
An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma

 , , and
, , and {kind=link}
Abstract
:1. Introduction
- Proneural—most common in younger patients. It presents an oligodendrocytic lineage associated with secondary GBM, and enhancing mutations in tumor protein 53 (TP53) and IDH1 genes;
- Neural—appears in older patients. Derived from astrocytes and oligodendrocytes, it expresses neuron-related genes and no specific mutations;
- Classical—with no TP53 mutations and enhancing expression of EGFR;
- Mesenchymal—presents an astroglial lineage, with mutations in neurofibromin 1, phosphatase, and tensin homolog (PTEN) and TP53 genes.
- completely ligand mediated, with no direct contact between the extracellular regions of the two receptors [36];
- completely receptor mediated, with no physical interaction between two activating ligands—as in the case of EGFR [37];
- ligand homodimers attach themselves to two receptor molecules and then interact across the dimer interface [38];
2. EGFR Biology
3. EGFR Pathway Activation
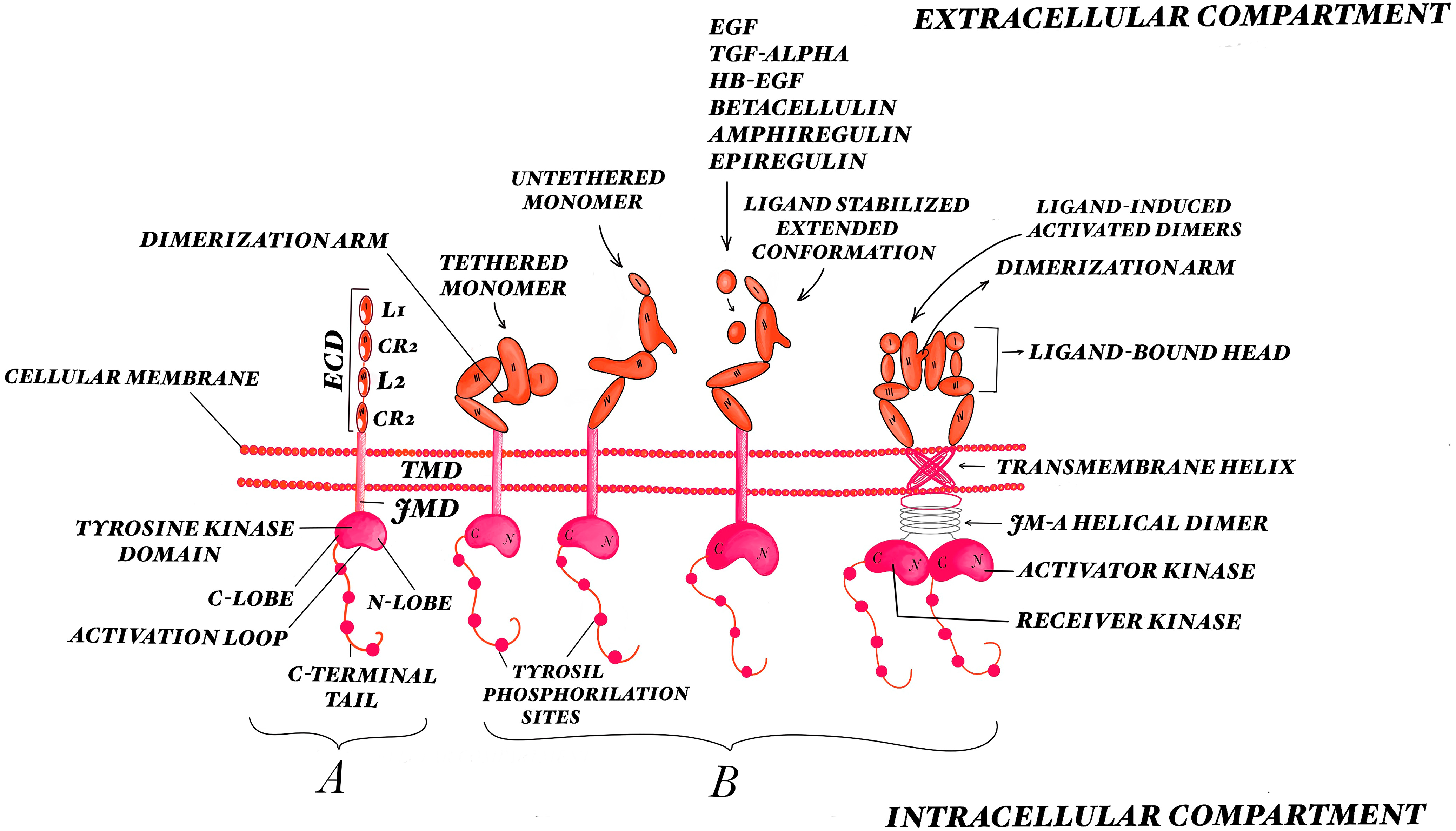
3.1. Activation of the Extracellular Domain
3.2. Activation of the Intracellular Domains
3.3. Downstream Signaling
4. Role of EGFR in the Molecular Pathogenesis of Glioblastoma
4.1. Oncogenic Activation of RTKs
- Gain of function mutations: these types of mutations lead to atypical downstream signal transduction. One of these types of mutations are the “driver mutations” that are able to give a selective growth advantage to the cells [81]. It is possible that the further study of these “driver mutations” could help us to understand how the cancer initiates and progresses and to bring new perspectives for targeted treatments. EGFR TKD is encoded by exons 18–24, while the EGFR mutations appear mostly in exons 18–21, close to the ATP binding pocket [82]. The kinase and the downstream signaling are hyperactivated by these mutations, giving them oncogenic properties [82,83,84]. It was demonstrated that patients with tumors that present somatic EGFR TKD mutations are more sensitive to EGFR TKIs [85,86,87,88,89,90,91]. Other types of mutations can appear in the extracellular domain (ECD), transmembrane domain (TMD), or juxtamembrane domain (JMD) of RTKs. In glioblastoma, missense mutations at the level of EGFR ECD were discovered and were associated with higher expression of EGFR protein, which undergoes phosphorylation when not stimulated by ligand [92,93,94]. Patients with EGFR ECD mutations had poor clinical results when under treatment with EGFR TKIs erlotinib and gefitinib [95,96]. The ECD mutations seem to adopt an inactive form, suggesting that they may be more responsive to EGFR targeted therapies that bind to the inactive form of the receptor [97]. It seems that both intra- and extracellular GBM mutations have, as a result, ligand-independent oncogenic activation.
- Genomic amplification: EGFR is the most commonly overexpressed RTK in GBM [98]. The prevalence of EGFR gene amplification and overexpression is higher in primary GBM than secondary GBM [99]. The consequence of overexpression is a higher local concentration of the receptor, resulting in elevated RTK signaling and overpowered antagonizing regulatory effects [100]. This overexpression is caused by multiple mechanisms, the most important being gene amplification, followed by transcriptional/translational enhancement [101,102], oncogenic viruses [103], degradation of normal regulatory mechanisms such as the loss of phosphatases [104], and other negative regulators. Gene amplification leads to an increase in the copy number of a specific region of the genome [105] in the form of extrachromosomal elements (double minutes) that are usually high-level amplifications with more than 25 copies, or repeated units at a single locus or throughout the genome (distributed insertions) characterized by low-level amplification with 5 to 25 copies [98,106]. These amplifications can be determined by flaws in the DNA replication, fragile sites at the chromosomal level, or telomere dysfunction [105]. The amplification pattern is quite different in the same tumor type [98].
- Chromosomal rearrangements: studies have shown that the formation of new tyrosine kinase fusion oncoproteins is caused by numerous chromosomal rearrangements [23,107,108]. It may be of significant importance to identify these fusion proteins, as they can be therapeutically targetable with small molecule inhibitors. Some risk factors are thought to participate in the gene fusion events—topoisomerase poisons [109], exposure to ionizing radiation [110,111], and oxidative stress [112]—but the exact way in which these mechanisms function is not yet known. Although a great number of tyrosine kinase fusions have been described, the structure of the fusion oncoproteins is quite similar. The fusion can arise in either the N-terminal or the C-terminal of the RTK, the tyrosine kinase domain being preserved either way. The genomic breakpoint can appear either downstream of the exons that encode the full kinase domain, in which case the ECD, TMD, and JMD are conserved and the resultant fusion protein will behave like a membrane-bound receptor, or upstream thereof, in which case loss of the ECD, TMD, and JMD occurs and the protein that appears as a result will not be membrane bound. The chimeric fusion proteins that appear as a result of the chromosomal rearrangements lead to oncogene addiction [113,114]. The use of target-specific TKIs against RTK fusions may be a good weapon in the battle against numerous types of RTK fusion-driven cancers.
- Autocrine activation: communication between cells is carried out with the help of messengers, like growth factors and cytokines, released by secretory cells. When the target cells are also the secretory cells, the signaling is called “autocrine” [115]. This type of autocrine activation can lead to tumor formation and clonal expansion [116]. In association with other autocrine growth pathways, the autocrine activation loop of RTKs can lead to tumor formation. The autocrine pathways can be used as a target for cancer therapy [117]. The wild-type EGFR ligands, like TGF-alpha and HB-EGF, are generally increased in glioblastoma, leading to an autocrine loop that results in the growth of glioma cells [118]. GBM expresses EGFRvIII, which does not bind ligands and is thought to be more tumorigenic than wild-type EGFR. TGF-α and HB-EGF induce the expression of EGFRvIII, implying that EGFRvIII may create an autocrine loop with wild-type EGFR, which plays an important role in signal transduction in glioblastoma cells [119].
4.2. EGFR Mutations
4.3. EGFR Wild-Type
4.4. EGFR Copy Number Alterations
4.5. EGFR Rearrangements
4.5.1. EGFRvIII
4.5.2. Other Deletion Variants
4.6. EGFR Fusions
4.7. MicroRNAs
4.8. Crosstalk
5. Targeted Therapy—EGFR as Therapeutic Agent
The Kinase-Independent Pro-Survival Function of EGFR in Cancer Cells
6. The tyrosine Kinase Inhibitors
6.1. First-Generation EGFR Inhibitors
6.2. Second-Generation EGFR Inhibitors
6.3. Third-Generation EGFR Inhibitors
6.4. Fourth-Generation EGFR Inhibitors
7. The Monoclonal Antibodies
8. Immunotherapy
9. Targeted Isotopes
10. Nanoparticles
11. Targeting the Regulation of EGFR Gene Expression
12. Challenges to Current Anti-EGFR Therapies
- The target independence. Not all of the glioblastoma cells express EGFR proteins; therefore, the EGFR inhibitors have no effect on them. A frequent situation encountered in this type of resistance is the loss of extra-chromosomally encoded EGFR. It can appear after the use of small molecule therapies. Small circular fragments of extra-chromosomal DNA act as regulators of dynamic EGFRvIII expression, and they may be involved in the resistance to inhibition. Studies have shown that after the treatment of GBM cells with erlotinib, mutant EGFR was reversibly blocked by producing extra-chromosomal DNA. By seizing the use of erlotinib, the mutations reappeared, resulting in the upregulation of EGFRvIII [234].
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [Green Version]
- Brandes, A.A.; Bartolotti, M.; Tosoni, A.; Franceschi, E. Nitrosoureas in the management of malignant gliomas. Curr. Neurol. Neurosci. Rep. 2016, 16, 13. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, S.M.B.; Staicu, G.-A.; Sevastre, A.-S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma Stem Cells—Useful Tools in the Battle against Cancer. Int. J. Mol. Sci. 2022, 23, 4602. [Google Scholar] [CrossRef] [PubMed]
- Sanai, N.; Alvarez-Buylla, A.; Berger, M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005, 353, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Lim, D.A.; Cha, S.; Mayo, M.C.; Chen, M.-H.; Keles, E.; VandenBerg, S.; Berger, M.S. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro-Oncology 2007, 9, 424–429. [Google Scholar] [CrossRef]
- Goffart, N.; Kroonen, J.; Rogister, B. Glioblastoma-initiating cells: Relationship with neural stem cells and the micro-environment. Cancers 2013, 5, 1049–1071. [Google Scholar] [CrossRef]
- Deleanu, R.; Ceafalan, L.C.; Dricu, A. Transcriptomic Crosstalk between Gliomas and Telencephalic Neural Stem and Progenitor Cells for Defining Heterogeneity and Targeted Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 13211. [Google Scholar] [CrossRef]
- Bastien, J.I.; McNeill, K.A.; Fine, H.A. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer 2015, 121, 502–516. [Google Scholar] [CrossRef]
- Rong, Y.; Durden, D.L.; Van Meir, E.G.; Brat, D.J. ‘Pseudopalisading’necrosis in glioblastoma: A familiar morphologic feature that links vascular pathology, hypoxia, and angiogenesis. J. Neuropathol. Exp. Neurol. 2006, 65, 529–539. [Google Scholar] [CrossRef]
- Artene, S.-A.; Turcu-Stiolica, A.; Ciurea, M.E.; Folcuti, C.; Tataranu, L.G.; Alexandru, O.; Purcaru, O.S.; Tache, D.E.; Boldeanu, M.V.; Silosi, C. Comparative effect of immunotherapy and standard therapy in patients with high grade glioma: A meta-analysis of published clinical trials. Sci. Rep. 2018, 8, 11800. [Google Scholar] [CrossRef]
- Alexandru, O.; Sevastre, A.-S.; Castro, J.; Artene, S.-A.; Tache, D.E.; Purcaru, O.S.; Sfredel, V.; Tataranu, L.G.; Dricu, A. Platelet-derived growth factor receptor and ionizing radiation in high grade glioma cell lines. Int. J. Mol. Sci. 2019, 20, 4663. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; Von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization classification of tumors of the central nervous system: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wesseling, P.; Kros, J.M.; Jeuken, J.W. The pathological diagnosis of diffuse gliomas: Towards a smart synthesis of microscopic and molecular information in a multidisciplinary context. Diagn. Histopathol. 2011, 17, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; LeBrun, D.G.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyd, N.H.; Tran, A.N.; Bernstock, J.D.; Etminan, T.; Jones, A.B.; Gillespie, G.Y.; Friedman, G.K.; Hjelmeland, A.B. Glioma stem cells and their roles within the hypoxic tumor microenvironment. Theranostics 2021, 11, 665. [Google Scholar] [CrossRef] [PubMed]
- Burger, P.C. Pathologie Anatomy and CT Correlations in the Glioblastoma Multif orme. Stereotact. Funct. Neurosurg. 1983, 46, 180–187. [Google Scholar] [CrossRef]
- Wu, Z.; Dai, L.; Tang, K.; Ma, Y.; Song, B.; Zhang, Y.; Li, J.; Lui, S.; Gong, Q.; Wu, M. Advances in magnetic resonance imaging contrast agents for glioblastoma-targeting theranostics. Regen. Biomater. 2021, 8, rbab062. [Google Scholar] [CrossRef]
- Galldiks, N.; Niyazi, M.; Grosu, A.L.; Kocher, M.; Langen, K.-J.; Law, I.; Minniti, G.; Kim, M.M.; Tsien, C.; Dhermain, F. Contribution of PET imaging to radiotherapy planning and monitoring in glioma patients-a report of the PET/RANO group. Neuro-Oncology 2021, 23, 881–893. [Google Scholar] [CrossRef]
- Verdugo, E.; Puerto, I.; Medina, M.Á. An update on the molecular biology of glioblastoma, with clinical implications and progress in its treatment. Cancer Commun. 2022, 42, 1083–1111. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.; Pfister, S.M.; Reifenberger, G. The 2021 WHO classification of tumors of the central nervous system: A summary. Neuro-Oncology 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- van den Bent, M.J.; Gao, Y.; Kerkhof, M.; Kros, J.M.; Gorlia, T.; Van Zwieten, K.; Prince, J.; van Duinen, S.; Sillevis Smitt, P.A.; Taphoorn, M. Changes in the EGFR amplification and EGFRvIII expression between paired primary and recurrent glioblastomas. Neuro-Oncology 2015, 17, 935–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Ceccarelli, M.; Barthel, F.P.; Malta, T.M.; Sabedot, T.S.; Salama, S.R.; Murray, B.A.; Morozova, O.; Newton, Y.; Radenbaugh, A.; Pagnotta, S.M. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 2016, 164, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [Green Version]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakada, M.; Nakada, S.; Demuth, T.; Tran, N.; Hoelzinger, D.; Berens, M. Molecular targets of glioma invasion. Cell. Mol. Life Sci. 2007, 64, 458–478. [Google Scholar] [CrossRef]
- Dricu, A. Oncogenic Signalling of Growth Factor Receptors in Cancer: Mechanisms and Therapeutic Opportunities. Int. J. Mol. Sci. 2022, 23, 7376. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R. Structural analysis of receptor tyrosine kinases. Prog. Biophys. Mol. Biol. 1999, 71, 343–358. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Lin, S.-F. The protein tyrosine kinase family of the human genome. Oncogene 2000, 19, 5548–5557. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, T. Tyrosine phosphorylation: Thirty years and counting. Curr. Opin. Cell Biol. 2009, 21, 140–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrman, T.; He, X.; Raab, B.; Dukipatti, A.; Blau, H.; Garcia, K.C. Structural and mechanistic insights into nerve growth factor interactions with the TrkA and p75 receptors. Neuron 2007, 53, 25–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuzawa, S.; Opatowsky, Y.; Zhang, Z.; Mandiyan, V.; Lax, I.; Schlessinger, J. Structural basis for activation of the receptor tyrosine kinase KIT by stem cell factor. Cell 2007, 130, 323–334. [Google Scholar] [CrossRef] [Green Version]
- Yayon, A.; Klagsbrun, M.; Esko, J.D.; Leder, P.; Ornitz, D.M. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991, 64, 841–848. [Google Scholar] [CrossRef]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Chung, I.; Akita, R.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Soos, M.A.; Field, C.; Siddle, K. Purified hybrid insulin/insulin-like growth factor-I receptors bind insulin-like growth factor-I, but not insulin, with high affinity. Biochem. J. 1993, 290, 419–426. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Shewchuk, L.M.; Hassell, A.M.; Ellis, B.; Holmes, W.; Davis, R.; Horne, E.L.; Kadwell, S.H.; McKee, D.D.; Moore, J.T. Structure of the Tie2 RTK domain: Self-inhibition by the nucleotide binding loop, activation loop, and C-terminal tail. Structure 2000, 8, 1105–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wybenga-Groot, L.E.; Baskin, B.; Ong, S.H.; Tong, J.; Pawson, T.; Sicheri, F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell 2001, 106, 745–757. [Google Scholar] [CrossRef] [Green Version]
- Brummer, T.; Schmitz-Peiffer, C.; Daly, R.J. Docking proteins. FEBS J. 2010, 277, 4356–4369. [Google Scholar] [CrossRef] [PubMed]
- Alexandru, O.; Horescu, C.; Sevastre, A.-S.; Cioc, C.; Baloi, C.; Oprita, A.; Dricu, A. Receptor tyrosine kinase targeting in glioblastoma: Performance, limitations and future approaches. Contemp. Oncol./Współczesna Onkol. 2020, 24, 55–66. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Walker, F.; Pouliot, N.; Garrett, T.P.; Ward, C.W.; Burgess, A.W. Epidermal growth factor receptor: Mechanisms of activation and signalling. EGF Recept. Fam. 2003, 284, 31–53. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib) relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef] [PubMed]
- Bogdan, S.; Klämbt, C. Epidermal growth factor receptor signaling. Curr. Biol. 2001, 11, R292–R295. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, S.R.; Till, J.H. Protein tyrosine kinase structure and function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelzinger, D.B.; Demuth, T.; Berens, M.E. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J. Natl. Cancer Inst. 2007, 99, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Thomas, R.; Weihua, Z. Rethink of EGFR in cancer with its kinase independent function on board. Front. Oncol. 2019, 9, 800. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Jureczek, J.; Feldmann, A.; Bergmann, R.; Arndt, C.; Berndt, N.; Koristka, S.; Loureiro, L.R.; Mitwasi, N.; Hoffmann, A.; Kegler, A. Highly efficient targeting of EGFR-expressing tumor cells with UNiCAR T cells via target modules based on cetuximab®. OncoTargets Ther. 2020, 13, 5515. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Li, X.; Wu, Y.; Zhang, G.; Liu, X.; Li, Y.; Bao, Y.; Yang, W.; Cui, H. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene 2020, 39, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, G.; Cohen, S. Epidermal growth factor. Annu. Rev. Biochem. 1979, 48, 193–216. [Google Scholar] [CrossRef] [PubMed]
- Maire, C.L.; Ligon, K.L. Molecular pathologic diagnosis of epidermal growth factor receptor. Neuro-Oncology 2014, 16, viii1–viii6. [Google Scholar] [CrossRef] [PubMed]
- Hervieu, A.; Kermorgant, S. The role of PI3K in Met driven cancer: A recap. Front. Mol. Biosci. 2018, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Dawson, J.P.; Berger, M.B.; Lin, C.-C.; Schlessinger, J.; Lemmon, M.A.; Ferguson, K.M. Epidermal growth factor receptor dimerization and activation require ligand-induced conformational changes in the dimer interface. Mol. Cell. Biol. 2005, 25, 7734–7742. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Kharbanda, A.; Walter, D.M.; Gudiel, A.A.; Schek, N.; Feldser, D.M.; Witze, E.S. Blocking EGFR palmitoylation suppresses PI3K signaling and mutant KRAS lung tumorigenesis. Sci. Signal. 2020, 13, eaax2364. [Google Scholar] [CrossRef]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT pathway as a target for cancer treatment. Annu. Rev. Med. 2016, 67, 11–28. [Google Scholar] [CrossRef]
- Mattoon, D.R.; Lamothe, B.; Lax, I.; Schlessinger, J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biol. 2004, 2, 24. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, E.Q.X.; Colόn, R.R.; Abounader, R. HGF/MET signaling in malignant brain tumors. Int. J. Mol. Sci. 2020, 21, 7546. [Google Scholar] [CrossRef]
- Kiyatkin, A.; Aksamitiene, E.; Markevich, N.I.; Borisov, N.M.; Hoek, J.B.; Kholodenko, B.N. Scaffolding protein Grb2-associated binder 1 sustains epidermal growth factor-induced mitogenic and survival signaling by multiple positive feedback loops. J. Biol. Chem. 2006, 281, 19925–19938. [Google Scholar] [CrossRef] [Green Version]
- Oprita, A.; Baloi, S.-C.; Staicu, G.-A.; Alexandru, O.; Tache, D.E.; Danoiu, S.; Micu, E.S.; Sevastre, A.-S. Updated insights on EGFR signaling pathways in glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef]
- Pawson, T. Specificity in signal transduction: From phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 2004, 116, 191–203. [Google Scholar] [CrossRef] [Green Version]
- Ward, A.F.; Braun, B.S.; Shannon, K.M. Targeting oncogenic Ras signaling in hematologic malignancies. Blood J. Am. Soc. Hematol. 2012, 120, 3397–3406. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.P.; Mark, K.G.; Leslie, K.; Pao, W.; Motoi, N.; Gerald, W.L.; Travis, W.D.; Bornmann, W.; Veach, D.; Clarkson, B. Mutations in the EGFR kinase domain mediate STAT3 activation via IL-6 production in human lung adenocarcinomas. J. Clin. Investig. 2007, 117, 3846–3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.P.; Chang, Q.; Mao, N.; Daly, L.A.; Vogel, R.; Chan, T.; Liu, S.H.; Bournazou, E.; Schori, E.; Zhang, H. JAK2 inhibition sensitizes resistant EGFR-mutant lung adenocarcinoma to tyrosine kinase inhibitors. Sci. Signal. 2016, 9, ra33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padfield, E.; Ellis, H.P.; Kurian, K.M. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front. Oncol. 2015, 5, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Östman, A.; Böhmer, F.-D. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 2001, 11, 258–266. [Google Scholar] [CrossRef]
- Serban, F.; Artene, S.-A.; Georgescu, A.M.; Purcaru, S.O.; Tache, D.E.; Alexandru, O.; Dricu, A. Epidermal growth factor, latrophilin, and seven transmembrane domain-containing protein 1 marker, a novel angiogenesis marker. OncoTargets Ther. 2015, 8, 3767–3774. [Google Scholar]
- McDonell, L.M.; Kernohan, K.D.; Boycott, K.M.; Sawyer, S.L. Receptor tyrosine kinase mutations in developmental syndromes and cancer: Two sides of the same coin. Hum. Mol. Genet. 2015, 24, R60–R66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Wang, Z.; Longo, P.A.; Tarrant, M.K.; Kim, K.; Head, S.; Leahy, D.J.; Cole, P.A. Mechanistic insights into the activation of oncogenic forms of EGF receptor. Nat. Struct. Mol. Biol. 2011, 18, 1388–1393. [Google Scholar] [CrossRef] [PubMed]
- Red Brewer, M.; Yun, C.-H.; Lai, D.; Lemmon, M.A.; Eck, M.J.; Pao, W. Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. USA 2013, 110, E3595–E3604. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.-H.; Boggon, T.J.; Li, Y.; Woo, M.S.; Greulich, H.; Meyerson, M.; Eck, M.J. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: Mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 2007, 11, 217–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I. Gefitinib or chemotherapy for non–small-cell lung cancer with mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Yang, J.C.-H.; Yamamoto, N.; O’Byrne, K.; Hirsh, V.; Mok, T.; Geater, S.L.; Orlov, S.; Tsai, C.-M.; Boyer, M. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J. Clin. Oncol. 2013, 31, 3327–3334. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L. AZD9291 in EGFR inhibitor–resistant non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T. Osimertinib in untreated EGFR-mutated advanced non–small-cell lung cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Vivanco, I.; Beroukhim, R.; Huang, J.H.Y.; Feng, W.L.; DeBiasi, R.M.; Yoshimoto, K.; King, J.C.; Nghiemphu, P.; Yuza, Y. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS Med. 2006, 3, e485. [Google Scholar] [CrossRef] [PubMed]
- Arjona, D.; Bello, M.J.; Alonso, M.E.; Aminoso, C.; Isla, A.; De Campos, J.; Sarasa, J.; Gutierrez, M.; Villalobo, A.; Rey, J. Molecular analysis of the EGFR gene in astrocytic gliomas: mRNA expression, quantitative-PCR analysis of non-homogeneous gene amplification and DNA sequence alterations. Neuropathol. Appl. Neurobiol. 2005, 31, 384–394. [Google Scholar] [CrossRef] [PubMed]
- Danciulescu, O.T.; Folcuti, R.; Dricu, A. Temozolomide and targeted therapy against epidermal growth factor receptor in glioma. Int. J. Clin. Exp. Med. 2016, 9, 15249–15261. [Google Scholar]
- van den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.-F. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, E.; Cavallo, G.; Lonardi, S.; Magrini, E.; Tosoni, A.; Grosso, D.; Scopece, L.; Blatt, V.; Urbini, B.; Pession, A. Gefitinib in patients with progressive high-grade gliomas: A multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO). Br. J. Cancer 2007, 96, 1047–1051. [Google Scholar] [CrossRef]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N. Differential sensitivity of glioma-versus lung cancer–specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gines, C.; Gil-Benso, R.; Ferrer-Luna, R.; Benito, R.; Serna, E.; Gonzalez-Darder, J.; Quilis, V.; Monleon, D.; Celda, B.; Cerdá-Nicolas, M. New pattern of EGFR amplification in glioblastoma and the relationship of gene copy number with gene expression profile. Mod. Pathol. 2010, 23, 856–865. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [Green Version]
- Carraway, K.L.; Sweeney, C. EGF receptor activation by heterologous mechanisms. Cancer Cell 2002, 1, 405–406. [Google Scholar] [CrossRef] [Green Version]
- Ludes-Meyers, J.H.; Subler, M.A.; Shivakumar, C.V.; Munoz, R.M.; Jiang, P.; Bigger, J.E.; Brown, D.R.; Deb, S.P.; Deb, S. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 1996, 16, 6009–6019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reznik, T.E.; Sang, Y.; Ma, Y.; Abounader, R.; Rosen, E.M.; Xia, S.; Laterra, J. Transcription-dependent epidermal growth factor receptor activation by hepatocyte growth factor. Mol. Cancer Res. 2008, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanawa, M.; Suzuki, S.; Dobashi, Y.; Yamane, T.; Kono, K.; Enomoto, N.; Ooi, A. EGFR protein overexpression and gene amplification in squamous cell carcinomas of the esophagus. Int. J. Cancer 2006, 118, 1173–1180. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Albertson, D.G. Gene amplification in cancer. Trend. Genet. 2006, 22, 447–455. [Google Scholar] [CrossRef]
- Albertson, D.G.; Collins, C.; McCormick, F.; Gray, J.W. Chromosome aberrations in solid tumors. Nat. Genet. 2003, 34, 369–376. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [Green Version]
- Network, C.G.A.R. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543. [Google Scholar]
- Mistry, A.R.; Felix, C.A.; Whitmarsh, R.J.; Mason, A.; Reiter, A.; Cassinat, B.; Parry, A.; Walz, C.; Wiemels, J.L.; Segal, M.R. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N. Engl. J. Med. 2005, 352, 1529–1538. [Google Scholar] [CrossRef]
- Ito, T.; Seyama, T.; Iwamoto, K.S.; Hayashi, T.; Mizuno, T.; Tsuyama, N.; Dohi, K.; Nakamura, N.; Akiyama, M. In vitro irradiation is able to cause RET oncogene rearrangement. Cancer Res. 1993, 53, 2940–2943. [Google Scholar]
- Mizuno, T.; Kyoizumi, S.; Suzuki, T.; Iwamoto, K.; Seyama, T. Continued expression of a tissue specific activated oncogene in the early steps of radiation-induced human thyroid carcinogenesis. Oncogene 1997, 15, 1455–1460. [Google Scholar] [CrossRef]
- Tsai, A.G.; Lieber, M.R. Mechanisms of chromosomal rearrangement in the human genome. BMC Genom. 2010, 11, S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konduri, K.; Gallant, J.-N.; Chae, Y.K.; Giles, F.J.; Gitlitz, B.J.; Gowen, K.; Ichihara, E.; Owonikoko, T.K.; Peddareddigari, V.; Ramalingam, S.S. EGFR Fusions as Novel Therapeutic Targets in Lung CancerTherapeutically Targetable EGFR Fusions in Lung Cancer. Cancer Discov. 2016, 6, 601–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovly, C.M.; Gupta, A.; Lipson, D.; Otto, G.; Brennan, T.; Chung, C.T.; Borinstein, S.C.; Ross, J.S.; Stephens, P.J.; Miller, V.A. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014, 4, 889–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell. Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef]
- Walsh, J.H.; Karnes, W.; Cuttitta, F.; Walker, A. Autocrine growth factors and solid tumor malignancy. West. J. Med. 1991, 155, 152. [Google Scholar]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5. [Google Scholar] [CrossRef] [Green Version]
- Ramnarain, D.B.; Park, S.; Lee, D.Y.; Hatanpaa, K.J.; Scoggin, S.O.; Otu, H.; Libermann, T.A.; Raisanen, J.M.; Ashfaq, R.; Wong, E.T. Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res. 2006, 66, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.-Y.; Brady, D.C.; Villanueva, J. Double trouble: Kinase domain duplication as a new path to drug resistance. Pigment Cell Melanoma Res. 2016, 29, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallant, J.-N.; Sheehan, J.H.; Shaver, T.M.; Bailey, M.; Lipson, D.; Chandramohan, R.; Brewer, M.R.; York, S.J.; Kris, M.G.; Pietenpol, J.A. EGFR Kinase Domain Duplication (EGFR-KDD) Is a Novel Oncogenic Driver in Lung Cancer That Is Clinically Responsive to AfatinibEGFR-KDD as a Therapeutic Target in Lung and Other Cancers. Cancer Discov. 2015, 5, 1155–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Idbaih, A.; Aimard, J.; Boisselier, B.; Marie, Y.; Paris, S.; Criniere, E.; Carvalho Silva, R.; Laigle-Donadey, F.; Rousseau, A.; Mokhtari, K. Epidermal growth factor receptor extracellular domain mutations in primary glioblastoma. Neuropathol. Appl. Neurobiol. 2009, 35, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Ekstrand, A.J.; James, C.D.; Cavenee, W.K.; Seliger, B.; Pettersson, R.F.; Collins, V.P. Genes for epidermal growth factor receptor, transforming growth factor α, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991, 51, 2164–2172. [Google Scholar]
- Gullick, W.J.; Marsden, J.J.; Whittle, N.; Ward, B.; Bobrow, L.; Waterfield, M.D. Expression of epidermal growth factor receptors on human cervical, ovarian, and vulval carcinomas. Cancer Res. 1986, 46, 285–292. [Google Scholar] [PubMed]
- Inal, C.; Yilmaz, E.; Piperdi, B.; Perez-Soler, R.; Cheng, H. Emerging treatment for advanced lung cancer with EGFR mutation. Expert Opin. Emerg. Drugs 2015, 20, 597–612. [Google Scholar] [CrossRef]
- Siegelin, M.D.; Borczuk, A.C. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab. Investig. 2014, 94, 129–137. [Google Scholar] [CrossRef] [Green Version]
- De Luca, A.; Normanno, N. Predictive biomarkers to tyrosine kinase inhibitors for the epidermal growth factor receptor in non-small-cell lung cancer. Curr. Drug Targets 2010, 11, 851–864. [Google Scholar] [CrossRef]
- Li, A.R.; Chitale, D.; Riely, G.J.; Pao, W.; Miller, V.A.; Zakowski, M.F.; Rusch, V.; Kris, M.G.; Ladanyi, M. EGFR mutations in lung adenocarcinomas: Clinical testing experience and relationship to EGFR gene copy number and immunohistochemical expression. J. Mol. Diagn. 2008, 10, 242–248. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.J.; Woo, C.G.; Lee, S.; Chin, S.; Kim, H.K.; Kwak, J.J.; Koh, E.S.; Lee, B.; Jang, K.-T.; Moon, A. Gene copy number variation and protein overexpression of EGFR and HER2 in distal extrahepatic cholangiocarcinoma. Pathology 2017, 49, 582–588. [Google Scholar] [CrossRef]
- Birkman, E.-M.; Ålgars, A.; Lintunen, M.; Ristamäki, R.; Sundström, J.; Carpén, O. EGFR gene amplification is relatively common and associates with outcome in intestinal adenocarcinoma of the stomach, gastro-oesophageal junction and distal oesophagus. BMC Cancer 2016, 16, 406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libermann, T.A.; Nusbaum, H.R.; Razon, N.; Kris, R.; LAX, I.; Soreq, H.; Whittle, N.; Waterfield, M.D.; Ullrich, A.; Schlessinger, J. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J. Cell Sci. 1985, 1985, 161–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandru, O.; Purcaru, S.O.; Tataranu, L.G.; Lucan, L.; Castro, J.; Folcuţi, C.; Artene, S.-A.; Tuţă, C.; Dricu, A. The influence of EGFR inactivation on the radiation response in high grade glioma. Int. J. Mol. Sci. 2018, 19, 229. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, A.J.; Sugawa, N.; James, C.D.; Collins, V.P. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N-and/or C-terminal tails. Proc. Natl. Acad. Sci. USA 1992, 89, 4309–4313. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Fukui, Y.; Ueyama, Y.; Tamaoki, N.; Kawamoto, T.; Taniguchi, S.; Shibuya, M. Amplification of the structurally and functionally altered epidermal growth factor receptor gene (c-erbB) in human brain tumors. Mol. Cell. Biol. 1988, 8, 1816–1820. [Google Scholar]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef]
- Kita, D.; Yonekawa, Y.; Weller, M.; Ohgaki, H. PIK3CA alterations in primary (de novo) and secondary glioblastomas. Acta Neuropathol. 2007, 113, 295–302. [Google Scholar] [CrossRef]
- Ohgaki, H.; Dessen, P.; Jourde, B.; Horstmann, S.; Nishikawa, T.; Di Patre, P.-L.; Burkhard, C.; Schüler, D.; Probst-Hensch, N.M.; Maiorka, P.C. Genetic pathways to glioblastoma: A population-based study. Cancer Res. 2004, 64, 6892–6899. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of Tumor Specimens at the Time of Acquired Resistance to EGFR-TKI Therapy in 155 Patients with EGFR-Mutant Lung CancersMechanisms of Acquired Resistance to EGFR-TKI Therapy. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Campo, M.; Gerber, D.; Gainor, J.F.; Heist, R.S.; Temel, J.S.; Shaw, A.T.; Fidias, P.; Muzikansky, A.; Engelman, J.A.; Sequist, L.V. Acquired resistance to first-line afatinib and the challenges of prearranged progression biopsies. J. Thorac. Oncol. 2016, 11, 2022–2026. [Google Scholar] [CrossRef] [Green Version]
- Barker II, F.G.; Simmons, M.L.; Chang, S.M.; Prados, M.D.; Larson, D.A.; Sneed, P.K.; Wara, W.M.; Berger, M.S.; Chen, P.; Israel, M.A. EGFR overexpression and radiation response in glioblastoma multiforme. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 410–418. [Google Scholar] [CrossRef]
- Feldkamp, M.M.; Lala, P.; Lau, N.; Roncari, L.; Guha, A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurg.-Baltim. 1999, 45, 1442–1453. [Google Scholar] [CrossRef]
- Shinojima, N.; Tada, K.; Shiraishi, S.; Kamiryo, T.; Kochi, M.; Nakamura, H.; Makino, K.; Saya, H.; Hirano, H.; Kuratsu, J.-I. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003, 63, 6962–6970. [Google Scholar] [PubMed]
- Xu, N.; Fang, W.; Mu, L.; Tang, Y.; Gao, L.; Ren, S.; Cao, D.; Zhou, L.; Zhang, A.; Liu, D. Overexpression of wildtype EGFR is tumorigenic and denotes a therapeutic target in non-small cell lung cancer. Oncotarget 2016, 7, 3884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velu, T.J.; Beguinot, L.; Vass, W.C.; Willingham, M.C.; Merlino, G.T.; Pastan, I.; Lowy, D.R. Epidermal-growth-factor-dependent transformation by a human EGF receptor proto-oncogene. Science 1987, 238, 1408–1410. [Google Scholar] [CrossRef]
- Greulich, H.; Chen, T.-H.; Feng, W.; Jänne, P.A.; Alvarez, J.V.; Zappaterra, M.; Bulmer, S.E.; Frank, D.A.; Hahn, W.C.; Sellers, W.R. Oncogenic transformation by inhibitor-sensitive and-resistant EGFR mutants. PLoS Med. 2005, 2, e313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef]
- Carlsson, J.; Wester, K.; De La Torre, M.; Malmström, P.-U.; Gårdmark, T. EGFR-expression in primary urinary bladder cancer and corresponding metastases and the relation to HER2-expression. On the possibility to target these receptors with radionuclides. Radiol. Oncol. 2015, 49, 50–58. [Google Scholar] [CrossRef] [Green Version]
- Biernat, W.; Huang, H.; Yokoo, H.; Kleihues, P.; Ohgaki, H. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol. 2004, 14, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Fregnani, E.R.; Fonseca, F.P.; Alves, F.A.; Pinto, C.A.L.; Kaminagakura, E. EGFR is not amplified in ameloblastoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2018, 125, 454–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuda, N.; Lim, B.; Wang, X.; Ueno, N.T. Early clinical development of epidermal growth factor receptor targeted therapy in breast cancer. Expert Opin. Investig. Drugs 2017, 26, 463–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, A.H.; Zanca, C.; Furnari, F. Epidermal growth factor receptor targeting and challenges in glioblastoma. Neuro-Oncology 2016, 18, 914–918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacco, A.G.; Worden, F.P. Molecularly targeted therapy for the treatment of head and neck cancer: A review of the ErbB family inhibitors. OncoTargets Ther. 2016, 9, 1927–1943. [Google Scholar]
- Sieghart, W.; Pinter, M.; Dauser, B.; Rohr-Udilova, N.; Piguet, A.-C.; Prager, G.; Hayden, H.; Dienes, H.-P.; Dufour, J.-F.; Peck-Radosavljevic, M. Erlotinib and sorafenib in an orthotopic rat model of hepatocellular carcinoma. J. Hepatol. 2012, 57, 592–599. [Google Scholar] [CrossRef]
- Ioannou, N.; Dalgleish, A.; Seddon, A.; Mackintosh, D.; Guertler, U.; Solca, F.; Modjtahedi, H. Anti-tumour activity of afatinib, an irreversible ErbB family blocker, in human pancreatic tumour cells. Br. J. Cancer 2011, 105, 1554–1562. [Google Scholar] [CrossRef] [Green Version]
- Weihua, Z.; Tsan, R.; Huang, W.-C.; Wu, Q.; Chiu, C.-H.; Fidler, I.J.; Hung, M.-C. Survival of cancer cells is maintained by EGFR independent of its kinase activity. Cancer Cell 2008, 13, 385–393. [Google Scholar] [CrossRef] [Green Version]
- Tsuchihashi, K.; Okazaki, S.; Ohmura, M.; Ishikawa, M.; Sampetrean, O.; Onishi, N.; Wakimoto, H.; Yoshikawa, M.; Seishima, R.; Iwasaki, Y. The EGF Receptor Promotes the Malignant Potential of Glioma by Regulating Amino Acid Transport System xc (−) EGFR Regulates xCT in Glioma. Cancer Res. 2016, 76, 2954–2963. [Google Scholar] [CrossRef] [Green Version]
- Nagy, P.; Arndt-Jovin, D.J.; Jovin, T.M. Small interfering RNAs suppress the expression of endogenous and GFP-fused epidermal growth factor receptor (erbB1) and induce apoptosis in erbB1-overexpressing cells. Exp. Cell Res. 2003, 285, 39–49. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.-S.; Pu, P.-Y.; Li, Y.-H.; Zhang, Z.-Y.; Qiu, M.-Z.; Huang, Q.; Wang, G.-X. An in vitro study on the suppressive effect of glioma cell growth induced by plasmid-based small interference RNA (siRNA) targeting human epidermal growth factor receptor. J. Neuro-Oncol. 2005, 74, 267–273. [Google Scholar] [CrossRef]
- Chen, G.; Kronenberger, P.; Teugels, E.; Umelo, I.A.; De Greve, J. Effect of siRNAs targeting the EGFR T790M mutation in a non-small cell lung cancer cell line resistant to EGFR tyrosine kinase inhibitors and combination with various agents. Biochem. Biophys. Res. Commun. 2013, 431, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Francis, J.M.; Zhang, C.-Z.; Maire, C.L.; Jung, J.; Manzo, V.E.; Adalsteinsson, V.A.; Homer, H.; Haidar, S.; Blumenstiel, B.; Pedamallu, C.S. EGFR Variant Heterogeneity in Glioblastoma Resolved through Single-Nucleus SequencingSingle-Nucleus Sequencing of GBM Mutational Heterogeneity. Cancer Discov. 2014, 4, 956–971. [Google Scholar] [CrossRef] [Green Version]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellinghoff, I.K.; Wang, M.Y.; Vivanco, I.; Haas-Kogan, D.A.; Zhu, S.; Dia, E.Q.; Lu, K.V.; Yoshimoto, K.; Huang, J.H.; Chute, D.J. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N. Engl. J. Med. 2005, 353, 2012–2024. [Google Scholar] [CrossRef] [Green Version]
- Wen, P.Y.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Norden, A.D.; Cloughesy, T.F.; Robins, H.I.; Lieberman, F.S.; Gilbert, M.R.; Mehta, M.P. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro-Oncology 2014, 16, 567–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelloski, C.E.; Ballman, K.V.; Furth, A.F.; Zhang, L.; Lin, E.; Sulman, E.P.; Bhat, K.; McDonald, J.M.; Yung, W.A.; Colman, H. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J. Clin. Oncol. 2007, 25, 2288–2294. [Google Scholar] [CrossRef]
- Nishikawa, R.; Sugiyama, T.; Narita, Y.; Furnari, F.; Cavenee, W.K.; Matsutani, M. Immunohistochemical analysis of the mutant epidermal growth factor, ΔEGFR, in glioblastoma. Brain Tumor Pathol. 2004, 21, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Frederick, L.; Wang, X.-Y.; Eley, G.; James, C.D. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000, 60, 1383–1387. [Google Scholar]
- Cho, J.; Pastorino, S.; Zeng, Q.; Xu, X.; Johnson, W.; Vandenberg, S.; Verhaak, R.; Cherniack, A.D.; Watanabe, H.; Dutt, A. Glioblastoma-Derived Epidermal Growth Factor Receptor Carboxyl-Terminal Deletion Mutants Are Transforming and Are Sensitive to EGFR-Directed TherapiesStudy of GBM-Derived EGFR C-Terminal Deletion Mutants. Cancer Res. 2011, 71, 7587–7596. [Google Scholar] [CrossRef] [Green Version]
- Frattini, V.; Trifonov, V.; Chan, J.M.; Castano, A.; Lia, M.; Abate, F.; Keir, S.T.; Ji, A.X.; Zoppoli, P.; Niola, F. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet. 2013, 45, 1141–1149. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.; Lankerovich, M.; Lee, H.; Yoon, J.-G.; Schroeder, B.; Foltz, G. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genom. 2013, 14, 818. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Dingerdissen, H.; Gupta, S.; Kahsay, R.; Shanker, V.; Wan, Q.; Yan, C.; Mazumder, R. Identification of key differentially expressed MicroRNAs in cancer patients through pan-cancer analysis. Comput. Biol. Med. 2018, 103, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Donzelli, S.; Cioce, M.; Muti, P.; Strano, S.; Yarden, Y.; Blandino, G. MicroRNAs: Non-coding fine tuners of receptor tyrosine kinase signalling in cancer. In Seminars in Cell & Developmental Biology; Academic Press: Cambridge, MA, USA, 2016; pp. 133–142. [Google Scholar]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-regulated cancer stem cells in the pathogenesis of human malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, J.; Lu, Z.; Ji, C.; Chen, Y.; Liu, Y.; Lei, Z.; Wang, L.; Zhang, H.-T.; Li, X. Melatonin inhibits proliferation and invasion via repression of miRNA-155 in glioma cells. Biomed. Pharmacother. 2017, 93, 969–975. [Google Scholar] [CrossRef]
- Xiong, W.; Ran, J.; Jiang, R.; Guo, P.; Shi, X.; Li, H.; Lv, X.; Li, J.; Chen, D. miRNA-320a inhibits glioma cell invasion and migration by directly targeting aquaporin 4. Oncol. Rep. 2018, 39, 1939–1947. [Google Scholar] [CrossRef] [Green Version]
- Acunzo, M.; Romano, G.; Wernicke, D.; Croce, C.M. MicroRNA and cancer–a brief overview. Adv. Biol. Regul. 2015, 57, 1–9. [Google Scholar] [CrossRef]
- Rao, S.A.M.; Arimappamagan, A.; Pandey, P.; Santosh, V.; Hegde, A.S.; Chandramouli, B.A.; Somasundaram, K. miR-219-5p inhibits receptor tyrosine kinase pathway by targeting EGFR in glioblastoma. PLoS ONE 2013, 8, e63164. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Kim, J.; Mueller, A.; Dey, B.; Yang, Y.; Lee, D.; Hachmann, J.; Finderle, S.; Park, D.; Christensen, J. Multiple receptor tyrosine kinases converge on microRNA-134 to control KRAS, STAT5B, and glioblastoma. Cell Death Differ. 2014, 21, 720–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.-L.; Han, L.; Chen, L.-Y.; Shi, Z.-D.; Yang, M.; Ren, Y.; Chen, L.-C.; Zhang, J.-X.; Pu, P.-Y.; Kang, C.-S. Blockage of a miR-21/EGFR regulatory feedback loop augments anti-EGFR therapy in glioblastomas. Cancer Lett. 2014, 342, 139–149. [Google Scholar] [CrossRef]
- Stommel, J.M.; Kimmelman, A.C.; Ying, H.; Nabioullin, R.; Ponugoti, A.H.; Wiedemeyer, R.; Stegh, A.H.; Bradner, J.E.; Ligon, K.L.; Brennan, C. Coactivation of receptor tyrosine kinases affects the response of tumor cells to targeted therapies. Science 2007, 318, 287–290. [Google Scholar] [CrossRef]
- Huang, P.H.; Cavenee, W.K.; Furnari, F.B.; White, F.M. Uncovering therapeutic targets for glioblastoma: A systems biology approach. Cell Cycle 2007, 6, 2750–2754. [Google Scholar] [CrossRef]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaoka, T.; Ohba, M.; Ohmori, T. Molecular-targeted therapies for epidermal growth factor receptor and its resistance mechanisms. Int. J. Mol. Sci. 2017, 18, 2420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Bang, Y.-J.; Camidge, D.R.; Shaw, A.T.; Solomon, B.; Maki, R.G.; Ou, S.-H.I.; Dezube, B.J.; Jänne, P.A.; Costa, D.B. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N. Engl. J. Med. 2010, 363, 1693–1703. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Li, D.; Ambrogio, L.; Shimamura, T.; Kubo, S.; Takahashi, M.; Chirieac, L.; Padera, R.; Shapiro, G.; Baum, A.; Himmelsbach, F. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene 2008, 27, 4702–4711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, E.L.; Sordella, R.; Bell, D.W.; Godin-Heymann, N.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Driscoll, D.R.; Fidias, P.; Lynch, T.J. Irreversible inhibitors of the EGF receptor may circumvent acquired resistance to gefitinib. Proc. Natl. Acad. Sci. USA 2005, 102, 7665–7670. [Google Scholar] [CrossRef] [PubMed]
- Finlay, M.R.V.; Anderton, M.; Ashton, S.; Ballard, P.; Bethel, P.A.; Box, M.R.; Bradbury, R.H.; Brown, S.J.; Butterworth, S.; Campbell, A. Discovery of a Potent and Selective EGFR Inhibitor (AZD9291) of Both Sensitizing and T790M Resistance Mutations That Spares the Wild Type Form of the Receptor; ACS Publications: Washington, DC, USA, 2014. [Google Scholar]
- Janjigian, Y.Y.; Smit, E.F.; Groen, H.J.; Horn, L.; Gettinger, S.; Camidge, D.R.; Riely, G.J.; Wang, B.; Fu, Y.; Chand, V.K. Dual Inhibition of EGFR with Afatinib and Cetuximab in Kinase Inhibitor–Resistant EGFR-Mutant Lung Cancer with and without T790M MutationsDual EGFR Inhibition in TKI-Resistant, EGFR-Mutant NSCLC. Cancer Discov. 2014, 4, 1036–1045. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Ercan, D.; Chen, L.; Yun, C.-H.; Li, D.; Capelletti, M.; Cortot, A.B.; Chirieac, L.; Iacob, R.E.; Padera, R. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.C.; Chooback, N.; Ho, C.; Melosky, B. Outcome differences between first-and second-generation EGFR inhibitors in advanced EGFR mutated NSCLC in a large population-based cohort. Clin. Lung Cancer 2019, 20, e576–e583. [Google Scholar] [CrossRef] [Green Version]
- Walter, A.O.; Sjin, R.T.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z. Discovery of a Mutant-Selective Covalent Inhibitor of EGFR that Overcomes T790M-Mediated Resistance in NSCLCDevelopment of Covalent EGFRT790M Inhibitor in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Song, Y.; Liu, D. EAI045: The fourth-generation EGFR inhibitor overcoming T790M and C797S resistance. Cancer Lett. 2017, 385, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Morelli, M.; Cascone, T.; Troiani, T.; De Vita, F.; Orditura, M.; Laus, G.; Eckhardt, S.; Pepe, S.; Tortora, G.; Ciardiello, F. Sequence-dependent antiproliferative effects of cytotoxic drugs and epidermal growth factor receptor inhibitors. Ann. Oncol. 2005, 16, iv61–iv68. [Google Scholar] [CrossRef] [PubMed]
- Jutten, B.; Rouschop, K. EGFR signaling and autophagy dependence for growth, survival, and therapy resistance. Cell Cycle 2014, 13, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Henson, E.; Chen, Y.; Gibson, S. EGFR family members’ regulation of autophagy is at a crossroads of cell survival and death in cancer. Cancers 2017, 9, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, J.; Bollu, L.R.; Su, F.; Gao, G.; Xu, L.; Huang, W.C.; Hung, M.C.; Weihua, Z. EGFR–SGLT1 interaction does not respond to EGFR modulators, but inhibition of SGLT1 sensitizes prostate cancer cells to EGFR tyrosine kinase inhibitors. Prostate 2013, 73, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Hegi, M.E.; Diserens, A.-C.; Bady, P.; Kamoshima, Y.; Kouwenhoven, M.C.; Delorenzi, M.; Lambiv, W.L.; Hamou, M.-F.; Matter, M.S.; Koch, A. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib—A phase II trial. Mol. Cancer Ther. 2011, 10, 1102–1112. [Google Scholar] [CrossRef] [Green Version]
- Uhm, J.H.; Ballman, K.V.; Wu, W.; Giannini, C.; Krauss, J.; Buckner, J.C.; James, C.; Scheithauer, B.W.; Behrens, R.J.; Flynn, P.J. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. Int. J. Radiat. Oncol. Biol. Phys. 2011, 80, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Raizer, J.J.; Abrey, L.E.; Lassman, A.B.; Chang, S.M.; Lamborn, K.R.; Kuhn, J.G.; Yung, W.A.; Gilbert, M.R.; Aldape, K.A.; Wen, P.Y. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-Oncology 2010, 12, 95–103. [Google Scholar] [CrossRef]
- Haas-Kogan, D.A.; Prados, M.D.; Tihan, T.; Eberhard, D.A.; Jelluma, N.; Arvold, N.D.; Baumber, R.; Lamborn, K.R.; Kapadia, A.; Malec, M. Epidermal growth factor receptor, protein kinase B/Akt, and glioma response to erlotinib. J. Natl. Cancer Inst. 2005, 97, 880–887. [Google Scholar] [CrossRef] [Green Version]
- Thiessen, B.; Stewart, C.; Tsao, M.; Kamel-Reid, S.; Schaiquevich, P.; Mason, W.; Easaw, J.; Belanger, K.; Forsyth, P.; McIntosh, L. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: Clinical outcomes, pharmacokinetics and molecular correlation. Cancer Chemother. Pharmacol. 2010, 65, 353–361. [Google Scholar] [CrossRef]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2015, 17, 430–439. [Google Scholar] [CrossRef] [Green Version]
- Kizilbash, S.H.; Gupta, S.K.; Parrish, K.E.; Laramy, J.K.; Kim, M.; Gampa, G.; Carlson, B.L.; Bakken, K.K.; Mladek, A.C.; Schroeder, M.A. In Vivo Efficacy of Tesevatinib in EGFR-Amplified Patient-Derived Xenograft Glioblastoma Models May Be Limited by Tissue Binding and Compensatory SignalingTesevatinib Brain PK and Efficacy in GBM PDX Models. Mol. Cancer Ther. 2021, 20, 1009–1018. [Google Scholar] [CrossRef]
- Sepúlveda, J.M.; Sánchez-Gómez, P.; Vaz Salgado, M.Á.; Gargini, R.; Balañá, C. Dacomitinib: An investigational drug for the treatment of glioblastoma. Expert Opin. Investig. Drugs 2018, 27, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Napier, T.S.; Udayakumar, N.; Jani, A.H.; Hartman, Y.E.; Houson, H.A.; Moore, L.; Amm, H.M.; van den Berg, N.S.; Sorace, A.G.; Warram, J.M. Comparison of Panitumumab-IRDye800CW and 5-Aminolevulinic Acid to Provide Optical Contrast in a Model of Glioblastoma MultiformePanitumumab-IRDye800CW versus 5-ALA for Optical Contrast. Mol. Cancer Ther. 2020, 19, 1922–1929. [Google Scholar] [CrossRef]
- Wang, Q.; Ni, J.; Jiang, T.; Choi, H.G.; Zhang, T.; Gray, N.; Zhao, J.J. CM93, a novel covalent small molecule inhibitor targeting lung cancer with mutant EGFR. BioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Yang, Y.; Wang, Q.; Bergholz, J.S.; Jiang, T.; Roberts, T.M.; Gray, N.S.; Zhao, J.J. Targeting EGFR in glioblastoma with a novel brain-penetrant small molecule EGFR-TKI. BioRxiv 2021. [Google Scholar] [CrossRef]
- Erasca. Erasca Granted FDA Orphan Drug Designation for CNS-Penetrant EGFR Inhibitor ERAS-801 for the Treatment of Malignant Glioma. Available online: https://investors.erasca.com/news-releases/news-release-details/erasca-granted-fda-orphan-drug-designation-cns-penetrant-egfr (accessed on 20 February 2023).
- Conage-Pough, J.E.; Stopka, S.A.; Oh, J.-H.; Mladek, A.C.; Burgenske, D.M.; Regan, M.S.; Baquer, G.; Decker, P.A.; Carlson, B.L.; Bakken, K.K. WSD-0922, a novel brain-penetrant inhibitor of epidermal growth factor receptor, promotes survival in glioblastoma mouse models. Neuro-Oncol. Adv. 2023, 5, vdad066. [Google Scholar] [CrossRef]
- Mateos, M.E.; López-Laso, E.; Izquierdo, L.; Pérez-Navero, J.L.; García, S.; Garzás, C. Response to nimotuzumab in a child with a progressive diffuse intrinsic pontine glioma. Pediatr. Int. 2011, 53, 261–263. [Google Scholar] [CrossRef]
- Lam, C.; Bouffet, E.; Bartels, U. Nimotuzumab in pediatric glioma. Future Oncol. 2009, 5, 1349–1361. [Google Scholar] [CrossRef] [PubMed]
- Belda-Iniesta, C.; de Castro Carpeño, J.; Saenz, E.C.; Gutiérrez, M.; Perona, R.; González-Barón, M. Long term responses with cetuximab therapy in glioblastoma multiforme. Cancer Biol. Ther. 2006, 5, 912–914. [Google Scholar] [CrossRef] [Green Version]
- Eller, J.L.; Longo, S.L.; Kyle, M.M.; Bassano, D.; Hicklin, D.J.; Canute, G.W. Anti-epidermal growth factor receptor monoclonal antibody cetuximab augments radiation effects in glioblastoma multiforme in vitro and in vivo. Neurosurgery 2005, 56, 155–162. [Google Scholar] [CrossRef]
- Neyns, B.; Sadones, J.; Joosens, E.a.; Bouttens, F.; Verbeke, L.; Baurain, J.-F.; D’Hondt, L.; Strauven, T.; Chaskis, C.; In’t Veld, P. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Ann. Oncol. 2009, 20, 1596–1603. [Google Scholar] [CrossRef]
- Choi, B.D.; Kuan, C.-T.; Cai, M.; Archer, G.E.; Mitchell, D.A.; Gedeon, P.C.; Sanchez-Perez, L.; Pastan, I.; Bigner, D.D.; Sampson, J.H. Systemic administration of a bispecific antibody targeting EGFRvIII successfully treats intracerebral glioma. Proc. Natl. Acad. Sci. USA 2013, 110, 270–275. [Google Scholar] [CrossRef]
- Orellana, L.; Thorne, A.H.; Lema, R.; Gustavsson, J.; Parisian, A.D.; Hospital, A.; Cordeiro, T.N.; Bernado, P.; Scott, A.M.; Brun-Heath, I. Oncogenic mutations at the EGFR ectodomain structurally converge to remove a steric hindrance on a kinase-coupled cryptic epitope. Proc. Natl. Acad. Sci. USA 2019, 116, 10009–10018. [Google Scholar] [CrossRef] [Green Version]
- Johns, T.G.; McKay, M.J.; Cvrljevic, A.N.; Gan, H.K.; Taylor, C.; Xu, H.; Smyth, F.E.; Scott, A.M. MAb 806 enhances the efficacy of ionizing radiation in glioma xenografts expressing the de2-7 epidermal growth factor receptor. Int. J. Radiat. Oncol. Biol. Phys. 2010, 78, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.A.; Scholler, J.; Ohkuri, T.; Kosaka, A.; Patel, P.R.; McGettigan, S.E.; Nace, A.K.; Dentchev, T.; Thekkat, P.; Loew, A. Rational development and characterization of humanized anti–EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 2015, 7, 275ra222. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Sun, R.; Shi, B.; Wang, Y.; Di, S.; Luo, H.; Sun, Y.; Li, Z.; Zhou, M.; Jiang, H. Antitumor efficacy of chimeric antigen receptor T cells against EGFRvIII-expressing glioblastoma in C57BL/6 mice. Biomed. Pharmacother. 2019, 113, 108734. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma. J. Immunother. 2019, 42, 126. [Google Scholar] [CrossRef] [PubMed]
- Binder, D.C.; Ladomersky, E.; Lenzen, A.; Zhai, L.; Lauing, K.L.; Otto-Meyer, S.D.; Lukas, R.V.; Wainwright, D.A. Lessons learned from rindopepimut treatment in patients with EGFRvIII-expressing glioblastoma. Transl. Cancer Res. 2018, 7, S510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, J.; Lai, R.K.; Recht, L.D.; Reardon, D.A.; Paleologos, N.A.; Groves, M.D.; Mrugala, M.M.; Jensen, R.; Baehring, J.M.; Sloan, A. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: The ACT III study. Neuro-Oncology 2015, 17, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Weller, M.; Butowski, N.; Tran, D.D.; Recht, L.D.; Lim, M.; Hirte, H.; Ashby, L.; Mechtler, L.; Goldlust, S.A.; Iwamoto, F. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): A randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017, 18, 1373–1385. [Google Scholar] [CrossRef] [Green Version]
- Iurlaro, R.; Waldhauer, I.; Planas-Rigol, E.; Bonfill-Teixidor, E.; Arias, A.; Nicolini, V.; Freimoser-Grundschober, A.; Cuartas, I.; Martínez-Moreno, A.; Martínez-Ricarte, F. A novel EGFRvIII T-cell bispecific antibody for the treatment of glioblastoma. Mol. Cancer Ther. 2022, 21, 1499–1509. [Google Scholar] [CrossRef]
- Li, L.; Quang, T.S.; Gracely, E.J.; Kim, J.H.; Emrich, J.G.; Yaeger, T.E.; Jenrette, J.M.; Cohen, S.C.; Black, P.; Brady, L.W. A phase II study of anti–epidermal growth factor receptor radioimmunotherapy in the treatment of glioblastoma multiforme. J. Neurosurg. 2010, 113, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, R.; Palazzo, C.; Evrard, B.; Piel, G. Nanocarriers for the treatment of glioblastoma multiforme: Current state-of-the-art. J. Control. Release 2016, 227, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, J.H.; Jeppesen, M.; Pilgaard, L.; Agger, R.; Duroux, M.; Zachar, V.; Moos, T. Targeted antiepidermal growth factor receptor (cetuximab) immunoliposomes enhance cellular uptake in vitro and exhibit increased accumulation in an intracranial model of glioblastoma multiforme. J. Drug Deliv. 2013, 2013, 209205. [Google Scholar] [CrossRef] [Green Version]
- Read, J.; Ingram, A.; Al Saleh, H.A.; Platko, K.; Gabriel, K.; Kapoor, A.; Pinthus, J.; Majeed, F.; Qureshi, T.; Al-Nedawi, K. Nuclear transportation of exogenous epidermal growth factor receptor and androgen receptor via extracellular vesicles. Eur. J. Cancer 2017, 70, 62–74. [Google Scholar] [CrossRef]
- Ricklefs, F.; Mineo, M.; Rooj, A.K.; Nakano, I.; Charest, A.; Weissleder, R.; Breakefield, X.O.; Chiocca, E.A.; Godlewski, J.; Bronisz, A. Extracellular Vesicles from High-Grade Glioma Exchange Diverse Pro-oncogenic Signals That Maintain Intratumoral HeterogeneityTumor Heterogeneity Is Enhanced by Extracellular Vesicles. Cancer Res. 2016, 76, 2876–2881. [Google Scholar] [CrossRef] [Green Version]
- Saleem, H.; Abdul, U.K.; Küçükosmanoglu, A.; Houweling, M.; Cornelissen, F.M.; Heiland, D.H.; Hegi, M.E.; Kouwenhoven, M.C.; Bailey, D.; Würdinger, T. The TICking clock of EGFR therapy resistance in glioblastoma: Target Independence or target Compensation. Drug Resist. Updat. 2019, 43, 29–37. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [Green Version]
- Husain, H.; Scur, M.; Murtuza, A.; Bui, N.; Woodward, B.; Kurzrock, R. Strategies to overcome bypass mechanisms mediating clinical resistance to EGFR tyrosine kinase inhibition in lung cancer. Mol. Cancer Ther. 2017, 16, 265–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Syn, N.L.; Cho, B.C.; Soo, R.A. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat. Rev. 2018, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Camidge, D.R.; Pao, W.; Sequist, L.V. Acquired resistance to TKIs in solid tumours: Learning from lung cancer. Nat. Rev. Clin. Oncol. 2014, 11, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Cortot, A.B.; Jänne, P.A. Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur. Respir. Rev. 2014, 23, 356–366. [Google Scholar] [CrossRef]
- Jones, S.; King, P.J.; Antonescu, C.N.; Sugiyama, M.G.; Bhamra, A.; Surinova, S.; Angelopoulos, N.; Kragh, M.; Pedersen, M.W.; Hartley, J.A. Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep. 2020, 10, 663. [Google Scholar] [CrossRef] [Green Version]
- Guo, G.; Narayan, R.N.; Horton, L.; Patel, T.R.; Habib, A.A. The role of EGFR-Met interactions in the pathogenesis of glioblastoma and resistance to treatment. Curr. Cancer Drug Targets 2017, 17, 297–302. [Google Scholar] [CrossRef]
- Wen, Y.; Grandis, J.R. Emerging drugs for head and neck cancer. Expert Opin. Emerg. Drugs 2015, 20, 313–329. [Google Scholar] [CrossRef]
- Del Vecchio, C.; Giacomini, C.; Vogel, H.; Jensen, K.; Florio, T.; Merlo, A.; Pollack, J.; Wong, A. EGFRvIII gene rearrangement is an early event in glioblastoma tumorigenesis and expression defines a hierarchy modulated by epigenetic mechanisms. Oncogene 2013, 32, 2670–2681. [Google Scholar] [CrossRef] [Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodriguez, S.M.B.; Kamel, A.; Ciubotaru, G.V.; Onose, G.; Sevastre, A.-S.; Sfredel, V.; Danoiu, S.; Dricu, A.; Tataranu, L.G. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. Int. J. Mol. Sci. 2023, 24, 11110. https://doi.org/10.3390/ijms241311110
Rodriguez SMB, Kamel A, Ciubotaru GV, Onose G, Sevastre A-S, Sfredel V, Danoiu S, Dricu A, Tataranu LG. An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma. International Journal of Molecular Sciences. 2023; 24(13):11110. https://doi.org/10.3390/ijms241311110
Chicago/Turabian StyleRodriguez, Silvia Mara Baez, Amira Kamel, Gheorghe Vasile Ciubotaru, Gelu Onose, Ani-Simona Sevastre, Veronica Sfredel, Suzana Danoiu, Anica Dricu, and Ligia Gabriela Tataranu. 2023. "An Overview of EGFR Mechanisms and Their Implications in Targeted Therapies for Glioblastoma" International Journal of Molecular Sciences 24, no. 13: 11110. https://doi.org/10.3390/ijms241311110