Urakami, K.; Kusuhara, M.; Yoshida, K.; et al. Integrated Analysis Identifies Different Metabolic Signatures for Tumor-Initiating Cells in a Murine Glioblastoma Model. Neuro-Oncol. 2014, 16, 1048–1056. [Google Scholar] [CrossRef] [Green Version]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem Cells, Cancer, and Cancer Stem Cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer Stem Cells—Perspectives on Current Status and Future Directions: AACR Workshop on Cancer Stem Cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, Y.D.; Kwon, Y.T. Molecular Mechanisms Controlling Asymmetric and Symmetric Self-Renewal of Cancer Stem Cells. J. Anal. Sci. Technol. 2015, 6, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokim Ahmed, K.; Li, J.J. NF-ΚB-Mediated Adaptive Resistance to Ionizing Radiation. Free Radic. Biol. Med. 2008, 44, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Najafi, M.; Mortezaee, K.; Majidpoor, J. Cancer Stem Cell (CSC) Resistance Drivers. Life Sci. 2019, 234, 116781. [Google Scholar] [CrossRef]
- Lee, Y.T.; Tan, Y.J.; Oon, C.E. Molecular Targeted Therapy: Treating Cancer with Specificity. Eur. J. Pharmacol. 2018, 834, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Abugomaa, A.; Elbadawy, M.; Yamawaki, H.; Usui, T.; Sasaki, K. Emerging Roles of Cancer Stem Cells in Bladder Cancer Progression, Tumorigenesis, and Resistance to Chemotherapy: A Potential Therapeutic Target for Bladder Cancer. Cells 2020, 9, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, K.S.; Volkmer, J.-P.; Weissman, I. Cancer Stem Cells in Bladder Cancer: A Revisited and Evolving Concept. Curr. Opin. Urol. 2010, 20, 393–397. [Google Scholar] [CrossRef] [Green Version]
- Xia, P.; Liu, D.-H.; Xu, Z.-J.; Ren, F. Cancer Stem Cell Markers for Urinary Carcinoma. Stem Cells Int. 2022, 2022, 3611677. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, H.; Zhang, J.; Zhu, L.; Wang, C.; Yang, Y. Unraveling the Roles of CD44/CD24 and ALDH1 as Cancer Stem Cell Markers in Tumorigenesis and Metastasis. Sci. Rep. 2017, 7, 13856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brugnoli, F.; Grassilli, S.; Al-Qassab, Y.; Capitani, S.; Bertagnolo, V. CD133 in Breast Cancer Cells: More than a Stem Cell Marker. J. Oncol. 2019, 2019, 7512632. [Google Scholar] [CrossRef] [Green Version]
- Fedyanin, M.; Anna, P.; Elizaveta, P.; Sergei, T. Role of Stem Cells in Colorectal Cancer Progression and Prognostic and Predictive Characteristics of Stem Cell Markers in Colorectal Cancer. Curr. Stem Cell. Res. Ther. 2017, 12, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xia, L.; Wang, H.; Oyang, L.; Su, M.; Liu, Q.; Lin, J.; Tan, S.; Tian, Y.; Liao, Q.; et al. Cancer Stem Cells in Progression of Colorectal Cancer. Oncotarget 2017, 9, 33403–33415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalantari, E.; Taheri, T.; Fata, S.; Abolhasani, M.; Mehrazma, M.; Madjd, Z.; Asgari, M. Significant Co-Expression of Putative Cancer Stem Cell Markers, EpCAM and CD166, Correlates with Tumor Stage and Invasive Behavior in Colorectal Cancer. World J. Surg. Oncol. 2022, 20, 15. [Google Scholar] [CrossRef] [PubMed]
- Razmi, M.; Ghods, R.; Vafaei, S.; Sahlolbei, M.; Saeednejad Zanjani, L.; Madjd, Z. Clinical and Prognostic Significances of Cancer Stem Cell Markers in Gastric Cancer Patients: A Systematic Review and Meta-Analysis. Cancer Cell. Int. 2021, 21, 139. [Google Scholar] [CrossRef]
- Jo, J.H.; Park, S.B.; Park, S.; Lee, H.S.; Kim, C.; Jung, D.E.; Song, S.Y. Novel Gastric Cancer Stem Cell-Related Marker LINGO2 Is Associated with Cancer Cell Phenotype and Patient Outcome. Int. J. Mol. Sci. 2019, 20, 555. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-L.; Chen, X.-Z.; Wang, Y.-G.; He, D.; Lu, Z.-H.; Liu, K.; Zhang, W.-H.; Wang, W.; Li, C.-C.; Xue, L.; et al. Clinical Significance of Putative Markers of Cancer Stem Cells in Gastric Cancer: A Retrospective Cohort Study. Oncotarget 2016, 7, 62049–62069. [Google Scholar] [CrossRef] [Green Version]
- Baroni, M.; Yi, C.; Choudhary, S.; Lei, X.; Kosti, A.; Grieshober, D.; Velasco, M.; Qiao, M.; Burns, S.S.; Araujo, P.R.; et al. Musashi1 Contribution to Glioblastoma Development via Regulation of a Network of DNA Replication, Cell Cycle and Division Genes. Cancers 2021, 13, 1494. [Google Scholar] [CrossRef]
- Chow, K.-H.; Park, H.J.; George, J.; Yamamoto, K.; Gallup, A.D.; Graber, J.H.; Chen, Y.; Jiang, W.; Steindler, D.A.; Neilson, E.G.; et al. S100A4 Is a Biomarker and Regulator of Glioma Stem Cells That Is Critical for Mesenchymal Transition in Glioblastoma. Cancer Res. 2017, 77, 5360–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalkan, R. Glioblastoma Stem Cells as a New Therapeutic Target for Glioblastoma. Clin. Med. Insights Oncol. 2015, 9, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.; Zuo, C.; Fang, P.; Liu, G.; Qiu, Y.; Huang, Y.; Tang, R. Targeting Glioblastoma Stem Cells: A Review on Biomarkers, Signal Pathways and Targeted Therapy. Front. Oncol. 2021, 11, 701291. [Google Scholar] [CrossRef]
- Wańkowicz, P.; Rogińska, D.; Machaliński, B.; Nowacki, P. Expression of Markers of Neural Stem and Progenitor Cells in Glioblastoma Multiforme in Relation to Tumor Recurrence and Overall Survival. Arch. Med. Sci. 2020, 16, 481–483. [Google Scholar] [CrossRef]
- Jeng, K.-S.; Chang, C.-F.; Sheen, I.-S.; Jeng, C.-J.; Wang, C.-H. Cellular and Molecular Biology of Cancer Stem Cells of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 1417. [Google Scholar] [CrossRef]
- Afify, S.M.; Sanchez Calle, A.; Hassan, G.; Kumon, K.; Nawara, H.M.; Zahra, M.H.; Mansour, H.M.; Khayrani, A.C.; Alam, M.J.; Du, J.; et al. A Novel Model of Liver Cancer Stem Cells Developed from Induced Pluripotent Stem Cells. Br. J. Cancer 2020, 122, 1378–1390. [Google Scholar] [CrossRef] [Green Version]
- Nio, K.; Yamashita, T.; Kaneko, S. The Evolving Concept of Liver Cancer Stem Cells. Mol. Cancer 2017, 16, 4. [Google Scholar] [CrossRef] [Green Version]
- Rebouissou, S.; Franconi, A.; Calderaro, J.; Letouzé, E.; Imbeaud, S.; Pilati, C.; Nault, J.-C.; Couchy, G.; Laurent, A.; Balabaud, C.; et al. Genotype-Phenotype Correlation of CTNNB1 Mutations Reveals Different ß-Catenin Activity Associated with Liver Tumor Progression. Hepatology 2016, 64, 2047–2061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sang, X.; Wu, F.; Wu, D.; Lin, S.; Li, J.; Zhao, N.; Chen, X.; Xu, A. Human Hepatic Cancer Stem Cells (HCSCs) Markers Correlated with Immune Infiltrates Reveal Prognostic Significance of Hepatocellular Carcinoma. Front. Genet. 2020, 11, 112. [Google Scholar] [CrossRef]
- Sun, J.-H.; Luo, Q.; Liu, L.-L.; Song, G.-B. Liver Cancer Stem Cell Markers: Progression and Therapeutic Implications. World J. Gastroenterol. 2016, 22, 3547–3557. [Google Scholar] [CrossRef] [PubMed]
- Tsui, Y.-M.; Chan, L.-K.; Ng, I.O.-L. Cancer Stemness in Hepatocellular Carcinoma: Mechanisms and Translational Potential. Br. J. Cancer 2020, 122, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.G.; Breen, E.; Cuffe, S.; Finn, S.P.; O’Byrne, K.J.; Barr, M.P. Lung Cancer Stem Cells: The Root of Resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, L.; Yin, L.; Yao, Z.; Tong, R.; Xue, J.; Lu, Y. Lung Cancer Stem Cell Markers as Therapeutic Targets: An Update on Signaling Pathways and Therapies. Front. Oncol. 2022, 12, 873994. [Google Scholar] [CrossRef]
- Yan, X.; Luo, H.; Zhou, X.; Zhu, B.; Wang, Y.; Bian, X. Identification of CD90 as a Marker for Lung Cancer Stem Cells in A549 and H446 Cell Lines. Oncol. Rep. 2013, 30, 2733–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, Y.; Bian, B.; Yang, X.; Cui, W.; Cui, H.; Cui, Y.; Zhang, X.; Xu, C.; Bian, X. POU5F1 Enhances the Invasiveness of Cancer Stem-Like Cells in Lung Adenocarcinoma by Upregulation of MMP-2 Expression. PLoS ONE 2013, 8, e83373. [Google Scholar] [CrossRef] [Green Version]
- Parmiani, G. Melanoma Cancer Stem Cells: Markers and Functions. Cancers 2016, 8, 34. [Google Scholar] [CrossRef] [Green Version]
- Pinc, A.; Somasundaram, R.; Wagner, C.; Hörmann, M.; Karanikas, G.; Jalili, A.; Bauer, W.; Brunner, P.; Grabmeier-Pfistershammer, K.; Gschaider, M.; et al. Targeting CD20 in Melanoma Patients at High Risk of Disease Recurrence. Mol. Ther. 2012, 20, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Setia, N.; Abbas, O.; Sousa, Y.; Garb, J.L.; Mahalingam, M. Profiling of ABC Transporters ABCB5, ABCF2 and Nestin-Positive Stem Cells in Nevi, in Situ and Invasive Melanoma. Mod. Pathol. 2012, 25, 1169–1175. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Annett, S.L.; Morgan, M.P.; Robson, T. The Cancer Stem Cell Niche in Ovarian Cancer and Its Impact on Immune Surveillance. Int. J. Mol. Sci. 2021, 22, 4091. [Google Scholar] [CrossRef] [PubMed]
- Keyvani, V.; Farshchian, M.; Esmaeili, S.-A.; Yari, H.; Moghbeli, M.; Nezhad, S.-R.K.; Abbaszadegan, M.R. Ovarian Cancer Stem Cells and Targeted Therapy. J. Ovarian Res. 2019, 12, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Królewska-Daszczyńska, P.; Wendlocha, D.; Smycz-Kubańska, M.; Stępień, S.; Mielczarek-Palacz, A. Cancer Stem Cells Markers in Ovarian Cancer: Clinical and Therapeutic Significance (Review). Oncol. Lett. 2022, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Motohara, T.; Yoshida, G.J.; Katabuchi, H. The Hallmarks of Ovarian Cancer Stem Cells and Niches: Exploring Their Harmonious Interplay in Therapy Resistance. Semin. Cancer Biol. 2021, 77, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Terraneo, N.; Jacob, F.; Dubrovska, A.; Grünberg, J. Novel Therapeutic Strategies for Ovarian Cancer Stem Cells. Front. Oncol. 2020, 10, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gzil, A.; Zarębska, I.; Bursiewicz, W.; Antosik, P.; Grzanka, D.; Szylberg, Ł. Markers of Pancreatic Cancer Stem Cells and Their Clinical and Therapeutic Implications. Mol. Biol. Rep. 2019, 46, 6629–6645. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of Human Pancreatic Cancer Stem Cells. In Cancer Stem Cells: Methods and Protocols; Yu, J.S., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2009; pp. 161–173. ISBN 978-1-59745-280-9. [Google Scholar]
- Lin, V.C.; Huang, S.-P.; Huang, C.-Y.; Yu, C.-C.; Yin, H.-L.; Huang, T.-Y.; Lee, C.-H.; Lu, T.-L.; Bao, B.-Y. Cancer Stem Cell Gene Variants Predict Disease Recurrence in Patients Treated with Radical Prostatectomy for Prostate Cancer. Int. J. Med. Sci. 2017, 14, 1301–1306. [Google Scholar] [CrossRef] [Green Version]
- Wolf, I.; Gratzke, C.; Wolf, P. Prostate Cancer Stem Cells: Clinical Aspects and Targeted Therapies. Front. Oncol. 2022, 12, 935715. [Google Scholar] [CrossRef]
- Houshmand, M.; Kazemi, A.; Anjam Najmedini, A.; Ali, M.S.; Gaidano, V.; Cignetti, A.; Fava, C.; Cilloni, D.; Saglio, G.; Circosta, P. Shedding Light on Targeting Chronic Myeloid Leukemia Stem Cells. J. Clin. Med. 2021, 10, 5805. [Google Scholar] [CrossRef]
- Mojtahedi, H.; Yazdanpanah, N.; Rezaei, N. Chronic Myeloid Leukemia Stem Cells: Targeting Therapeutic Implications. Stem Cell Res. Ther. 2021, 12, 603. [Google Scholar] [CrossRef]
- Soverini, S.; De Santis, S.; Monaldi, C.; Bruno, S.; Mancini, M. Targeting Leukemic Stem Cells in Chronic Myeloid Leukemia: Is It Worth the Effort? Int. J. Mol. Sci. 2021, 22, 7093. [Google Scholar] [CrossRef]
- Arrigoni, E.; Del Re, M.; Galimberti, S.; Restante, G.; Rofi, E.; Crucitta, S.; Baratè, C.; Petrini, M.; Danesi, R.; Di Paolo, A. Concise Review: Chronic Myeloid Leukemia: Stem Cell Niche and Response to Pharmacologic Treatment. Stem Cells Transl. Med. 2018, 7, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Gao, H.; Zhang, Q. The Biomarkers of Leukemia Stem Cells in Acute Myeloid Leukemia. Stem Cell Investig. 2017, 4, 19. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, G.; Zhang, J.; Chen, X.; Xu, H.; Heng, G.; Chen, J.; Zhao, Y.; Li, J.; Ni, Y.; et al. CD9, a Potential Leukemia Stem Cell Marker, Regulates Drug Resistance and Leukemia Development in Acute Myeloid Leukemia. Stem Cell Res. Ther. 2021, 12, 86. [Google Scholar] [CrossRef]
- Picot, T.; Aanei, C.M.; Fayard, A.; Flandrin-Gresta, P.; Tondeur, S.; Gouttenoire, M.; Tavernier-Tardy, E.; Wattel, E.; Guyotat, D.; Campos, L. Expression of Embryonic Stem Cell Markers in Acute Myeloid Leukemia. Tumour Biol. 2017, 39, 1010428317716629. [Google Scholar] [CrossRef] [Green Version]
- Kakiuchi, S.; Minami, Y.; Miyata, Y.; Mizutani, Y.; Goto, H.; Kawamoto, S.; Yakushijin, K.; Kurata, K.; Matsuoka, H.; Minami, H. NANOG Expression as a Responsive Biomarker during Treatment with Hedgehog Signal Inhibitor in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2017, 18, 486. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Yao, R.; Wang, H. Update of ALDH as a Potential Biomarker and Therapeutic Target for AML. Biomed. Res. Int. 2018, 2018, 9192104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, W.H. Cancer Stem Cell Signaling Pathways. Medicine 2016, 95, S8–S19. [Google Scholar] [CrossRef]
- Akbar Samadani, A.; Keymoradzdeh, A.; Shams, S.; Soleymanpour, A.; Elham Norollahi, S.; Vahidi, S.; Rashidy-Pour, A.; Ashraf, A.; Mirzajani, E.; Khanaki, K.; et al. Mechanisms of Cancer Stem Cell Therapy. Clin. Chim. Acta 2020, 510, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Guerrero, R.; Belmonte-Fernández, A.; Flores, M.L.; González-Moreno, M.; Pérez-Valderrama, B.; Romero, F.; Japón, M.Á.; Sáez, C. Wnt/β-Catenin Signaling Contributes to Paclitaxel Resistance in Bladder Cancer Cells with Cancer Stem Cell-Like Properties. Int. J. Mol. Sci. 2021, 23, 450. [Google Scholar] [CrossRef] [PubMed]
- Dolatabadi, S.; Jonasson, E.; Lindén, M.; Fereydouni, B.; Bäcksten, K.; Nilsson, M.; Martner, A.; Forootan, A.; Fagman, H.; Landberg, G.; et al. JAK–STAT Signalling Controls Cancer Stem Cell Properties Including Chemotherapy Resistance in Myxoid Liposarcoma. Int. J. Cancer 2019, 145, 435–449. [Google Scholar] [CrossRef] [Green Version]
- Hallis, S.P.; Kim, J.M.; Kwak, M.-K. Emerging Role of NRF2 Signaling in Cancer Stem Cell Phenotype. Mol. Cells 2023, 46, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Giuli, M.V.; Mancusi, A.; Giuliani, E.; Screpanti, I.; Checquolo, S. Notch Signaling in Female Cancers: A Multifaceted Node to Overcome Drug Resistance. Cancer Drug. Resist. 2021, 4, 805–836. [Google Scholar] [CrossRef]
- Manni, W.; Min, W. Signaling Pathways in the Regulation of Cancer Stem Cells and Associated Targeted Therapy. MedComm 2022, 3, e176. [Google Scholar] [CrossRef] [PubMed]
- Seydi, H.; Nouri, K.; Rezaei, N.; Tamimi, A.; Hassan, M.; Mirzaei, H.; Vosough, M. Autophagy Orchestrates Resistance in Hepatocellular Carcinoma Cells. Biomed. Pharm. 2023, 161, 114487. [Google Scholar] [CrossRef]
- Araki, K.; Miyoshi, Y. Mechanism of Resistance to Endocrine Therapy in Breast Cancer: The Important Role of PI3K/Akt/MTOR in Estrogen Receptor-Positive, HER2-Negative Breast Cancer. Breast Cancer 2018, 25, 392–401. [Google Scholar] [CrossRef]
- Safa, A.R. Drug and Apoptosis Resistance in Cancer Stem Cells: A Puzzle with Many Pieces. Cancer Drug. Resist. 2022, 5, 850–872. [Google Scholar] [CrossRef]
- Al Bitar, S.; El-Sabban, M.; Doughan, S.; Abou-Kheir, W. Molecular Mechanisms Targeting Drug-Resistance and Metastasis in Colorectal Cancer: Updates and Beyond. World J. Gastroenterol. 2023, 29, 1395–1426. [Google Scholar] [CrossRef]
- Verma, P.; Shukla, N.; Kumari, S.; Ansari, M.S.; Gautam, N.K.; Patel, G.K. Cancer Stem Cell in Prostate Cancer Progression, Metastasis and Therapy Resistance. Biochim. Biophys. Acta Rev. Cancer 2023, 1878, 188887. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Zhao, H.; Wei, Y.; Zhou, Y.; Zhang, S.; Zhao, J.; Li, X.; Lin, Y.; Liu, K. The Roles of the SOX2 Protein in the Development of Esophagus and Esophageal Squamous Cell Carcinoma, and Pharmacological Target for Therapy. Biomed. Pharm. 2023, 163, 114764. [Google Scholar] [CrossRef] [PubMed]
- Deldar Abad Paskeh, M.; Mirzaei, S.; Ashrafizadeh, M.; Zarrabi, A.; Sethi, G. Wnt/β-Catenin Signaling as a Driver of Hepatocellular Carcinoma Progression: An Emphasis on Molecular Pathways. J. Hepatocell. Carcinoma 2021, 8, 1415–1444. [Google Scholar] [CrossRef] [PubMed]
- Furlan, G.; Huyghe, A.; Combémorel, N.; Lavial, F. Molecular Versatility during Pluripotency Progression. Nat. Commun. 2023, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Bakyt, L.; Akhmetkaliyev, A.; Toktarkhanova, D.; Bulanin, D. Re-Sensitizing Cancer Stem Cells to Conventional Chemotherapy Agents. Int. J. Mol. Sci. 2023, 24, 2122. [Google Scholar] [CrossRef] [PubMed]
- Park, H.W.; Kim, Y.C.; Yu, B.; Moroishi, T.; Mo, J.-S.; Plouffe, S.W.; Meng, Z.; Lin, K.C.; Yu, F.-X.; Alexander, C.M.; et al. Alternative Wnt Signaling Activates YAP/TAZ. Cell 2015, 162, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt Signaling in Cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Tang, P.; Li, S.; Qin, X.; Yang, H.; Wu, C.; Liu, Y. Notch Signaling Pathway Networks in Cancer Metastasis: A New Target for Cancer Therapy. Med. Oncol. 2017, 34, 180. [Google Scholar] [CrossRef]
- Zhou, B.; Lin, W.; Long, Y.; Yang, Y.; Zhang, H.; Wu, K.; Chu, Q. Notch Signaling Pathway: Architecture, Disease, and Therapeutics. Signal Transduct. Target. Ther. 2022, 7, 1–33. [Google Scholar] [CrossRef]
- Hanna, A.; Shevde, L.A. Hedgehog Signaling: Modulation of Cancer Properies and Tumor Mircroenvironment. Mol. Cancer 2016, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Hao, K.; Tian, X.-D.; Qin, C.-F.; Xie, X.-H.; Yang, Y.-M. Hedgehog Signaling Pathway Regulates Human Pancreatic Cancer Cell Proliferation and Metastasis. Oncol. Rep. 2013, 29, 1124–1132. [Google Scholar] [CrossRef] [Green Version]
- Yao, Z.; Han, L.; Chen, Y.; He, F.; Sun, B.; Kamar, S.; Zhang, Y.; Yang, Y.; Wang, C.; Yang, Z. Hedgehog Signalling in the Tumourigenesis and Metastasis of Osteosarcoma, and Its Potential Value in the Clinical Therapy of Osteosarcoma. Cell Death Dis. 2018, 9, 701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.J. YAP/TAZ: Drivers of Tumor Growth, Metastasis, and Resistance to Therapy. Bioessays 2020, 42, e1900162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Taouk, G.M. A Potential Role of YAP/TAZ in the Interplay between Metastasis and Metabolic Alterations. Front. Oncol. 2020, 10, 928. [Google Scholar] [CrossRef] [PubMed]
- Weigmann, A.; Corbeil, D.; Hellwig, A.; Huttner, W.B. Prominin, a Novel Microvilli-Specific Polytopic Membrane Protein of the Apical Surface of Epithelial Cells, Is Targeted to Plasmalemmal Protrusions of Non-Epithelial Cells. Proc. Natl. Acad. Sci. USA 1997, 94, 12425–12430. [Google Scholar] [CrossRef] [Green Version]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A Novel Five-Transmembrane Hematopoietic Stem Cell Antigen: Isolation, Characterization, and Molecular Cloning. Blood 1997, 90, 5013–5021. [Google Scholar] [CrossRef]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a Novel Marker for Human Hematopoietic Stem and Progenitor Cells. Blood 1997, 90, 5002–5012. [Google Scholar] [CrossRef] [Green Version]
- Schmohl, J.; Vallera, D. CD133, Selectively Targeting the Root of Cancer. Toxins 2016, 8, 165. [Google Scholar] [CrossRef] [Green Version]
- Akbari, M.; Shanehbandi, D.; Asadi, M.; Shomali, N.; Faraji, A.; Khaze, V.; Pakdel, A.; Mokhtarzadeh, A.; Ebrahimi, A.A.; Shabani, A.; et al. Effects of CD133 Silencing on Survival and Migration of HT-29 Colorectal Cancer Cells. Iran. J. Immunol. 2019, 16. [Google Scholar] [CrossRef]
- Brendel, C.; Goebel, B.; Daniela, A.; Brugman, M.; Kneissl, S.; Schwäble, J.; Kaufmann, K.B.; Müller-Kuller, U.; Kunkel, H.; Chen-Wichmann, L.; et al. CD133-Targeted Gene Transfer into Long-Term Repopulating Hematopoietic Stem Cells. Mol. Ther. 2015, 23, 63–70. [Google Scholar] [CrossRef]
- Hefni, A.M.; Sayed, A.M.; Hussien, M.T.; Abdalla, A.Z.; Gabr, A.G. CD133 Is an Independent Predictive and Prognostic Marker in Metastatic Breast Cancer. Cancer Biomark. 2022, 35, 207–215. [Google Scholar] [CrossRef]
- Ikram, D.; Masadah, R.; Nelwan, B.J.; Zainuddin, A.A.; Ghaznawie, M.; Wahid, S. CD133 Act as an Essential Marker in Ovarian Carcinogenesis. Asian Pac. J. Cancer Prev. 2021, 22, 3525–3531. [Google Scholar] [CrossRef]
- Kim, J.; Shin, K.; Lee, S.H.; Kim, I.-H. Slug and CD133 Expression Are Associated with Peritoneal Carcinomatosis and Survival in Gastric Cancer. J. Gastrointest. Oncol. 2021, 12, 1326–1337. [Google Scholar] [CrossRef] [PubMed]
- Koren, A.; Rijavec, M.; Kern, I.; Sodja, E.; Korosec, P.; Cufer, T. BMI1, ALDH1A1, and CD133 Transcripts Connect Epithelial-Mesenchymal Transition to Cancer Stem Cells in Lung Carcinoma. Stem Cells Int. 2016, 2016, 9714315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.Y.; An, C.H.; Oh, S.T.; Chang, E.D.; Lee, J. Expression of CD133 Is Associated with Poor Prognosis in Stage II Colorectal Carcinoma. Medicine 2019, 98, e16709. [Google Scholar] [CrossRef]
- Pustovalova, M.; Blokhina, T.; Alhaddad, L.; Chigasova, A.; Chuprov-Netochin, R.; Veviorskiy, A.; Filkov, G.; Osipov, A.N.; Leonov, S. CD44+ and CD133+ Non-Small Cell Lung Cancer Cells Exhibit DNA Damage Response Pathways and Dormant Polyploid Giant Cancer Cell Enrichment Relating to Their P53 Status. Int. J. Mol. Sci. 2022, 23, 4922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Hua, R.; Wang, X.; Huang, M.; Gan, L.; Wu, Z.; Zhang, J.; Wang, H.; Cheng, Y.; Li, J.; et al. Identification of Stem-like Cells and Clinical Significance of Candidate Stem Cell Markers in Gastric Cancer. Oncotarget 2016, 7, 9815–9831. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Dai, C.; Yang, Y.; Wu, X.; Wang, L.; Wang, P. The Expression Levels and Clinical Significance of MFG-E8 and CD133 in Epithelial Ovarian Cancer. Gynecol. Endocrinol. 2020, 36, 803–807. [Google Scholar] [CrossRef]
- Wei, F.; Zhang, T.; Deng, S.-C.; Wei, J.-C.; Yang, P.; Wang, Q.; Chen, Z.-P.; Li, W.-L.; Chen, H.-C.; Hu, H.; et al. PD-L1 Promotes Colorectal Cancer Stem Cell Expansion by Activating HMGA1-Dependent Signaling Pathways. Cancer Lett. 2019, 450, 1–13. [Google Scholar] [CrossRef]
- PROM1 Prominin 1 [Homo Sapiens (Human)]-Gene-NCBI. Available online: https://www.ncbi.nlm.nih.gov/gene/8842 (accessed on 22 January 2023).
- PROM1-Prominin-1-Homo Sapiens (Human)|UniProtKB|UniProt. Available online: https://www.uniprot.org/uniprotkb/O43490/entry#structure (accessed on 22 January 2023).
- Irollo, E.; Pirozzi, G. CD133: To Be or Not to Be, Is This the Real Question? Am. J. Transl. Res. 2013, 5, 563–581. [Google Scholar]
- Glumac, P.M.; LeBeau, A.M. The Role of CD133 in Cancer: A Concise Review. Clin. Transl. Med. 2018, 7, 18. [Google Scholar] [CrossRef]
- Peh, G.S.-L.; Lang, R.J.; Pera, M.F.; Hawes, S.M. CD133 Expression by Neural Progenitors Derived from Human Embryonic Stem Cells and Its Use for Their Prospective Isolation. Stem Cells Dev. 2009, 18, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Opdenakker, G.; Abu El-Asrar, A. Metalloproteinases Mediate Diabetes-Induced Retinal Neuropathy and Vasculopathy. Cell. Mol. Life Sci. 2019, 76, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Jászai, J.; Fargeas, C.A.; Florek, M.; Huttner, W.B.; Corbeil, D. Focus on Molecules: Prominin-1 (CD133). Exp. Eye Res. 2007, 85, 585–586. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; O’Bara, M.A.; Pol, S.U.; Sim, F.J. CD133/CD140a-Based Isolation of Distinct Human Multipotent Neural Progenitor Cells and Oligodendrocyte Progenitor Cells. Stem Cells Dev. 2013, 22, 2121–2131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, D.K.; Bonaguidi, M.A.; Ming, G.; Song, H. Adult Neural Stem Cells in the Mammalian Central Nervous System. Cell Res. 2009, 19, 672–682. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.B.; Ji, X.Y.; Huang, Q.; Dong, J.; Zhu, Y.D.; Lan, Q. Differentiation Profile of Brain Tumor Stem Cells: A Comparative Study with Neural Stem Cells. Cell Res. 2006, 16, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Fontán-Lozano, Á.; Morcuende, S.; Davis-López de Carrizosa, M.A.; Benítez-Temiño, B.; Mejías, R.; Matarredona, E.R. To Become or Not to Become Tumorigenic: Subventricular Zone Versus Hippocampal Neural Stem Cells. Front. Oncol. 2020, 10, 602217. [Google Scholar] [CrossRef]
- Brossa, A.; Papadimitriou, E.; Collino, F.; Incarnato, D.; Oliviero, S.; Camussi, G.; Bussolati, B. Role of CD133 Molecule in Wnt Response and Renal Repair. Stem Cells Transl. Med. 2018, 7, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Grange, C.; Iampietro, C.; Camussi, G.; Bussolati, B. Human CD133+ Renal Progenitor Cells Induce Erythropoietin Production and Limit Fibrosis after Acute Tubular Injury. Sci. Rep. 2016, 6, 37270. [Google Scholar] [CrossRef]
- Liu, F.; Qian, Y. The Role of CD133 in Hepatocellular Carcinoma. Cancer Biol. Ther. 2021, 22, 291–300. [Google Scholar] [CrossRef]
- Kordes, C.; Sawitza, I.; Müller-Marbach, A.; Ale-Agha, N.; Keitel, V.; Klonowski-Stumpe, H.; Häussinger, D. CD133+ Hepatic Stellate Cells Are Progenitor Cells. Biochem. Biophys. Res. Commun. 2007, 352, 410–417. [Google Scholar] [CrossRef] [Green Version]
- Boulter, L.; Lu, W.-Y.; Forbes, S.J. Differentiation of Progenitors in the Liver: A Matter of Local Choice. J. Clin. Investig. 2013, 123, 1867–1873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, S.; Russell, J.O.; Molina, L.M.; Monga, S.P. Liver Progenitors and Adult Cell Plasticity in Hepatic Injury and Repair: Knowns and Unknowns. Annu. Rev. Pathol. 2020, 15, 23–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadd, V.L.; Aleksieva, N.; Forbes, S.J. Epithelial Plasticity during Liver Injury and Regeneration. Cell Stem Cell 2020, 27, 557–573. [Google Scholar] [CrossRef]
- Ma, S. Biology and Clinical Implications of CD133+ Liver Cancer Stem Cells. Exp. Cell Res. 2013, 319, 126–132. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, X.; Wang, C.; Wang, M.; Wang, Y.; Ye, M.; Liu, Y. Peptide-Based 68Ga-PET Radiotracer for Imaging CD133 Expression in Colorectal Cancer. Nucl. Med. Commun. 2021, 42, 1144–1150. [Google Scholar] [CrossRef]
- Lakowski, J.; Welby, E.; Budinger, D.; Di Marco, F.; Di Foggia, V.; Bainbridge, J.W.B.; Wallace, K.; Gamm, D.M.; Ali, R.R.; Sowden, J.C. Isolation of Human Photoreceptor Precursors via a Cell Surface Marker Panel from Stem Cell-Derived Retinal Organoids and Fetal Retinae. Stem Cells 2018, 36, 709–722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handgretinger, R.; Kuçi, S. CD133-Positive Hematopoietic Stem Cells: From Biology to Medicine. In Prominin-1 (CD133): New Insights on Stem & Cancer Stem Cell Biology; Corbeil, D., Ed.; Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2013; Volume 777, pp. 99–111. ISBN 978-1-4614-5893-7. [Google Scholar]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/MTOR Pathway in Breast Cancer: Targets, Trials and Biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Y.; Wang, Y.; Zhou, C.; Mei, W.; Zeng, C. PI3K/Akt/MTOR Pathway and Its Role in Cancer Therapeutics: Are We Making Headway? Front. Oncol. 2022, 12, 819128. [Google Scholar] [CrossRef]
- Xia, P.; Xu, X.-Y. PI3K/Akt/MTOR Signaling Pathway in Cancer Stem Cells: From Basic Research to Clinical Application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Mladinich, M.; Ruan, D.; Chan, C.-H. Tackling Cancer Stem Cells via Inhibition of EMT Transcription Factors. Stem Cells Int. 2016, 2016, 5285892. [Google Scholar] [CrossRef] [Green Version]
- Rinkenbaugh, A.L.; Baldwin, A.S. The NF-ΚB Pathway and Cancer Stem Cells. Cells 2016, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-κB Signaling in Inflammation and Cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Shi, X.; Jiang, M.; Liu, H. Cross-Talk between Cancer Stem Cells and Immune Cells: Potential Therapeutic Targets in the Tumor Immune Microenvironment. Mol. Cancer 2023, 22, 38. [Google Scholar] [CrossRef]
- Alisson-Silva, F.; de Carvalho Rodrigues, D.; Vairo, L.; Asensi, K.D.; Vasconcelos-dos-Santos, A.; Mantuano, N.R.; Dias, W.B.; Rondinelli, E.; Goldenberg, R.C.d.S.; Urmenyi, T.P.; et al. Evidences for the Involvement of Cell Surface Glycans in Stem Cell Pluripotency and Differentiation. Glycobiology 2014, 24, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Barkeer, S.; Chugh, S.; Batra, S.K.; Ponnusamy, M.P. Glycosylation of Cancer Stem Cells: Function in Stemness, Tumorigenesis, and Metastasis. Neoplasia 2018, 20, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Mallard, B.W.; Tiralongo, J. Cancer Stem Cell Marker Glycosylation: Nature, Function and Significance. Glycoconj. J. 2017, 34, 441–452. [Google Scholar] [CrossRef]
- Khan, T.; Cabral, H. Abnormal Glycosylation of Cancer Stem Cells and Targeting Strategies. Front. Oncol. 2021, 11, 649338. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-K.; Wu, J.-C.; Wang, H.; Zhou, L.; Zhang, C.; Cheng, R.; Kan, D.; Zhang, X.; Yu, C. Turning Solid into Gel for High-Efficient Persistent Luminescence-Sensitized Photodynamic Therapy. Biomaterials 2019, 218, 119328. [Google Scholar] [CrossRef] [PubMed]
- Starbuck, K.; Al-Alem, L.; Eavarone, D.A.; Hernandez, S.F.; Bellio, C.; Prendergast, J.M.; Stein, J.; Dransfield, D.T.; Zarrella, B.; Growdon, W.B.; et al. Treatment of Ovarian Cancer by Targeting the Tumor Stem Cell-Associated Carbohydrate Antigen, Sialyl-Thomsen-Nouveau. Oncotarget 2018, 9, 23289–23305. [Google Scholar] [CrossRef] [Green Version]
- Tivadar, S.T.; McIntosh, R.S.; Chua, J.X.; Moss, R.; Parsons, T.; Zaitoun, A.M.; Madhusudan, S.; Durrant, L.G.; Vankemmelbeke, M. Monoclonal Antibody Targeting Sialyl-Di-Lewisa–Containing Internalizing and Noninternalizing Glycoproteins with Cancer Immunotherapy Development Potential. Mol. Cancer Ther. 2020, 19, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Berois, N.; Pittini, A.; Osinaga, E. Targeting Tumor Glycans for Cancer Therapy: Successes, Limitations, and Perspectives. Cancers 2022, 14, 645. [Google Scholar] [CrossRef] [PubMed]
- Diniz, F.; Coelho, P.; Duarte, H.O.; Sarmento, B.; Reis, C.A.; Gomes, J. Glycans as Targets for Drug Delivery in Cancer. Cancers 2022, 14, 911. [Google Scholar] [CrossRef]
- Tang, R.; Xu, Z. Gene Therapy: A Double-Edged Sword with Great Powers. Mol. Cell. Biochem. 2020, 474, 73–81. [Google Scholar] [CrossRef]
- Anliker, B.; Abel, T.; Kneissl, S.; Hlavaty, J.; Caputi, A.; Brynza, J.; Schneider, I.C.; Münch, R.C.; Petznek, H.; Kontermann, R.E.; et al. Specific Gene Transfer to Neurons, Endothelial Cells and Hematopoietic Progenitors with Lentiviral Vectors. Nat. Methods 2010, 7, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Bayin, N.S.; Modrek, A.S.; Dietrich, A.; Lebowitz, J.; Abel, T.; Song, H.-R.; Schober, M.; Zagzag, D.; Buchholz, C.J.; Chao, M.V.; et al. Selective Lentiviral Gene Delivery to CD133-Expressing Human Glioblastoma Stem Cells. PLoS ONE 2014, 9, e116114. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mansoori, B.; Mohammadi, A.; Kazemi, T.; Mokhtarzadeh, A.; Shanehbandi, D.; Hemmat, N.; Derakhshani, A.; Brunetti, O.; Safaei, S.; et al. The Combination Effect of Prominin1 (CD133) Suppression and Oxaliplatin Treatment in Colorectal Cancer Therapy. Biomed. Pharmacother. 2021, 137, 111364. [Google Scholar] [CrossRef] [PubMed]
- Nygren, P. What Is Cancer Chemotherapy? Acta Oncol. 2001, 40, 166–174. [Google Scholar] [CrossRef]
- Zhou, G.; Da Won Bae, S.; Nguyen, R.; Huo, X.; Han, S.; Zhang, Z.; Hebbard, L.; Duan, W.; Eslam, M.; Liddle, C.; et al. An Aptamer-Based Drug Delivery Agent (CD133-Apt-Dox) Selectively and Effectively Kills Liver Cancer Stem-like Cells. Cancer Lett. 2021, 501, 124–132. [Google Scholar] [CrossRef]
- Yin, W.; Pham, C.V.; Wang, T.; Al Shamaileh, H.; Chowdhury, R.; Patel, S.; Li, Y.; Kong, L.; Hou, Y.; Zhu, Y.; et al. Inhibition of Autophagy Promotes the Elimination of Liver Cancer Stem Cells by CD133 Aptamer-Targeted Delivery of Doxorubicin. Biomolecules 2022, 12, 1623. [Google Scholar] [CrossRef]
- Wang, C.-H.; Chiou, S.-H.; Chou, C.-P.; Chen, Y.-C.; Huang, Y.-J.; Peng, C.-A. Photothermolysis of Glioblastoma Stem-like Cells Targeted by Carbon Nanotubes Conjugated with CD133 Monoclonal Antibody. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 69–79. [Google Scholar] [CrossRef]
- Kim, J.S.; Shin, D.H.; Kim, J.-S. Dual-Targeting Immunoliposomes Using Angiopep-2 and CD133 Antibody for Glioblastoma Stem Cells. J. Control. Release 2018, 269, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Shigdar, S.; Qiao, L.; Zhou, S.-F.; Xiang, D.; Wang, T.; Li, Y.; Lim, L.Y.; Kong, L.; Li, L.; Duan, W. RNA Aptamers Targeting Cancer Stem Cell Marker CD133. Cancer Lett. 2013, 330, 84–95. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Sun, Y.; Ye, G.; Zhao, Y.; Wu, J. Effects of CD133 Expression on Chemotherapy and Drug Sensitivity of Adenoid Cystic Carcinoma. Mol. Med. Rep. 2022, 25, 18. [Google Scholar] [CrossRef]
- Xi, G.; Li, Y.D.; Grahovac, G.; Rajaram, V.; Wadhwani, N.; Pundy, T.; Mania-Farnell, B.; James, C.D.; Tomita, T. Targeting CD133 Improves Chemotherapeutic Efficacy of Recurrent Pediatric Pilocytic Astrocytoma Following Prolonged Chemotherapy. Mol. Cancer 2017, 16, 21. [Google Scholar] [CrossRef] [Green Version]
- Schuster, M.; Nechansky, A.; Kircheis, R. Cancer Immunotherapy. Biotechnol. J. 2006, 1, 138–147. [Google Scholar] [CrossRef]
- Itai, S.; Fujii, Y.; Nakamura, T.; Chang, Y.-W.; Yanaka, M.; Saidoh, N.; Handa, S.; Suzuki, H.; Harada, H.; Yamada, S.; et al. Establishment of CMab-43, a Sensitive and Specific Anti-CD133 Monoclonal Antibody, for Immunohistochemistry. Monoclon. Antibodies Immunodiagn. Immunother. 2017, 36, 231–235. [Google Scholar] [CrossRef]
- Kato, Y.; Ohishi, T.; Yamada, S.; Itai, S.; Furusawa, Y.; Sano, M.; Nakamura, T.; Kawada, M.; Kaneko, M.K. Anti-CD133 Monoclonal Antibody CMab-43 Exerts Antitumor Activity in a Mouse Xenograft Model of Colon Cancer. Monoclon. Antibodies Immunodiagn. Immunother. 2019, 38, 75–78. [Google Scholar] [CrossRef]
- Vora, P.; Venugopal, C.; Salim, S.K.; Tatari, N.; Bakhshinyan, D.; Singh, M.; Seyfrid, M.; Upreti, D.; Rentas, S.; Wong, N.; et al. The Rational Development of CD133-Targeting Immunotherapies for Glioblastoma. Cell Stem Cell 2020, 26, 832–844.e6. [Google Scholar] [CrossRef]
- Sangsuwannukul, T.; Supimon, K.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; Yenchitsomanus, P. Anti-Tumour Effect of the Fourth-Generation Chimeric Antigen Receptor T Cells Targeting CD133 against Cholangiocarcinoma Cells. Int. Immunopharmacol. 2020, 89, 107069. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, M.; Wu, Z.; Tong, C.; Dai, H.; Guo, Y.; Liu, Y.; Huang, J.; Lv, H.; Luo, C.; et al. CD133-Directed CAR T Cells for Advanced Metastasis Malignancies: A Phase I Trial. OncoImmunology 2018, 7, e1440169. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.; Tong, C.; Shi, D.; Chen, M.; Guo, Y.; Chen, D.; Han, X.; Wang, H.; Wang, Y.; Shen, P. Efficacy and Biomarker Analysis of CD133-Directed CAR T Cells in Advanced Hepatocellular Carcinoma: A Single-Arm, Open-Label, Phase II Trial. OncoImmunology 2020, 9, 1846926. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Zou, Y.; Zhang, L.; Tang, J.; Niedermann, G.; Firat, E.; Huang, X.; Zhu, X. Nucleofection with Plasmid DNA for CRISPR/Cas9-Mediated Inactivation of Programmed Cell Death Protein 1 in CD133-Specific CAR T Cells. Hum. Gene Ther. 2019, 30, 446–458. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.; Guo, Y.; Liu, Y.; Dai, H.; Wang, Y.; Lv, H.; Huang, J.; Yang, Q.; Han, W. Cocktail Treatment with EGFR-Specific and CD133-Specific Chimeric Antigen Receptor-Modified T Cells in a Patient with Advanced Cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef] [Green Version]
- Hak, A.; Ravasaheb Shinde, V.; Rengan, A.K. A Review of Advanced Nanoformulations in Phototherapy for Cancer Therapeutics. Photodiagnosis Photodyn. Ther. 2021, 33, 102205. [Google Scholar] [CrossRef]
- Bartusik-Aebisher, D.; Ożóg, Ł.; Aebisher, D. Alternative Methods of Photodynamic Therapy and Oxygen Consumption Measurements—A Review. Biomed. Pharmacother. 2021, 134, 111095. [Google Scholar] [CrossRef]
- Yan, S.; Tang, D.; Hong, Z.; Wang, J.; Yao, H.; Lu, L.; Yi, H.; Fu, S.; Zheng, C.; He, G.; et al. CD133 Peptide-Conjugated Pyropheophorbide-a as a Novel Photosensitizer for Targeted Photodynamic Therapy in Colorectal Cancer Stem Cells. Biomater. Sci. 2021, 9, 2020–2031. [Google Scholar] [CrossRef]
- Jerjes, W.; Theodossiou, T.A.; Hirschberg, H.; Høgset, A.; Weyergang, A.; Selbo, P.K.; Hamdoon, Z.; Hopper, C.; Berg, K. Photochemical Internalization for Intracellular Drug Delivery. From Basic Mechanisms to Clinical Research. J. Clin. Med. 2020, 9, 528. [Google Scholar] [CrossRef] [Green Version]
- Berg, K. Photochemical Internalization (PCI)-an Intracellular Drug Delivery Technology for Treatment of Solid Tumors. Photodiagnosis Photodyn. Ther. 2023, 41, 103453. [Google Scholar] [CrossRef]
- Olsen, C.E.; Cheung, L.H.; Weyergang, A.; Berg, K.; Vallera, D.A.; Rosenblum, M.G.; Selbo, P.K. Design, Characterization, and Evaluation of ScFvCD133/RGelonin: A CD133-Targeting Recombinant Immunotoxin for Use in Combination with Photochemical Internalization. J. Clin. Med. 2020, 9, 68. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Sun, M.; Li, W.; Fan, L.; Zhou, Y.; Hu, Z. A Novel CD133- and EpCAM-Targeted Liposome with Redox-Responsive Properties Capable of Synergistically Eliminating Liver Cancer Stem Cells. Front. Chem. 2020, 8, 649. [Google Scholar] [CrossRef]
- Lee, H.H.; Seo, K.J.; An, C.H.; Kim, J.S.; Jeon, H.M. CD133 Expression Is Correlated with Chemoresistance and Early Recurrence of Gastric Cancer. J. Surg. Oncol. 2012, 106, 999–1004. [Google Scholar] [CrossRef]
- Joseph, C.; Arshad, M.; Kurozomi, S.; Althobiti, M.; Miligy, I.M.; Al-izzi, S.; Toss, M.S.; Goh, F.Q.; Johnston, S.J.; Martin, S.G.; et al. Overexpression of the Cancer Stem Cell Marker CD133 Confers a Poor Prognosis in Invasive Breast Cancer. Breast Cancer Res. Treat. 2019, 174, 387–399. [Google Scholar] [CrossRef]
- Yamashita, N.; Oyama, T.; So, T.; Miyata, T.; Yoshimatsu, T.; Nakano, R.; Matsunaga, W.; Gotoh, A. Association Between CD133 Expression and Prognosis in Human Lung Adenocarcinoma. Anticancer. Res. 2021, 41, 905–910. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, X.; Chang, D.Y.; Rosen, D.G.; Mercado-Uribe, I.; Liu, J. CD133 Expression Associated with Poor Prognosis in Ovarian Cancer. Mod. Pathol. 2012, 25, 456–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdoli Shadbad, M.; Hosseinkhani, N.; Asadzadeh, Z.; Brunetti, O.; Silvestris, N.; Baradaran, B. The Prognostic Value of CD133 in Predicting the Relapse and Recurrence Pattern of High-Grade Gliomas on MRI: A Meta-Analysis. Front. Oncol. 2021, 11, 722833. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, N.; Yan, Z.; Li, C.; Zhao, Z. MiR-29a-Mediated CD133 Expression Contributes to Cisplatin Resistance in CD133+ Glioblastoma Stem Cells. J. Mol. Neurosci. 2018, 66, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly Tumorigenic Lung Cancer CD133+ Cells Display Stem-like Features and Are Spared by Cisplatin Treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Shi, S.; Yen, Y.; Brown, J.; Ta, J.Q.; Le, A.D. A Subpopulation of CD133+ Cancer Stem-like Cells Characterized in Human Oral Squamous Cell Carcinoma Confer Resistance to Chemotherapy. Cancer Lett. 2010, 289, 151–160. [Google Scholar] [CrossRef]
- Paschall, A.V.; Yang, D.; Lu, C.; Redd, P.S.; Choi, J.-H.; Heaton, C.M.; Lee, J.R.; Nayak-Kapoor, A.; Liu, K. CD133+CD24lo Defines a 5-Fluorouracil-Resistant Colon Cancer Stem Cell-like Phenotype. Oncotarget 2016, 7, 78698–78712. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.-H.; Ro, E.J.; Yoon, J.-S.; Mizutani, T.; Kang, D.-W.; Park, J.-C.; Il Kim, T.; Clevers, H.; Choi, K.-Y. 5-FU Promotes Stemness of Colorectal Cancer via P53-Mediated WNT/β-Catenin Pathway Activation. Nat. Commun. 2020, 11, 5321. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Kanwar, S.S.; Patel, B.B.; Nautiyal, J.; Sarkar, F.H.; Majumdar, A.P.N. Elimination of Colon Cancer Stem-Like Cells by the Combination of Curcumin and FOLFOX. Transl. Oncol. 2009, 2, 321–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.-P.; Yang, C.-J.; Huang, M.-S.; Yeh, C.-T.; Wu, A.T.H.; Lee, Y.-C.; Lai, T.-C.; Lee, C.-H.; Hsiao, Y.-W.; Lu, J.; et al. Cisplatin Selects for Multidrug-Resistant CD133+ Cells in Lung Adenocarcinoma by Activating Notch Signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Zeng, H.; Ying, K. The Combination of Stem Cell Markers CD133 and ABCG2 Predicts Relapse in Stage I Non-Small Cell Lung Carcinomas. Med. Oncol. 2011, 28, 1458–1462. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, R.; Deng, L.; Shuai, Z.; Chen, M. The Effect of Metformin on the Proliferation, Apoptosis and CD133 MRNA Expression of Colon Cancer Stem Cells by Upregulation of MiR 342-3p. Drug. Des. Dev. Ther. 2021, 15, 4633–4647. [Google Scholar] [CrossRef]
- Gou, S.; Cui, P.; Li, X.; Shi, P.; Liu, T.; Wang, C. Low Concentrations of Metformin Selectively Inhibit CD133+ Cell Proliferation in Pancreatic Cancer and Have Anticancer Action. PLoS ONE 2013, 8, e63969. [Google Scholar] [CrossRef] [PubMed]
- Chai, X.; Chu, H.; Yang, X.; Meng, Y.; Shi, P.; Gou, S. Metformin Increases Sensitivity of Pancreatic Cancer Cells to Gemcitabine by Reducing CD133+ Cell Populations and Suppressing ERK/P70S6K Signaling. Sci. Rep. 2015, 5, 14404. [Google Scholar] [CrossRef] [Green Version]
- Maehara, O.; Ohnishi, S.; Asano, A.; Suda, G.; Natsuizaka, M.; Nakagawa, K.; Kobayashi, M.; Sakamoto, N.; Takeda, H. Metformin Regulates the Expression of CD133 Through the AMPK-CEBPβ Pathway in Hepatocellular Carcinoma Cell Lines. Neoplasia 2019, 21, 545–556. [Google Scholar] [CrossRef]
- Brown, J.R.; Chan, D.K.; Shank, J.J.; Griffith, K.A.; Fan, H.; Szulawski, R.; Yang, K.; Reynolds, R.K.; Johnston, C.; McLean, K.; et al. Phase II Clinical Trial of Metformin as a Cancer Stem Cell–Targeting Agent in Ovarian Cancer. JCI Insight 2020, 5, e133247. [Google Scholar] [CrossRef]
- Song, Y.; Kim, I.-K.; Choi, I.; Kim, S.-H.; Seo, H.R. Oxytetracycline Have the Therapeutic Efficiency in CD133+ HCC Population through Suppression CD133 Expression by Decreasing of Protein Stability of CD133. Sci. Rep. 2018, 8, 16100. [Google Scholar] [CrossRef] [Green Version]
- Moon, C.M.; Kwon, J.-H.; Kim, J.S.; Oh, S.-H.; Jin Lee, K.; Park, J.J.; Pil Hong, S.; Cheon, J.H.; Kim, T.I.; Kim, W.H. Nonsteroidal Anti-Inflammatory Drugs Suppress Cancer Stem Cells via Inhibiting PTGS2 (Cyclooxygenase 2) and NOTCH/HES1 and Activating PPARG in Colorectal Cancer. Int. J. Cancer 2014, 134, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, T.; Hui, Z. Aspirin Overcomes Cisplatin Resistance in Lung Cancer by Inhibiting Cancer Cell Stemness. Thorac. Cancer 2020, 11, 3117–3125. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Su, Q.; Mo, J.; Fu, X.; Zhang, Y.; Lin, E.H. Celecoxib Downregulates CD133 Expression through Inhibition of the Wnt Signaling Pathway in Colon Cancer Cells. Cancer Investig. 2013, 31, 97–102. [Google Scholar] [CrossRef]
- Carlo, F.D.; Witte, T.R.; Hardman, W.E.; Claudio, P.P. Omega-3 Eicosapentaenoic Acid Decreases CD133 Colon Cancer Stem-Like Cell Marker Expression While Increasing Sensitivity to Chemotherapy. PLoS ONE 2013, 8, e69760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moro, M.; Bertolini, G.; Pastorino, U.; Roz, L.; Sozzi, G. Combination Treatment with All-Trans Retinoic Acid Prevents Cisplatin-Induced Enrichment of CD133+ Tumor-Initiating Cells and Reveals Heterogeneity of Cancer Stem Cell Compartment in Lung Cancer. J. Thorac. Oncol. 2015, 10, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Kim, S.; Lee, H.; No, J.H.; Ryu, H.C.; Kim, J.; Lim, J.W.; Kim, M.; Choi, I.; Seo, H.R. Chromenopyrimidinone Controls Stemness and Malignancy by Suppressing CD133 Expression in Hepatocellular Carcinoma. Cancers 2020, 12, 1193. [Google Scholar] [CrossRef]
- Kim, N.; Kim, S.; Song, Y.; Choi, I.; Lee, S.-Y.; Kim, K.m.; Rhu, H.C.; Lee, J.Y.; Seo, H.R. Chromenopyrimidinone Exhibit Antitumor Effects through Inhibition of CAP1 (Adenylyl Cyclase-Associated Protein 1) Expression in Hepatocellular Carcinoma. Chem. Biol. Interact. 2022, 365, 110066. [Google Scholar] [CrossRef]
- Lan, X.; Wu, Y.-Z.; Wang, Y.; Wu, F.-R.; Zang, C.-B.; Tang, C.; Cao, S.; Li, S.-L. CD133 Silencing Inhibits Stemness Properties and Enhances Chemoradiosensitivity in CD133-Positive Liver Cancer Stem Cells. Int. J. Mol. Med. 2013, 31, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Zihan, X.; Yizhe, W.; Yanyang, L.; Zhixi, L.; Xi, Y. Trilobatin Induces Apoptosis and Attenuates Stemness Phenotype of Acquired Gefitinib Resistant Lung Cancer Cells via Suppression of NF-ΚB Pathway. Nutr. Cancer 2022, 74, 735–746. [Google Scholar] [CrossRef]
- Duan, Y.; Mi, X.; Su, W.; Tang, S.; Jiang, S.; Wang, Z.; Zhao, L.-C.; Li, W. Trilobatin, an Active Dihydrochalcone from Lithocarpus Polystachyus, Prevents Cisplatin-Induced Nephrotoxicity via Mitogen-Activated Protein Kinase Pathway-Mediated Apoptosis in Mice. ACS Omega 2022, 7, 37401–37409. [Google Scholar] [CrossRef]
- Patel, D.K. Biological Importance and Therapeutic Potential of Trilobatin in the Management of Human Disorders and Associated Secondary Complications. Pharmacol. Res. Mod. Chin. Med. 2022, 5, 100185. [Google Scholar] [CrossRef]
- Liu, M.; Wang, L.; Li, X.; Wu, Y.; Yin, F.; Liu, J. Trilobatin Ameliorates Insulin Resistance through IRS-AKT-GLUT4 Signaling Pathway in C2C12 Myotubes and Ob/Ob Mice. Chin. Med. 2020, 15, 110. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, Y.; Dong, H.; Wang, B.; Ji, H.; Liu, X. Trilobatin Attenuates the LPS-Mediated Inflammatory Response by Suppressing the NF-ΚB Signaling Pathway. Food Chem. 2015, 166, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Li, W.; Liu, Z. Preparative Isolation, Quantification and Antioxidant Activity of Dihydrochalcones from Sweet Tea (Lithocarpus Polystachyus Rehd.). J. Chromatogr. B. 2015, 1002, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Muhammad, K.; Waheed, Y. Nanotechnology: A Revolution in Modern Industry. Molecules 2023, 28, 661. [Google Scholar] [CrossRef] [PubMed]
- Bayda, S.; Adeel, M.; Tuccinardi, T.; Cordani, M.; Rizzolio, F. The History of Nanoscience and Nanotechnology: From Chemical-Physical Applications to Nanomedicine. Molecules 2019, 25, 112. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.-H.; Kim, A.-R.; Kim, S.-H.; Lee, S.-J.; Chung, H.; Yoon, M.-Y. Development of a Novel Imaging Agent Using Peptide-Coated Gold Nanoparticles toward Brain Glioma Stem Cell Marker CD133. Acta Biomater. 2017, 47, 182–192. [Google Scholar] [CrossRef]
- Poonaki, E.; Nickel, A.-C.; Shafiee Ardestani, M.; Rademacher, L.; Kaul, M.; Apartsin, E.; Meuth, S.G.; Gorji, A.; Janiak, C.; Kahlert, U.D. CD133-Functionalized Gold Nanoparticles as a Carrier Platform for Telaglenastat (CB-839) against Tumor Stem Cells. Int. J. Mol. Sci. 2022, 23, 5479. [Google Scholar] [CrossRef]
- Mohd-Zahid, M.H.; Zulkifli, S.N.; Che Abdullah, C.A.; Lim, J.; Fakurazi, S.; Wong, K.K.; Zakaria, A.D.; Ismail, N.; Uskoković, V.; Mohamud, R.; et al. Gold Nanoparticles Conjugated with Anti-CD133 Monoclonal Antibody and 5-Fluorouracil Chemotherapeutic Agent as Nanocarriers for Cancer Cell Targeting. RSC Adv. 2021, 11, 16131–16141. [Google Scholar] [CrossRef]
- Crous, A.; Abrahamse, H. Effective Gold Nanoparticle-Antibody-Mediated Drug Delivery for Photodynamic Therapy of Lung Cancer Stem Cells. Int. J. Mol. Sci. 2020, 21, 3742. [Google Scholar] [CrossRef]
- Tan, H.; Hou, N.; Liu, Y.; Liu, B.; Cao, W.; Zheng, D.; Li, W.; Liu, Y.; Xu, B.; Wang, Z.; et al. CD133 Antibody Targeted Delivery of Gold Nanostars Loading IR820 and Docetaxel for Multimodal Imaging and Near-Infrared Photodynamic/Photothermal/Chemotherapy against Castration Resistant Prostate Cancer. Nanomed. Nanotechnol. Biol. Med. 2020, 27, 102192. [Google Scholar] [CrossRef]
- Dadashi Noshahr, K.; Shamsi, F.; Valtchev, P.; Kokhaei, P.; Hemati, M.; Reza Akbari Eidgahi, M.; Khaleghian, A. Optimization of Post-Insertion Method to Conjugate Doxil with Anti-CD133 Monoclonal Antibodies: Investigating the Specific Binding and Cytotoxicity to Colorectal Cancer Cells in Vitro. Saudi Pharm. J. 2020, 28, 1392–1401. [Google Scholar] [CrossRef]
- Yu, Z.; Ni, M.; Xiong, M.; Zhang, X.; Cai, G.; Chen, H.; Zeng, Q. Poly(Lactic-Co-Glycolic Acid) Nanoparticles Conjugated with CD133 Aptamers for Targeted Salinomycin Delivery to CD133+ Osteosarcoma Cancer Stem Cells. Int. J. Nanomed. 2015, 10, 2537–2554. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Huang, J.; Leng, D.; Yang, S.; Yao, Q.; Sun, J.; Hu, J. Gefitinib-Loaded DSPE-PEG2000 Nanomicelles with CD133 Aptamers Target Lung Cancer Stem Cells. World J. Surg. Onc. 2017, 15, 167. [Google Scholar] [CrossRef] [Green Version]
- Farokhzad, O.C.; Langer, R. Impact of Nanotechnology on Drug Delivery. ACS Nano 2009, 3, 16–20. [Google Scholar] [CrossRef]
- Ghafelehbashi, R.; Farshbafnadi, M.; Aghdam, N.S.; Amiri, S.; Salehi, M.; Razi, S. Nanoimmunoengineering Strategies in Cancer Diagnosis and Therapy. Clin. Transl. Oncol. 2023, 25, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Aghebati-Maleki, A.; Dolati, S.; Ahmadi, M.; Baghbanzhadeh, A.; Asadi, M.; Fotouhi, A.; Yousefi, M.; Aghebati-Maleki, L. Nanoparticles and Cancer Therapy: Perspectives for Application of Nanoparticles in the Treatment of Cancers. J. Cell. Physiol. 2020, 235, 1962–1972. [Google Scholar] [CrossRef]
- Alavi, M.; Hamidi, M. Passive and Active Targeting in Cancer Therapy by Liposomes and Lipid Nanoparticles. Drug. Metab. Pers. Ther. 2019, 34, 20180032. [Google Scholar] [CrossRef]
- Grosse-Gehling, P.; Fargeas, C.A.; Dittfeld, C.; Garbe, Y.; Alison, M.R.; Corbeil, D.; Kunz-Schughart, L.A. CD133 as a Biomarker for Putative Cancer Stem Cells in Solid Tumours: Limitations, Problems and Challenges. J. Pathol. 2013, 229, 355–378. [Google Scholar] [CrossRef]
- Reyes, E.E.; Kunovac, S.K.; Duggan, R.; Kregel, S.; Vander Griend, D.J. Growth Kinetics of CD133-Positive Prostate Cancer Cells. Prostate 2013, 73, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Xu, W.; Tan, J.; Liu, Z.; Huang, G.; Wang, S.; He, Z. Fluorescence Detection of Cancer Stem Cell Markers Using a Sensitive Nano-Aptamer Sensor. Front. Chem. 2022, 10, 920123. [Google Scholar] [CrossRef]
- Zhang, F.R.; Lu, J.Y.; Yao, Q.F.; Zhu, Q.Y.; Zhang, X.X.; Huang, W.T.; Xia, L.Q.; Ding, X.Z. Matter, Energy and Information Network of a Graphene-Peptide-Based Fluorescent Sensing System for Molecular Logic Computing, Detection and Imaging of Cancer Stem Cell Marker CD133 in Cells and Tumor Tissues. Analyst 2019, 144, 1881–1891. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; Clair, R.S.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 Expression Is Not Restricted to Stem Cells, and Both CD133+ and CD133– Metastatic Colon Cancer Cells Initiate Tumors. J. Clin. Investig. 2008, 118, 2111–2120. [Google Scholar] [CrossRef]
- Wang, J.; Sakariassen, P.Ø.; Tsinkalovsky, O.; Immervoll, H.; Bøe, S.O.; Svendsen, A.; Prestegarden, L.; Røsland, G.; Thorsen, F.; Stuhr, L.; et al. CD133 Negative Glioma Cells Form Tumors in Nude Rats and Give Rise to CD133 Positive Cells. Int. J. Cancer 2008, 122, 761–768. [Google Scholar] [CrossRef]
- Yuan, N.; Wang, L.; Xi, Q.; Zou, N.; Zhang, X.; Lu, X.; Zhang, Z. ITGA7, CD133, ALDH1 Are Inter-Correlated, and Linked with Poor Differentiation, Lymph Node Metastasis as Well as Worse Survival in Surgical Cervical Cancer. J. Obstet. Gynaecol. Res. 2022, 48, 1011–1018. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, H.; Lv, S.; Yang, H. High CD133 Expression Is Associated with Worse Prognosis in Patients with Glioblastoma. Mol. Neurobiol. 2016, 53, 2354–2360. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Location | Marker | Refs. |
|---|---|---|---|
| Bladder | Extracellular/Surface | CD44, CD47, CEACAM-6 | [20,21,22] |
| Intracellular | ALDH1A1, SOX4 | ||
| Breast | Extracellular/Surface | CD44, CD24, CD133, CD49f, SSEA-3, CD70, PROCR, CD326, CD29, CD25, CD49, CD61, CD90E | [23,24] |
| Intracellular | TSPAN8, ALDH1A1, BMI1, FOXO3, NANOG, NOTCH 1-3, OCT4, SOX2 | ||
| Colorectal | Extracellular/Surface | CD24, CD44, CD49, CD90, CD133, CD166, CD326, LGR5 CD29 | [25,26,27] |
| Intracellular | ALDH1A1, LETM1, NANOG, OCT4, SALL4, SOX2 | ||
| Gastric | Extracellular/Surface | CD24, CD44, CD90, CD133, CD326, LGR5, LINGO2 | [28,29,30] |
| Intracellular | ALDH1A1, NANOG OCT4, SOX2 | ||
| Glioblastoma | Extracellular/Surface | CD44, CD133, CD15, CD70 | [31,32,33,34,35,36] |
| Intracellular | S100A4, ALDH1A3, NANOG, OCT4, SOX2, Nestin, Musashi-1 | ||
| Liver | Extracellular/Surface | CD24, CD44, CD90, CD133, CD326 | [37,38,39,40,41,42,43] |
| Intracellular | AFP, NANOG, NOTCH 1-3, OCT4, SOX2 | ||
| Lung | Extracellular/Surface | CD44, CD87, CD133, CD166, CD326, CD117, CD90 | [44,45,46,47] |
| Intracellular | ALDH1A1, NANOG, OCT4 | ||
| Melanoma | Extracellular/Surface | CD20, CD133, CD166, CD279, ABCB5, ABCG2 | [48,49,50] |
| Ovarian | Extracellular/Surface | CD24, CD44, CD117, CD133, CD326 | [51,52,53,54,55] |
| Intracellular | OCT4, NANOG, SOX2, ALDH1 | ||
| Pancreatic | Extracellular/Surface | CD24, CD44, CD133, CD326, ABCB1 | [56,57] |
| Intracellular | DCLK1, CXCR4, OCT4, | ||
| Prostate | Extracellular/Surface | CD166, CD44, CD133, CD326, CD117, TACSTD2 | [22,58,59] |
| Intracellular | ALDH1A1, TGM2 | ||
| CML Leukemia | Extracellular/Surface | CD33, CD34, CD36, CD117, CD123, CD114 CD56, CD135 CD93 | [60,61,62,63] |
| Intracellular | ALDH | ||
| AML Leukemia | Extracellular/Surface | CD33, CD34, CD123, CD244, CLL1, CD9, CD96, CD25, CD32 | [64,65,66,67,68] |
| Intracellular | ALDH1A1, NANOG, OCT4, SOX2 |
| Molecule Type | Therapy Type | Target | Model | Ref. |
|---|---|---|---|---|
| Gold nanoparticles | Imaging agent | CD133 | In vitro and in vivo | [212] |
| Gold nanoparticles | Drug delivery | CD133 | In vitro | [213] |
| Gold nanoparticles | Drug delivery | CD133 | In vitro | [214] |
| Gold nanoparticles | Photodynamic therapy | Lung CSC | In vitro | [215] |
| Gold nanoparticles | Multimodal therapy | CRPC | In vitro and in vivo | [216] |
| Nanoliposomes | Drug delivery | CD133 | In vitro | [217] |
| Liposomes | Drug delivery | CD133 | In vitro | [175] |
| Polymeric nanoparticles | Drug delivery | CD133 | In vitro | [218] |
| Nanomicelles | Drug delivery | CD133 | In vitro and in vivo | [219] |
| Carbon nanotubes | Photothermal therapy | CD133 | In vitro and in vivo | [155] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pospieszna, J.; Dams-Kozlowska, H.; Udomsak, W.; Murias, M.; Kucinska, M. Unmasking the Deceptive Nature of Cancer Stem Cells: The Role of CD133 in Revealing Their Secrets. Int. J. Mol. Sci. 2023, 24, 10910. https://doi.org/10.3390/ijms241310910
Pospieszna J, Dams-Kozlowska H, Udomsak W, Murias M, Kucinska M. Unmasking the Deceptive Nature of Cancer Stem Cells: The Role of CD133 in Revealing Their Secrets. International Journal of Molecular Sciences. 2023; 24(13):10910. https://doi.org/10.3390/ijms241310910
Chicago/Turabian StylePospieszna, Julia, Hanna Dams-Kozlowska, Wachirawit Udomsak, Marek Murias, and Malgorzata Kucinska. 2023. "Unmasking the Deceptive Nature of Cancer Stem Cells: The Role of CD133 in Revealing Their Secrets" International Journal of Molecular Sciences 24, no. 13: 10910. https://doi.org/10.3390/ijms241310910