
Selective Killing of BRCA2-Deficient Ovarian Cancer Cells via MRE11 Blockade
 , , , , ,
, , , , ,  ,
,
Abstract
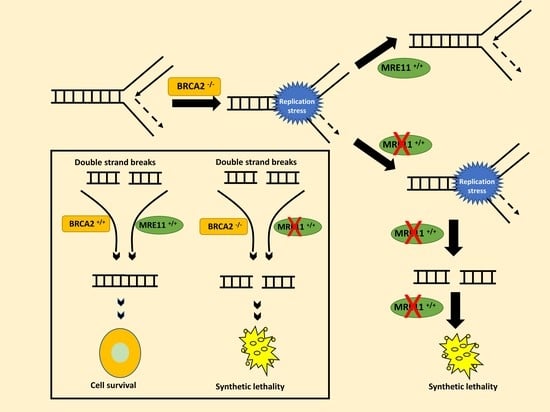
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. MRE11 Blockade Is Synthetically Lethal in BRCA2-Deficient Cancer Cells
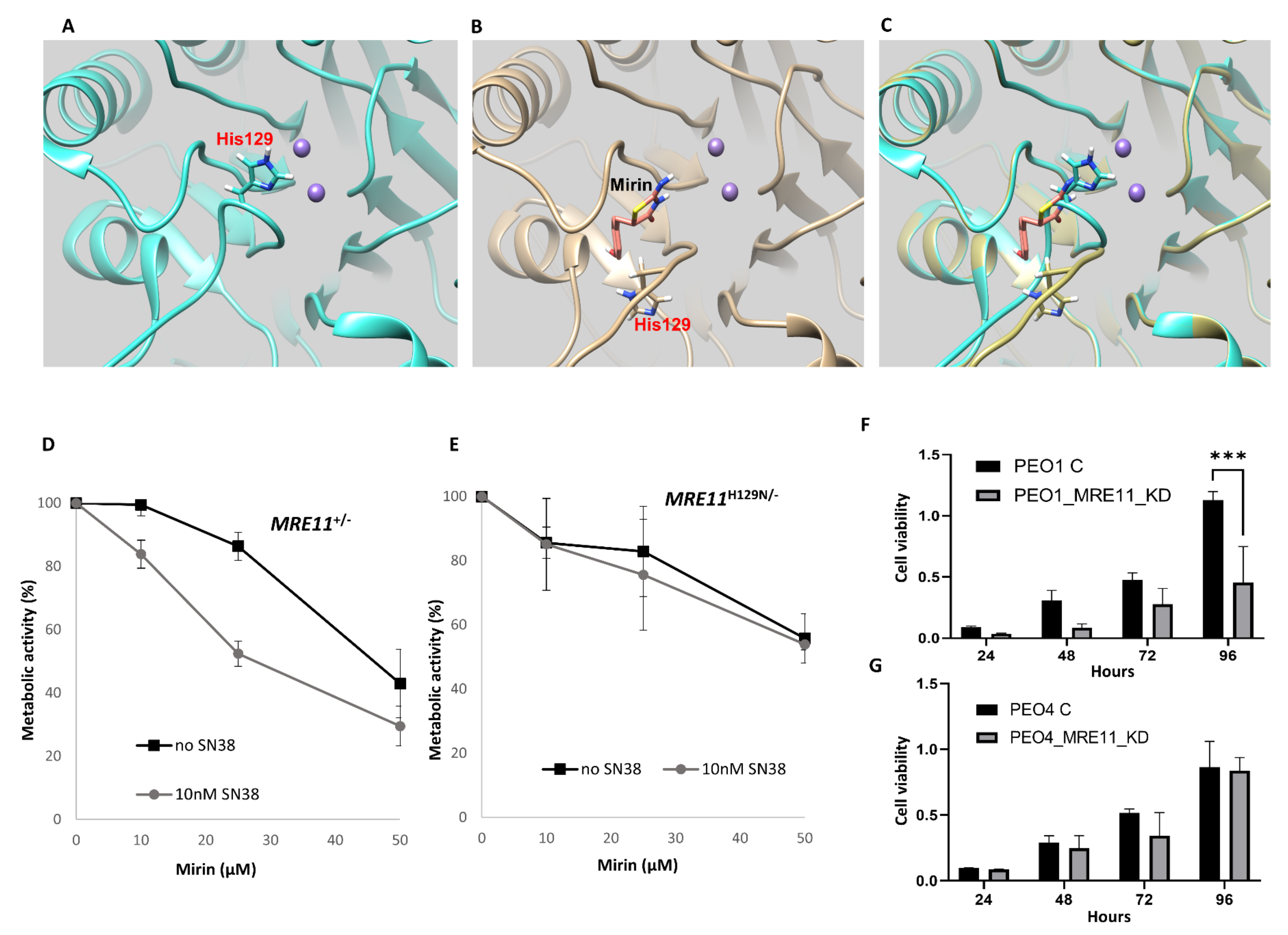
2.2. In Silico Docking of Mirin onto MRE11
2.3. Mirin Is Cytotoxic in DT40 MRE11+/− but Not in Nuclease-Dead MRE11H129N/− Cells
2.4. MRE11 Depletion Reduces Cell Viability in BRCA2-Deficient Cells
2.5. Mechanism of Resistance to MRE11 Blockade in BRCA2-Deficient Cells
In Vivo Xenograft Studies
2.6. MRE11-BRCA2 Co-Expression in Human Ovarian Cancers
3. Discussion
4. Materials and Methods
4.1. Pre-Clinical Study
4.1.1. Cell Lines and Tissue Culture
4.1.2. Western Blot Analysis
4.1.3. Transient Knockdowns of MRE11
4.1.4. MTS Assay
4.1.5. Clonogenic Assays
4.1.6. Functional Studies
4.1.7. Generation of 3D Spheroids
4.1.8. Immunofluorescence Staining
4.1.9. Real-Time PCR
4.1.10. Development of Mirin-Resistant PEO1R Cell Line
4.1.11. Tumour Xenograft Studies
4.1.12. RNA Seq Analyses
4.1.13. Gene Set Enrichment Analysis
4.1.14. Statistical Analysis
4.1.15. In Silico Mirin Docking Studies
4.2. Clinical Study
4.2.1. MRE11 Expression Levels in Ovarian Cancers
4.2.2. Tissue Microarray (TMA) and Immunohistochemistry (IHC)
4.2.3. Evaluation of Immune Staining
4.2.4. Statistical Analysis
4.2.5. Genomic and Transcriptomic Analysis in the TCGA Data Set
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Le, H.P.; Heyer, W.D.; Liu, J. Guardians of the Genome: BRCA2 and Its Partners. Genes 2021, 12, 1229. [Google Scholar]
- Shulman, L.P. Hereditary breast and ovarian cancer (HBOC): Clinical features and counseling for BRCA1 and BRCA2, Lynch syndrome, Cowden syndrome, and Li-Fraumeni syndrome. Obstet. Gynecol. Clin. N. Am. 2010, 37, 109–133. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary breast and ovarian cancer due to mutations in BRCA1 and BRCA2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramus, S.J.; Gayther, S.A. The contribution of BRCA1 and BRCA2 to ovarian cancer. Mol. Oncol. 2009, 3, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Elias, K.M. Synthetic Lethality in Ovarian Cancer. Mol. Cancer Ther. 2021, 20, 2117–2128. [Google Scholar] [CrossRef]
- Biegala, L.; Gajek, A.; Marczak, A.; Rogalska, A. PARP inhibitor resistance in ovarian cancer: Underlying mechanisms and therapeutic approaches targeting the ATR/CHK1 pathway. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188633. [Google Scholar]
- Ray-Coquard, I.; Vanacker, H.; Le Saux, O.; Tredan, O. Overcoming resistance to PARP inhibitor in epithelial ovarian cancer, are we ready? EBioMedicine 2020, 61, 103046. [Google Scholar] [CrossRef]
- McMullen, M.; Karakasis, K.; Madariaga, A.; Oza, A.M. Overcoming Platinum and PARP-Inhibitor Resistance in Ovarian Cancer. Cancers 2020, 12, 1607. [Google Scholar] [CrossRef]
- Feng, Z.; Scott, S.P.; Bussen, W.; Sharma, G.G.; Guo, G.; Pandita, T.K.; Powell, S.N. Rad52 inactivation is synthetically lethal with BRCA2 deficiency. Proc. Natl. Acad. Sci. USA 2011, 108, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef]
- Al-Mugotir, M.; Lovelace, J.J.; George, J.; Bessho, M.; Pal, D.; Struble, L.; Kolar, C.; Rana, S.; Natarajan, A.; Bessho, T.; et al. Selective killing of homologous recombination-deficient cancer cell lines by inhibitors of the RPA:RAD52 protein-protein interaction. PLoS ONE 2021, 16, e0248941. [Google Scholar] [CrossRef]
- Hanamshet, K.; Mazin, A.V. The function of RAD52 N-terminal domain is essential for viability of BRCA-deficient cells. Nucleic Acids Res. 2020, 48, 12778–12791. [Google Scholar] [CrossRef]
- Mahajan, S.; Raina, K.; Verma, S.; Rao, B.J. Human RAD52 protein regulates homologous recombination and checkpoint function in BRCA2-deficient cells. Int. J. Biochem. Cell Biol. 2019, 107, 128–139. [Google Scholar] [CrossRef]
- Biswas, K.; Sharan, S.K. RAD52 S346X variant reduces breast cancer risk in BRCA2 mutation carriers. Mol. Oncol. 2020, 14, 1121–1123. [Google Scholar] [CrossRef] [Green Version]
- Hamid, A.B.; Frank, L.E.; Bouley, R.A.; Petreaca, R.C. Pan-cancer analysis of co-occurring mutations in RAD52 and the BRCA1-BRCA2-PALB2 axis in human cancers. PLoS ONE 2022, 17, e0273736. [Google Scholar] [CrossRef]
- Adamson, A.W.; Ding, Y.C.; Mendez-Dorantes, C.; Bailis, A.M.; Stark, J.M.; Neuhausen, S.L. The RAD52 S346X variant reduces risk of developing breast cancer in carriers of pathogenic germline BRCA2 mutations. Mol. Oncol. 2020, 14, 1124–1133. [Google Scholar] [CrossRef] [Green Version]
- Bian, L.; Meng, Y.; Zhang, M.; Li, D. MRE11-RAD50-NBS1 complex alterations and DNA damage response: Implications for cancer treatment. Mol. Cancer 2019, 18, 169. [Google Scholar] [CrossRef] [Green Version]
- Rein, K.; Stracker, T.H. The MRE11 complex: An important source of stress relief. Exp. Cell Res. 2014, 329, 162–169. [Google Scholar] [CrossRef]
- Syed, A.; Tainer, J.A. The MRE11-RAD50-NBS1 Complex Conducts the Orchestration of Damage Signaling and Outcomes to Stress in DNA Replication and Repair. Annu. Rev. Biochem. 2018, 87, 263–294. [Google Scholar] [CrossRef]
- Costanzo, V. Brca2, Rad51 and Mre11: Performing balancing acts on replication forks. DNA Repair. 2011, 10, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Hamdy, F.C.; Helleday, T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012, 72, 2814–2821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemacon, D.; Jackson, J.; Quinet, A.; Brickner, J.R.; Li, S.; Yazinski, S.; You, Z.; Ira, G.; Zou, L.; Mosammaparast, N.; et al. MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nat. Commun. 2017, 8, 860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlacher, K.; Christ, N.; Siaud, N.; Egashira, A.; Wu, H.; Jasin, M. Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 2011, 145, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Alblihy, A.; Ali, R.; Algethami, M.; Shoqafi, A.; Toss, M.S.; Brownlie, J.; Tatum, N.J.; Hickson, I.; Moran, P.O.; Grabowska, A.; et al. Targeting Mre11 overcomes platinum resistance and induces synthetic lethality in XRCC1 deficient epithelial ovarian cancers. NPJ Precis. Oncol. 2022, 6, 51. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Jasin, M. Homologous Recombination and Replication Fork Protection: BRCA2 and More! Cold Spring Harb. Symp. Quant. Biol. 2017, 82, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Dupre, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A forward chemical genetic screen reveals an inhibitor of the Mre11-Rad50-Nbs1 complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef]
- Park, Y.B.; Chae, J.; Kim, Y.C.; Cho, Y. Crystal structure of human MRE11: Understanding tumorigenic mutations. Structure 2011, 19, 1591–1602. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef] [Green Version]
- Moreau, S.; Ferguson, J.R.; Symington, L.S. The nuclease activity of Mre11 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol. Cell. Biol. 1999, 19, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Fradet-Turcotte, A.; Sitz, J.; Grapton, D.; Orthwein, A. BRCA2 functions: From DNA repair to replication fork stabilization. Endocr. Relat. Cancer 2016, 23, T1–T17. [Google Scholar] [CrossRef] [Green Version]
- Holloman, W.K. Unraveling the mechanism of BRCA2 in homologous recombination. Nat. Struct. Mol. Biol. 2011, 18, 748–754. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T.; Lee, J.H. The Mre11/Rad50/Nbs1 complex and its role as a DNA double-strand break sensor for ATM. Cell Cycle 2005, 4, 737–740. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Boeckemeier, L.; Kraehenbuehl, R.; Keszthelyi, A.; Gasasira, M.U.; Vernon, E.G.; Beardmore, R.; Vagbo, C.B.; Chaplin, D.; Gollins, S.; Krokan, H.E.; et al. Mre11 exonuclease activity removes the chain-terminating nucleoside analog gemcitabine from the nascent strand during DNA replication. Sci. Adv. 2020, 6, eaaz4126. [Google Scholar] [CrossRef]
- Ridpath, J.R.; Takeda, S.; Swenberg, J.A.; Nakamura, J. Convenient, multi-well plate-based DNA damage response analysis using DT40 mutants is applicable to a high-throughput genotoxicity assay with characterization of modes of action. Environ. Mol. Mutagen. 2011, 52, 153–160. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauffmann, A.; Rosselli, F.; Lazar, V.; Winnepenninckx, V.; Mansuet-Lupo, A.; Dessen, P.; van den Oord, J.J.; Spatz, A.; Sarasin, A. High expression of DNA repair pathways is associated with metastasis in melanoma patients. Oncogene 2008, 27, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a Shared Vision for Cancer Genomic Data. N. Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alblihy, A.; Ali, R.; Algethami, M.; Ritchie, A.A.; Shoqafi, A.; Alqahtani, S.; Mesquita, K.A.; Toss, M.S.; Ordóñez-Morán, P.; Jeyapalan, J.N.; et al. Selective Killing of BRCA2-Deficient Ovarian Cancer Cells via MRE11 Blockade. Int. J. Mol. Sci. 2023, 24, 10966. https://doi.org/10.3390/ijms241310966
Alblihy A, Ali R, Algethami M, Ritchie AA, Shoqafi A, Alqahtani S, Mesquita KA, Toss MS, Ordóñez-Morán P, Jeyapalan JN, et al. Selective Killing of BRCA2-Deficient Ovarian Cancer Cells via MRE11 Blockade. International Journal of Molecular Sciences. 2023; 24(13):10966. https://doi.org/10.3390/ijms241310966
Chicago/Turabian StyleAlblihy, Adel, Reem Ali, Mashael Algethami, Alison A. Ritchie, Ahmed Shoqafi, Shatha Alqahtani, Katia A. Mesquita, Michael S. Toss, Paloma Ordóñez-Morán, Jennie N. Jeyapalan, and et al. 2023. "Selective Killing of BRCA2-Deficient Ovarian Cancer Cells via MRE11 Blockade" International Journal of Molecular Sciences 24, no. 13: 10966. https://doi.org/10.3390/ijms241310966