
Influence of Inflammatory Pain and Dopamine on Synaptic Transmission in the Mouse ACC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
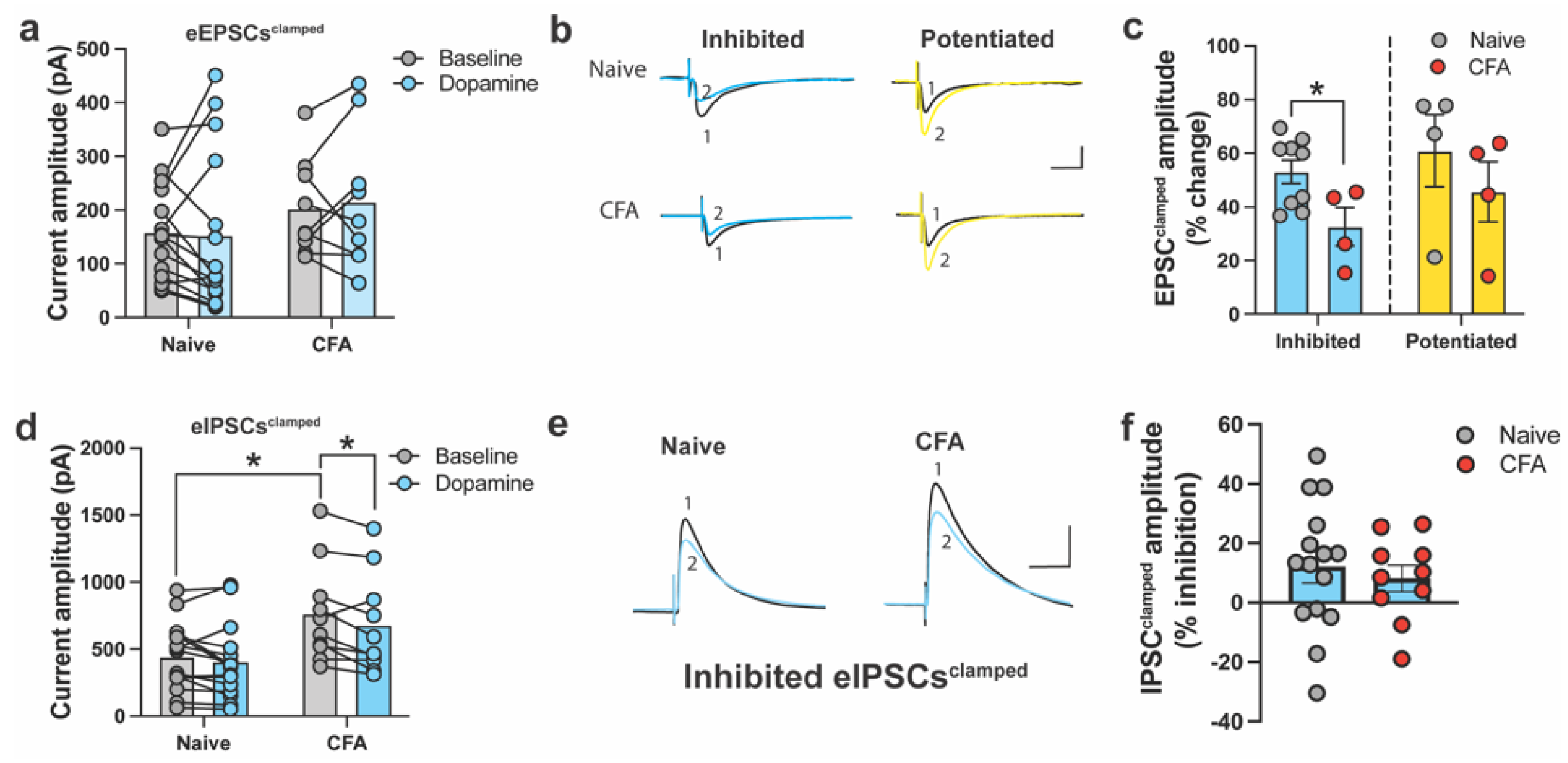
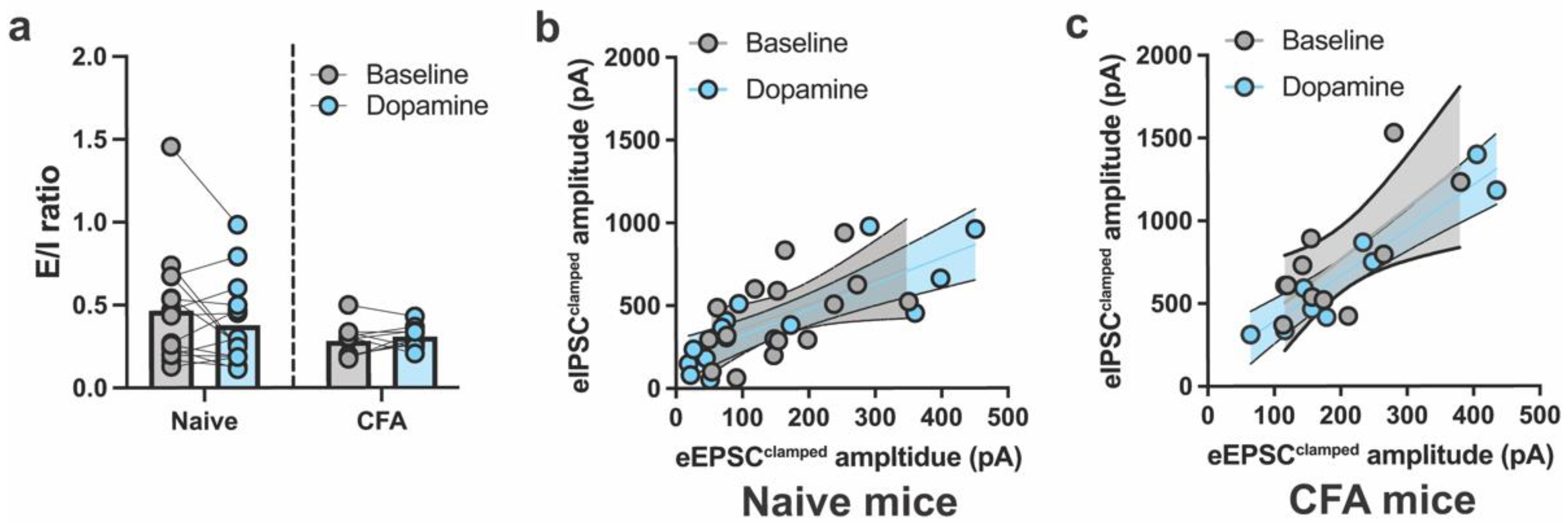
2.1. DA and CFA-Induced Inflammation Do Not Alter E/I Balance in the ACC
2.2. DA Modulates Spontaneous Excitatory and Inhibitory Currents in the ACC
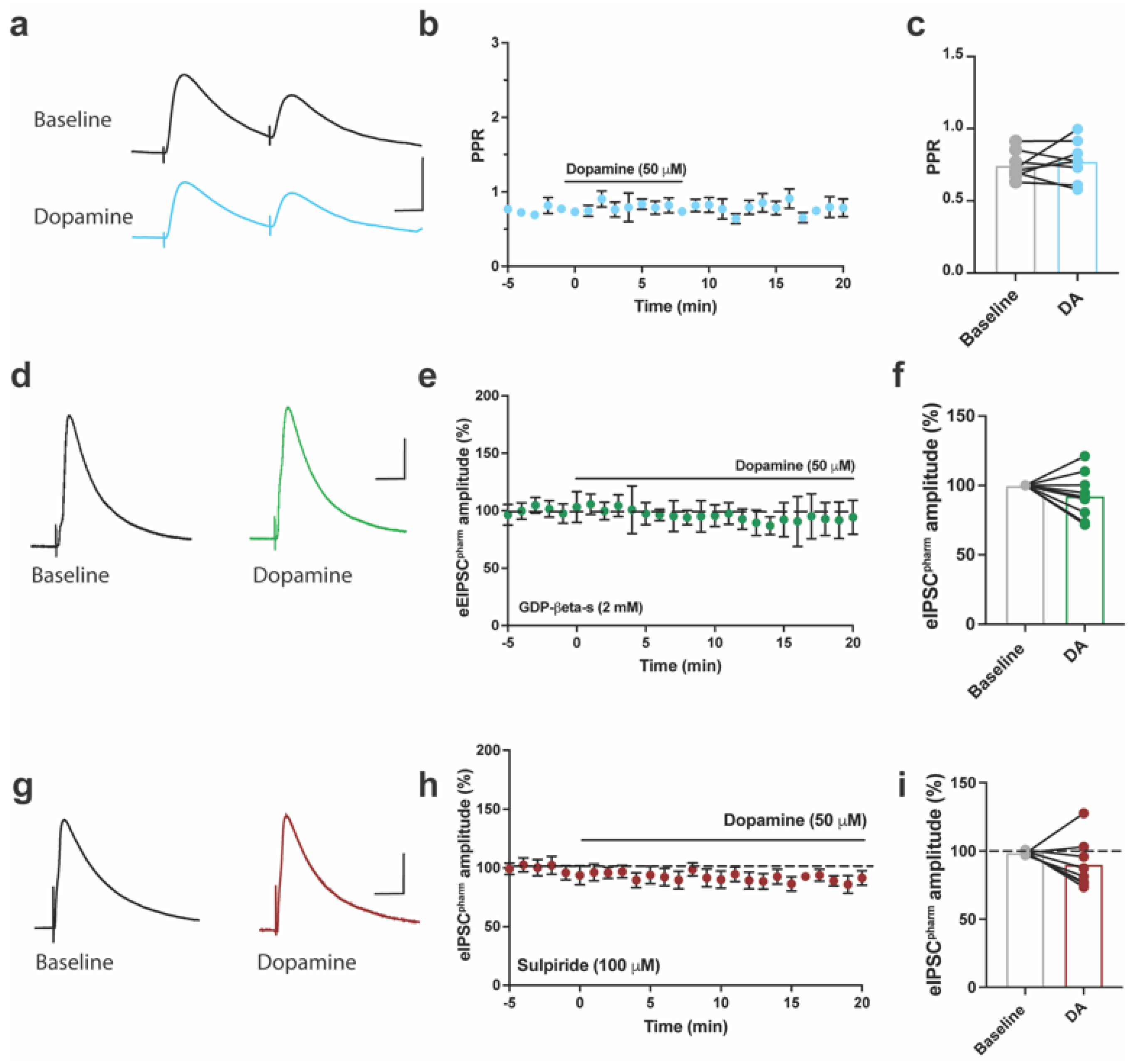
2.3. DA inhibits Pharmacologically Isolated Evoked GABAergic Currents in ACC of Mice
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Tissue Preparation for Electrophysiology
4.3. Whole-Cell Patch-Clamp Recording
4.4. Synaptic Drive
4.5. Complete Freund’s Adjuvant (CFA) Model of Inflammatory Pain
4.6. Drugs and Solutions
4.7. Data and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breivik, H.; Collett, B.; Ventafridda, V.; Cohen, R.; Gallacher, D. Survey of chronic pain in Europe: Prevalence, impact on daily life, and treatment. Eur. J. Pain 2006, 10, 287–333. [Google Scholar] [CrossRef]
- Elliott, A.M.; Smith, B.H.; Penny, K.I.; Smith, W.C.; Chambers, W.A. The epidemiology of chronic pain in the community. Lancet 1999, 354, 1248–1252. [Google Scholar] [CrossRef]
- Goldberg, D.S.; McGee, S.J. Pain as a global public health priority. BMC Public Health 2011, 11, 770. [Google Scholar] [CrossRef] [Green Version]
- Schopflocher, D.; Taenzer, P.; Jovey, R. The prevalence of chronic pain in Canada. Pain Res. Manag. 2011, 16, 445–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, M.E.; Taddio, A.; Katz, J.; Shah, V.; Krahn, M. Incremental health care costs for chronic pain in Ontario, Canada: A population-based matched cohort study of adolescents and adults using administrative data. Pain 2016, 157, 1626–1633. [Google Scholar] [CrossRef]
- Moulin, D.E.; Clark, A.J.; Speechley, M.; Morley-Forster, P.K. Chronic pain in Canada--prevalence, treatment, impact and the role of opioid analgesia. Pain Res. Manag. 2002, 7, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouhassira, D.; Lantéri-Minet, M.; Attal, N.; Laurent, B.; Touboul, C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008, 136, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bliss, T.V.; Collingridge, G.L.; Kaang, B.-K.; Zhuo, M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 2016, 17, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Apkarian, A.V.; Bushnell, M.C.; Treede, R.-D.; Zubieta, J.-K. Human brain mechanisms of pain perception and regulation in health and disease. Eur. J. Pain 2005, 9, 463–484. [Google Scholar] [CrossRef]
- Zhuo, M. Cortical excitation and chronic pain. Trends Neurosci. 2008, 31, 199–207. [Google Scholar] [CrossRef]
- Johansen, J.P.; Fields, H.L.; Manning, B.H. The affective component of pain in rodents: Direct evidence for a contribution of the anterior cingulate cortex. Proc. Natl. Acad. Sci. USA 2001, 98, 8077–8082. [Google Scholar] [CrossRef]
- Meda, K.S.; Patel, T.; Braz, J.M.; Malik, R.; Turner, M.L.; Seifikar, H.; Basbaum, A.I.; Sohal, V.S. Microcircuit mechanisms through which mediodorsal thalamic input to anterior cingulate cortex exacerbates pain-related aversion. Neuron 2019, 102, 944–959.e943. [Google Scholar] [CrossRef]
- Cheriyan, J.; Sheets, P.L. Peripheral nerve injury reduces the excitation-inhibition balance of basolateral amygdala inputs to prelimbic pyramidal neurons projecting to the periaqueductal gray. Mol. Brain 2020, 13, 100. [Google Scholar] [CrossRef]
- Potter, L.E.; Paylor, J.W.; Suh, J.S.; Tenorio, G.; Caliaperumal, J.; Colbourne, F.; Baker, G.; Winship, I.; Kerr, B.J. Altered excitatory-inhibitory balance within somatosensory cortex is associated with enhanced plasticity and pain sensitivity in a mouse model of multiple sclerosis. J. Neuroinflamm. 2016, 13, 142. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Li, X.; Tjia, M.; Thapliyal, S. Homeostatic plasticity and excitation-inhibition balance: The good, the bad, and the ugly. Curr. Opin. Neurobiol. 2022, 75, 102553. [Google Scholar] [CrossRef] [PubMed]
- Reckziegel, D.; Raschke, F.; Cottam, W.J.; Auer, D.P. Cingulate GABA levels inversely correlate with the intensity of ongoing chronic knee osteoarthritis pain. Mol. Pain 2016, 12, 1744806916650690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blom, S.M.; Pfister, J.P.; Santello, M.; Senn, W.; Nevian, T. Nerve injury-induced neuropathic pain causes disinhibition of the anterior cingulate cortex. J. Neurosci. 2014, 34, 5754–5764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, K.; Matsuzaki, Y.; Honda, K.; Eto, F.; Furukawa, T.; Migita, K.; Irie, K.; Mishima, K.; Ueno, S. Activations of muscarinic M1 receptors in the anterior cingulate cortex contribute to the antinociceptive effect via GABAergic transmission. Mol. Pain 2017, 13, 1744806917692330. [Google Scholar] [CrossRef] [Green Version]
- Juarez-Salinas, D.L.; Braz, J.M.; Etlin, A.; Gee, S.; Sohal, V.; Basbaum, A.I. GABAergic cell transplants in the anterior cingulate cortex reduce neuropathic pain aversiveness. Brain 2019, 142, 2655–2669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, K.; Shimoyama, S.; Yamada, A.; Furukawa, T.; Nikaido, Y.; Furue, H.; Nakamura, K.; Ueno, S. Chronic inflammatory pain induced GABAergic synaptic plasticity in the adult mouse anterior cingulate cortex. Mol. Pain 2018, 14, 1744806918783478. [Google Scholar] [CrossRef] [Green Version]
- Mitsi, V.; Zachariou, V. Modulation of pain, nociception, and analgesia by the brain reward center. Neuroscience 2016, 338, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elman, I.; Borsook, D. Common Brain Mechanisms of Chronic Pain and Addiction. Neuron 2016, 89, 11–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafini, R.A.; Pryce, K.D.; Zachariou, V. The Mesolimbic Dopamine System in Chronic Pain and Associated Affective Comorbidities. Biol. Psychiatry 2020, 87, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navratilova, E.; Xie, J.Y.; Okun, A.; Qu, C.; Eyde, N.; Ci, S.; Ossipov, M.H.; King, T.; Fields, H.L.; Porreca, F. Pain relief produces negative reinforcement through activation of mesolimbic reward-valuation circuitry. Proc. Natl. Acad. Sci. USA 2012, 109, 20709–20713. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Becker, S.; Schweinhardt, P.; Cahill, C. Mesolimbic dopamine signaling in acute and chronic pain: Implications for motivation, analgesia, and addiction. Pain 2016, 157, 1194–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, T.A.; Weintraub, N.C.; Lu, D.; Phelps, C.E.; Navratilova, E.; Heien, M.L.; Porreca, F. A pain-induced tonic hypodopaminergic state augments phasic dopamine release in the nucleus accumbens. Pain 2020, 161, 2376–2384. [Google Scholar] [CrossRef]
- Borsook, D.; Linnman, C.; Faria, V.; Strassman, A.M.; Becerra, L.; Elman, I. Reward deficiency and anti-reward in pain chronification. Neurosci. Biobehav. Rev. 2016, 68, 282–297. [Google Scholar] [CrossRef]
- Lopez-Avila, A.; Coffeen, U.; Ortega-Legaspi, J.M.; del Angel, R.; Pellicer, F. Dopamine and NMDA systems modulate long-term nociception in the rat anterior cingulate cortex. Pain 2004, 111, 136–143. [Google Scholar] [CrossRef]
- Darvish-Ghane, S.; Quintana, C.; Beaulieu, J.M.; Martin, L.J. D1 receptors in the anterior cingulate cortex modulate basal mechanical sensitivity threshold and glutamatergic synaptic transmission. Mol. Brain 2020, 13, 121. [Google Scholar] [CrossRef]
- Darvish-Ghane, S.; Yamanaka, M.; Zhuo, M. Dopaminergic Modulation of Excitatory Transmission in the Anterior Cingulate Cortex of Adult Mice. Mol. Pain 2016, 12, 1744806916648153. [Google Scholar] [CrossRef]
- Darvish-Ghane, S.; Lyver, B.; Facciol, A.; Chatterjee, D.; Martin, L.J. Inflammatory Pain Alters Dopaminergic Modulation of Excitatory Synapses in the Anterior Cingulate Cortex of Mice. Neuroscience 2022, 498, 249–259. [Google Scholar] [CrossRef]
- Lançon, K.; Navratilova, E.; Porreca, F.; Séguéla, P. Neuropathic pain linked to defective dopaminergic inhibition in anterior cingulate cortex. bioRxiv 2020. [Google Scholar] [CrossRef]
- Sohal, V.S.; Rubenstein, J.L.R. Excitation-inhibition balance as a framework for investigating mechanisms in neuropsychiatric disorders. Mol. Psychiatry 2019, 24, 1248–1257. [Google Scholar] [CrossRef]
- Kato, N. Dependence of long-term depression on postsynaptic metabotropic glutamate receptors in visual cortex. Proc. Natl. Acad. Sci. USA 1993, 90, 3650–3654. [Google Scholar] [CrossRef] [PubMed]
- Lidhar, N.K.; Darvish-Ghane, S.; Sivaselvachandran, S.; Khan, S.; Wasif, F.; Turner, H.; Sivaselvachandran, M.; Fournier, N.M.; Martin, L.J. Prelimbic cortex glucocorticoid receptors regulate the stress-mediated inhibition of pain contagion in male mice. Neuropsychopharmacology 2021, 46, 1183–1193. [Google Scholar] [CrossRef]
- Lancon, K.; Qu, C.; Navratilova, E.; Porreca, F.; Seguela, P. Decreased dopaminergic inhibition of pyramidal neurons in anterior cingulate cortex maintains chronic neuropathic pain. Cell Rep. 2021, 37, 109933. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Smith, B.N. Presynaptic ionotropic glutamate receptors modulate GABA release in the mouse dorsal motor nucleus of the vagus. Neuroscience 2015, 308, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satake, S.; Saitow, F.; Rusakov, D.; Konishi, S. AMPA receptor-mediated presynaptic inhibition at cerebellar GABAergic synapses: A characterization of molecular mechanisms. Eur. J. Neurosci. 2004, 19, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Fattorini, G.; Ripoli, C.; Cocco, S.; Spinelli, M.; Mattera, A.; Grassi, C.; Conti, F. Glutamate/GABA co-release selectively influences postsynaptic glutamate receptors in mouse cortical neurons. Neuropharmacology 2019, 161, 107737. [Google Scholar] [CrossRef]
- Momiyama, T.; Sim, J.A. Modulation of inhibitory transmission by dopamine in rat basal forebrain nuclei: Activation of presynaptic D1-like dopaminergic receptors. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 7505–7512. [Google Scholar] [CrossRef] [Green Version]
- Jijon-Lorenzo, R.; Caballero-Floran, I.H.; Recillas-Morales, S.; Cortes, H.; Avalos-Fuentes, J.A.; Paz-Bermudez, F.J.; Erlij, D.; Floran, B. Presynaptic Dopamine D2 Receptors Modulate [(3)H]GABA Release at StriatoPallidal Terminals via Activation of PLC --> IP3 --> Calcineurin and Inhibition of AC --> cAMP --> PKA Signaling Cascades. Neuroscience 2018, 372, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Snyder, G.L.; Allen, P.B.; Fienberg, A.A.; Valle, C.G.; Huganir, R.L.; Nairn, A.C.; Greengard, P. Regulation of phosphorylation of the GluR1 AMPA receptor in the neostriatum by dopamine and psychostimulants in vivo. J. Neurosci. Off. J. Soc. Neurosci. 2000, 20, 4480–4488. [Google Scholar] [CrossRef] [Green Version]
- Koga, K.; Descalzi, G.; Chen, T.; Ko, H.G.; Lu, J.; Li, S.; Son, J.; Kim, T.; Kwak, C.; Huganir, R.L.; et al. Coexistence of two forms of LTP in ACC provides a synaptic mechanism for the interactions between anxiety and chronic pain. Neuron 2015, 85, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.Q.; Puente, N.; Grandes, P.; Castillo, P.E. Dopaminergic modulation of endocannabinoid-mediated plasticity at GABAergic synapses in the prefrontal cortex. J. Neurosci. 2010, 30, 7236–7248. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.J.; Toyoda, H.; Zhao, M.G.; Lee, Y.S.; Tang, J.; Ko, S.W.; Jia, Y.H.; Shum, F.W.; Zerbinatti, C.V.; Bu, G.; et al. Upregulation of forebrain NMDA NR2B receptors contributes to behavioral sensitization after inflammation. J. Neurosci. 2005, 25, 11107–11116. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wu, L.J.; Kim, S.S.; Lee, F.J.; Gong, B.; Toyoda, H.; Ren, M.; Shang, Y.Z.; Xu, H.; Liu, F.; et al. FMRP acts as a key messenger for dopamine modulation in the forebrain. Neuron 2008, 59, 634–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, T.; Taniguchi, W.; Chen, Q.Y.; Tozaki-Saitoh, H.; Song, Q.; Liu, R.H.; Koga, K.; Matsuda, T.; Kaito-Sugimura, Y.; Wang, J.; et al. Top-down descending facilitation of spinal sensory excitatory transmission from the anterior cingulate cortex. Nat. Commun. 2018, 9, 1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darvish-Ghane, S.; Baumbach, J.; Martin, L.J. Influence of Inflammatory Pain and Dopamine on Synaptic Transmission in the Mouse ACC. Int. J. Mol. Sci. 2023, 24, 11113. https://doi.org/10.3390/ijms241311113
Darvish-Ghane S, Baumbach J, Martin LJ. Influence of Inflammatory Pain and Dopamine on Synaptic Transmission in the Mouse ACC. International Journal of Molecular Sciences. 2023; 24(13):11113. https://doi.org/10.3390/ijms241311113
Chicago/Turabian StyleDarvish-Ghane, Soroush, Jennet Baumbach, and Loren J. Martin. 2023. "Influence of Inflammatory Pain and Dopamine on Synaptic Transmission in the Mouse ACC" International Journal of Molecular Sciences 24, no. 13: 11113. https://doi.org/10.3390/ijms241311113
APA StyleDarvish-Ghane, S., Baumbach, J., & Martin, L. J. (2023). Influence of Inflammatory Pain and Dopamine on Synaptic Transmission in the Mouse ACC. International Journal of Molecular Sciences, 24(13), 11113. https://doi.org/10.3390/ijms241311113