

Preparation of Xanthene-TEMPO Dyads: Synthesis and Study of the Radical Enhanced Intersystem Crossing

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
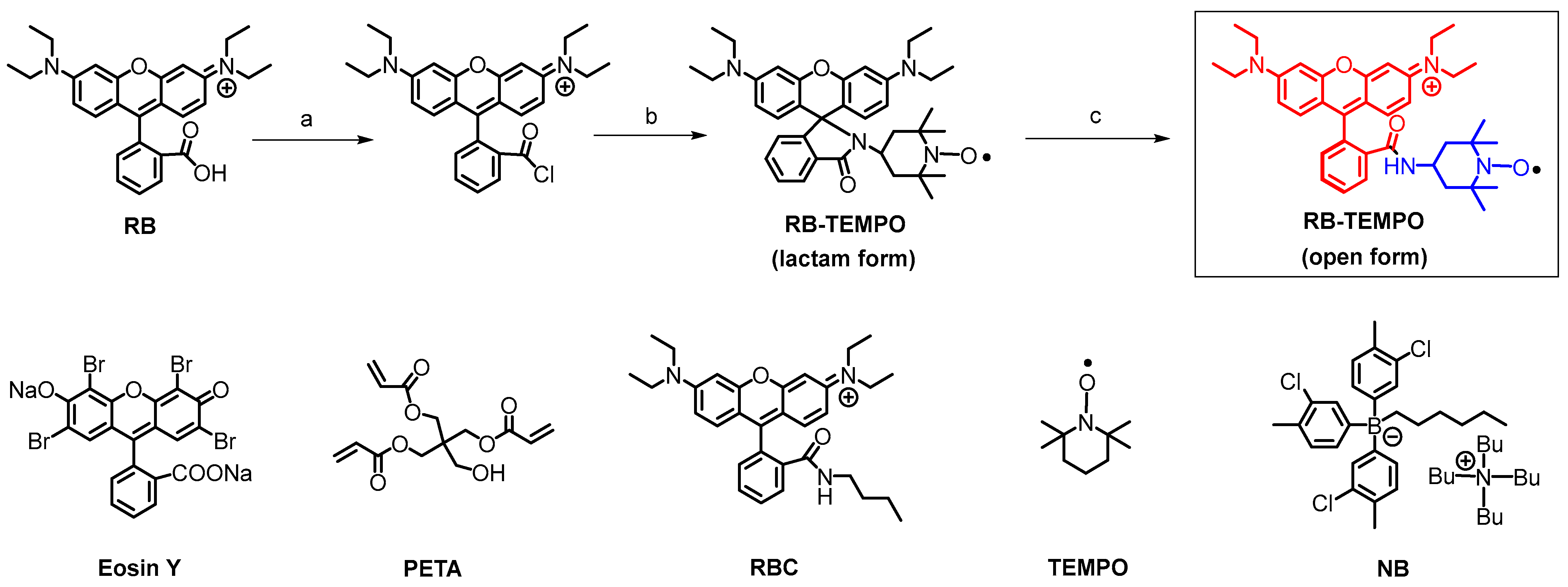
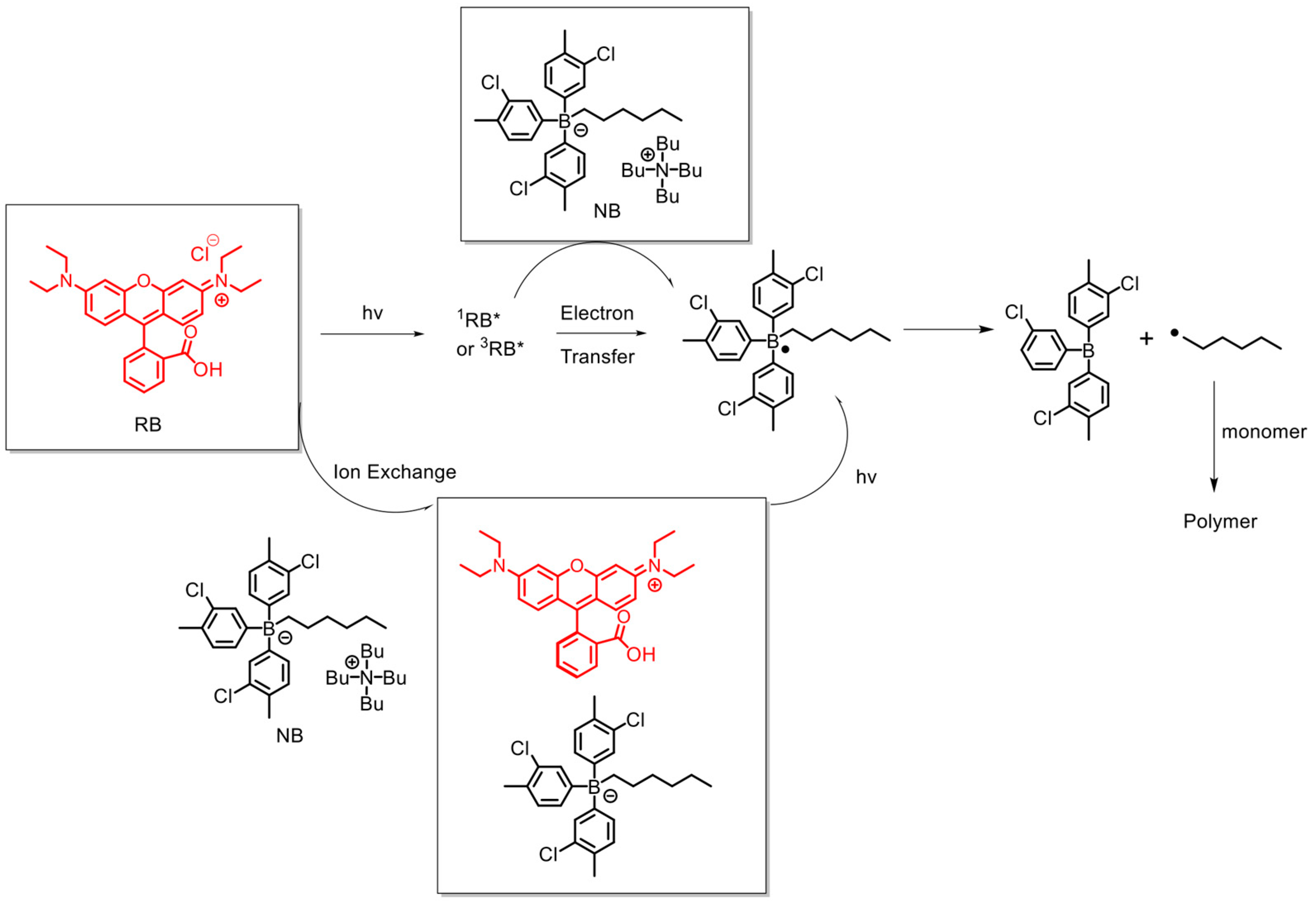
2.1. Molecular Structure Design Rationales
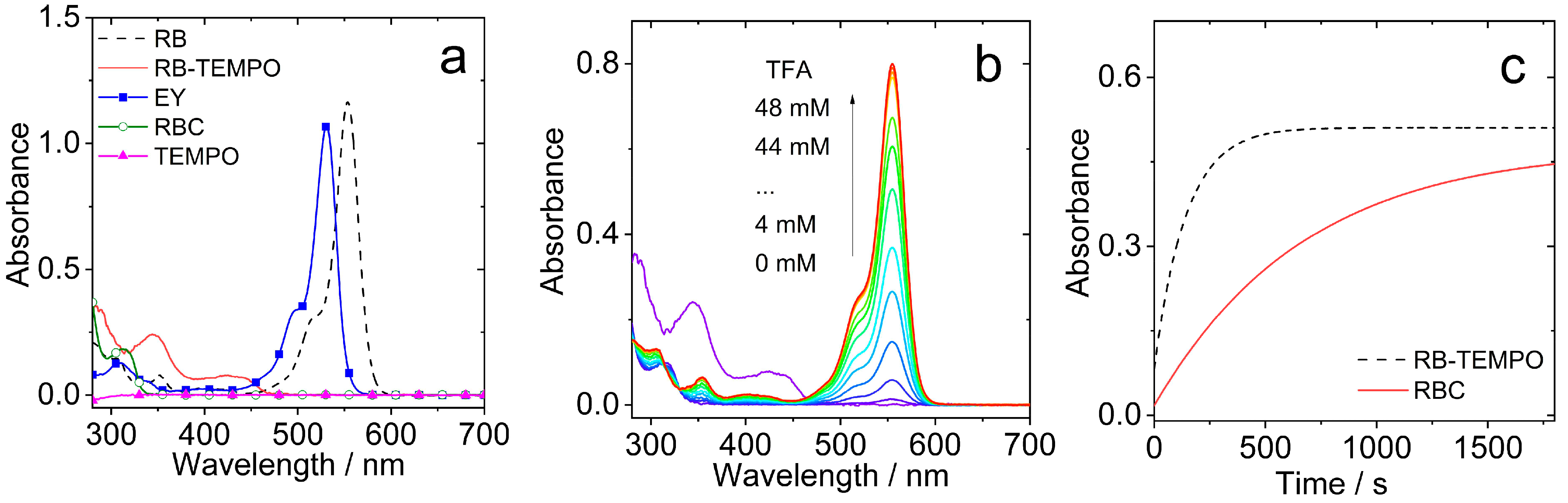
2.2. UV-Vis Absorption and Fluorescence Spectra
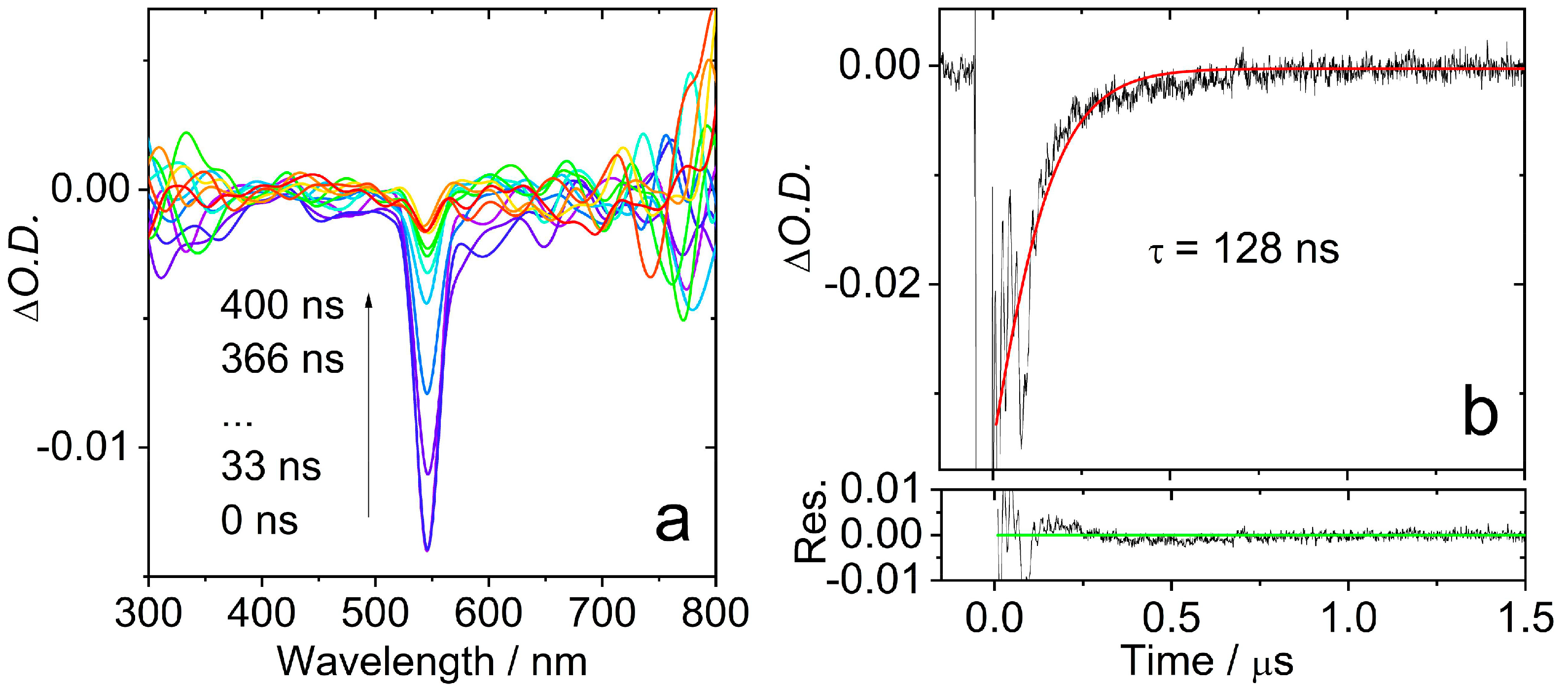
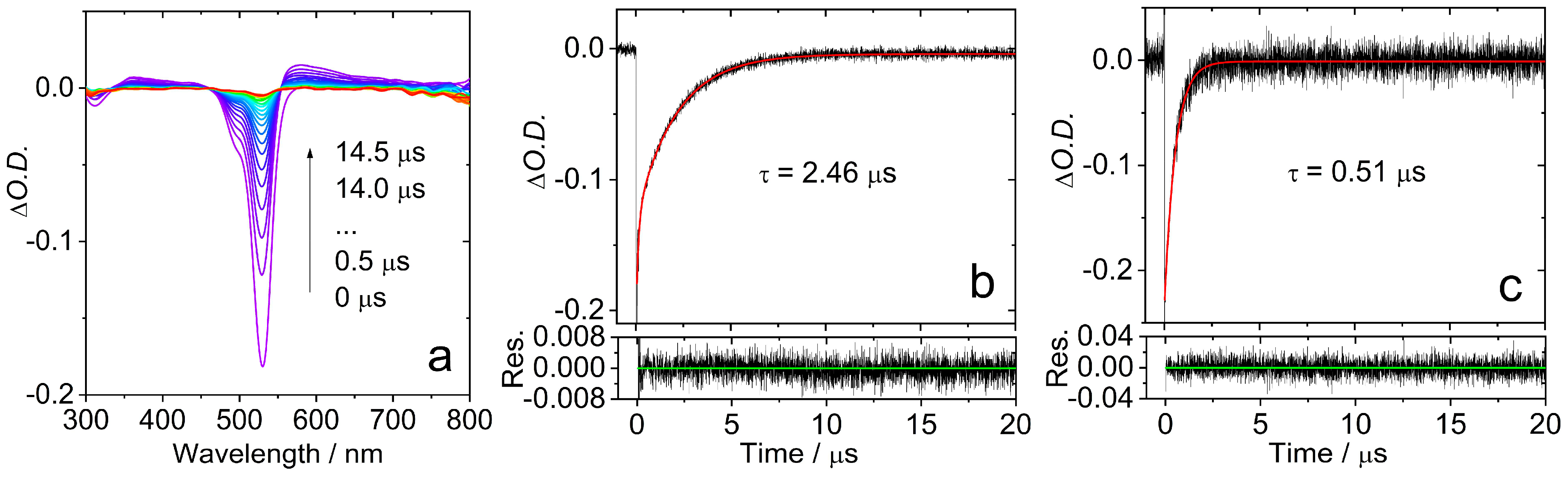
2.3. Nanosecond Transient Absorption (ns-TA) Spectroscopy: Properties of the Triplet States
2.4. Pulsed Laser Excited Time-Resolved Electron Paramagnetic Resonance (TREPR) Studies
2.5. Photopolymerization and Photobleaching
3. Materials and Methods
3.1. General Methods and Material
3.2. Compound RB-TEMPO
3.3. Nanosecond Transient Absorption (ns TA) Spectroscopy
3.4. Time-Resolved Electron Paramagnetic Resonance (TREPR) Spectra
3.5. Photopolymerization and Photobleaching Experiments
3.5.1. Photopolymerization
3.5.2. Photobleaching Experiments
3.6. Density Functional Theory (DFT) Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Awuah, S.G.; You, Y. Boron Dipyrromethene (Bodipy)-Based Photosensitizers for Photodynamic Therapy. RSC Adv. 2012, 2, 11169–11183. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, W.; Sun, J.; Guo, S. Triplet Photosensitizers: From Molecular Design to Applications. Chem. Soc. Rev. 2013, 42, 5323–5351. [Google Scholar] [CrossRef]
- Pham, T.C.; Nguyen, V.-N.; Choi, Y.; Lee, S.; Yoon, J. Recent Strategies to Develop Innovative Photosensitizers for Enhanced Photodynamic Therapy. Chem. Rev. 2021, 121, 13454–13619. [Google Scholar] [CrossRef] [PubMed]
- Bassan, E.; Gualandi, A.; Cozzi, P.G.; Ceroni, P. Design of Bodipy Dyes as Triplet Photosensitizers: Electronic Properties Tailored for Solar Energy Conversion, Photoredox Catalysis and Photodynamic Therapy. Chem. Sci. 2021, 12, 6607–6628. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, K.; Yang, W.; Wang, Z.; Zhong, F. The Triplet Excited State of Bodipy: Formation, Modulation and Application. Chem. Soc. Rev. 2015, 44, 8904–8939. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Tian, W.; Imran, M.; Cao, H.; Zhao, J. Controlling the Triplet States and Their Application in External Stimuli-Responsive Triplet–Triplet-Annihilation Photon Upconversion: From the Perspective of Excited State Photochemistry. Chem. Soc. Rev. 2021, 50, 9686–9714. [Google Scholar] [CrossRef]
- Kamkaew, A.; Lim, S.H.; Lee, H.B.; Kiew, L.V.; Chung, L.Y.; Burgess, K. Bodipy Dyes in Photodynamic Therapy. Chem. Soc. Rev. 2013, 42, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, V.-N.; Yan, Y.; Zhao, J.; Yoon, J. Heavy-Atom-Free Photosensitizers: From Molecular Design to Applications in the Photodynamic Therapy of Cancer. Acc. Chem. Res. 2021, 54, 207–220. [Google Scholar] [CrossRef]
- Shi, L.; Xia, W. Photoredox Functionalization of C–H Bonds Adjacent to a Nitrogen Atom. Chem. Soc. Rev. 2012, 41, 7687–7697. [Google Scholar] [CrossRef]
- Sideri, I.K.; Voutyritsa, E.; Kokotos, C.G. Photoorganocatalysis, Small Organic Molecules and Light in the Service of Organic Synthesis: The Awakening of a Sleeping Giant. Org. Biomol. Chem. 2018, 16, 4596–4614. [Google Scholar] [CrossRef] [PubMed]
- De Bonfils, P.; Peault, L.; Nun, P.; Coeffard, V. State of the Art of Bodipy-Based Photocatalysts in Organic Synthesis. Eur. J. Org. Chem. 2021, 2021, 1809–1824. [Google Scholar] [CrossRef]
- Dai, F.R.; Zhan, H.M.; Liu, Q.; Fu, Y.Y.; Li, J.H.; Wang, Q.W.; Xie, Z.Y.; Wang, L.X.; Yan, F.; Wong, W.Y. Platinum(II)-Bis(Aryleneethynylene) Complexes for Solution-Processible Molecular Bulk Heterojunction Solar Cells. Chem. Eur. J. 2012, 18, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Tehfe, M.-A.; Lalevée, J.; Telitel, S.; Sun, J.; Zhao, J.; Graff, B.; Morlet-Savary, F.; Fouassier, J.-P. Iridium Complexes Incorporating Coumarin Moiety as Catalyst Photoinitiators: Towards Household Green Led Bulb and Halogen Lamp Irradiation. Polymer 2012, 53, 2803–2808. [Google Scholar] [CrossRef]
- Zhao, J.Z.; Ji, S.M.; Guo, H.M. Triplet-Triplet Annihilation Based Upconversion: From Triplet Sensitizers and Triplet Acceptors to Upconversion Quantum Yields. RSC. Adv. 2011, 1, 937–950. [Google Scholar] [CrossRef]
- Simon, Y.C.; Weder, C. Low-Power Photon Upconversion through Triplet–Triplet Annihilation in Polymers. J. Mater. Chem. 2012, 22, 20817–20830. [Google Scholar] [CrossRef]
- Monguzzi, A.; Tubino, R.; Hoseinkhani, S.; Campione, M.; Meinardi, F. Low Power, Non-Coherent Sensitized Photon up-Conversion: Modelling and Perspectives. Phys. Chem. Chem. Phys. 2012, 14, 4322–4332. [Google Scholar] [CrossRef]
- Ye, C.; Zhou, L.; Wang, X.; Liang, Z. Photon Upconversion: From Two-Photon Absorption (TPA) to Triplet–Triplet Annihilation (TTA). Phys. Chem. Chem. Phys. 2016, 18, 10818–10835. [Google Scholar] [CrossRef]
- Yanai, N.; Kimizuka, N. New Triplet Sensitization Routes for Photon Upconversion: Thermally Activated Delayed Fluorescence Molecules, Inorganic Nanocrystals, and Singlet-to-Triplet Absorption. Acc. Chem. Res. 2017, 50, 2487–2495. [Google Scholar] [CrossRef]
- Seo, S.E.; Choe, H.-S.; Cho, H.; Kim, H.-I.; Kim, J.-H.; Kwon, O.S. Recent Advances in Materials for and Applications of Triplet–Triplet Annihilation-Based Upconversion. J. Mater. Chem. C. 2022, 10, 4483–4496. [Google Scholar] [CrossRef]
- Turro, N.J.; Ramamurthy, V.; Scaiano, J.C. Principles of Molecular Photochemistry: An Introduction; University Science Books: Herndon, VA, USA, 2009. [Google Scholar]
- Chi, Y.; Chou, P.-T. Contemporary Progresses on Neutral, Highly Emissive Os(II) and Ru(II) Complexes. Chem. Soc. Rev. 2007, 36, 1421–1431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campagna, S.; Puntoriero, F.; Nastasi, F.; Bergamini, G.; Balzani, V. Photochemistry and Photophysics of Coordination Compounds: Ruthenium. Top. Curr. Chem. 2007, 280, 117–214. [Google Scholar]
- Williams, J.A.G. Photochemistry and Photophysics of Coordination Compounds: Platinum. Top. Curr. Chem. 2007, 281, 205–268. [Google Scholar]
- Fernández-Moreira, V.; Thorp-Greenwood, F.L.; Coogan, M.P. Application of d6 Transition Metal Complexes in Fluorescence Cell Imaging. Chem. Commun. 2010, 46, 186–202. [Google Scholar] [CrossRef] [PubMed]
- Herberger, J.; Winter, R.F. Platinum Emitters with Dye-Based σ-Aryl Ligands. Coord. Chem. Rev. 2019, 400, 213048. [Google Scholar] [CrossRef]
- Tilley, A.J.; Pensack, R.D.; Lee, T.S.; Djukic, B.; Scholes, G.D.; Seferos, D.S. Ultrafast Triplet Formation in Thionated Perylene Diimides. J. Phys. Chem. C 2014, 118, 9996–10004. [Google Scholar] [CrossRef]
- Hussain, M.; Zhao, J.; Yang, W.; Zhong, F.; Karatay, A.; Yaglioglu, H.G.; Yildiz, E.A.; Hayvali, M. Intersystem Crossing and Triplet Excited State Properties of Thionated Naphthalenediimide Derivatives. J. Lumin. 2017, 192, 211–217. [Google Scholar] [CrossRef]
- Yang, W.; Zhao, J.; Sonn, C.; Escudero, D.; Karatay, A.; Yaglioglu, H.G.; Küçüköz, B.; Hayvali, M.; Li, C.; Jacquemin, D. Efficient Intersystem Crossing in Heavy-Atom-Free Perylenebisimide Derivatives. J. Phys. Chem. C 2016, 120, 10162–10175. [Google Scholar] [CrossRef]
- Yu, Z.Y.; Wu, Y.S.; Peng, Q.; Sun, C.L.; Chen, J.W.; Yao, J.N.; Fu, H.B. Accessing the Triplet State in Heavy-Atom-Free Perylene Diimides. Chem. –A Eur. J. 2016, 22, 4717–4722. [Google Scholar] [CrossRef]
- Yan, Y.X.; Sukhanov, A.A.; Bousquet, M.H.E.; Guan, Q.L.; Zhao, J.Z.; Voronkova, V.K.; Escudero, D.; Barbon, A.; Xing, Y.H.; Gurzadyan, G.G.; et al. Does Twisted π-Conjugation Framework Always Induce Efficient Intersystem Crossing? A Case Study with Benzo[b]- and [a]Phenanthrene-Fused BODIPY Derivatives and Identification of a Dark State. J. Phys. Chem. B 2021, 125, 6280–6295. [Google Scholar] [CrossRef]
- Zhang, X.; Sukhanov, A.A.; Liu, X.; Taddei, M.; Zhao, J.; Harriman, A.; Voronkova, V.K.; Wan, Y.; Dick, B.; Di Donato, M. Origin of Intersystem Crossing in Highly Distorted Organic Molecules: A Case Study with Red Light-Absorbing N,N,O,O-Boron-Chelated Bodipys. Chem. Sci. 2023, 14, 5014–5027. [Google Scholar] [CrossRef]
- Hou, Y.; Zhang, X.; Chen, K.; Liu, D.; Wang, Z.; Liu, Q.; Zhao, J.; Barbon, A. Charge Separation, Charge Recombination, Long-Lived Charge Transfer State Formation and Intersystem Crossing in Organic Electron Donor/Acceptor Dyads. J. Mater. Chem. C 2019, 7, 12048–12074. [Google Scholar] [CrossRef]
- Filatov, M.A. Heavy-Atom-Free Bodipy Photosensitizers with Intersystem Crossing Mediated by Intramolecular Photoinduced Electron Transfer. Org. Biomol. Chem. 2020, 18, 10–27. [Google Scholar] [CrossRef] [Green Version]
- Gibbons, D.J.; Farawar, A.; Mazzella, P.; Leroy-Lhez, S.; Williams, R.M. Making Triplets from Photo-Generated Charges: Observations, Mechanisms and Theory. Photochem. Photobiol. Sci. 2020, 19, 136–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Yu, X.; Wu, W.; Zhao, J. Styryl Bodipy-C60 Dyads as Efficient Heavy-Atom-Free Organic Triplet Photosensitizers. Org. Lett. 2012, 14, 2594–2597. [Google Scholar] [CrossRef]
- Wu, W.; Zhao, J.; Sun, J.; Guo, S. Light-Harvesting Fullerene Dyads as Organic Triplet Photosensitizers for Triplet–Triplet Annihilation Upconversions. J. Org. Chem. 2012, 77, 5305–5312. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhou, M.; Zhou, Q.; Zhou, X.; Liu, S.; Zhang, S.; Zhang, B. Triplet–Triplet Annihilation Upconversion Kinetics of C60–Bodipy Dyads as Organic Triplet Photosensitizers. Phys. Chem. Chem. Phys. 2017, 19, 22049–22060. [Google Scholar] [CrossRef] [PubMed]
- Swedin, R.K.; Healy, A.T.; Schaffner, J.W.; Kuzmin, I.A.; Zatsikha, Y.V.; Nemykin, V.N.; Blank, D.A. Outsourcing Intersystem Crossing without Heavy Atoms: Energy Transfer Dynamics in Pyridonebodipy–C60 Complexes. J. Phys. Chem. Lett. 2022, 13, 8845–8850. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, J.; Barbon, A.; Toffoletti, A.; Liu, Y.; An, Y.; Xu, L.; Karatay, A.; Yaglioglu, H.G.; Yildiz, E.A.; et al. Radical-Enhanced Intersystem Crossing in New Bodipy Derivatives and Application for Efficient Triplet–Triplet Annihilation Upconversion. J. Am. Chem. Soc. 2017, 139, 7831–7842. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, Y.; Hussain, M.; Kundu, S.; Rane, V.; Hayvali, M.; Yildiz, E.A.; Zhao, J.; Yaglioglu, H.G.; Das, R.; et al. Efficient Radical-Enhanced Intersystem Crossing in an Ndi-Tempo Dyad: Photophysics, Electron Spin Polarization, and Application in Photodynamic Therapy. Chem. Eur. J. 2018, 24, 18663–18675. [Google Scholar] [CrossRef]
- Kawai, A.; Shibuya, K. Electron Spin Dynamics in a Pair Interaction between Radical and Electronically-Excited Molecule as Studied by a Time-Resolved Esr Method. J. Photochem. Photobiol. C 2006, 7, 89–103. [Google Scholar] [CrossRef]
- Colvin, M.T.; Giacobbe, E.M.; Cohen, B.; Miura, T.; Scott, A.M.; Wasielewski, M.R. Competitive Electron Transfer and Enhanced Intersystem Crossing in Photoexcited Covalent Tempo−Perylene-3,4:9,10-Bis(Dicarboximide) Dyads: Unusual Spin Polarization Resulting from the Radical−Triplet Interaction. J. Phys. Chem. A 2010, 114, 1741–1748. [Google Scholar] [CrossRef] [PubMed]
- Jiao, L.; Song, F.; Cui, J.; Peng, X. A near-Infrared Heptamethine Aminocyanine Dye with a Long-Lived Excited Triplet State for Photodynamic Therapy. Chem. Commun. 2018, 54, 9198–9201. [Google Scholar] [CrossRef]
- Xu, Z.; Huang, Y.; Cao, Y.; Jin, T.; Miller, K.A.; Kaledin, A.L.; Musaev, D.G.; Lian, T.; Egap, E. Enhanced Intersystem Crossing of Boron Dipyrromethene by Tempo Radical. J. Chem. Phys. 2020, 153, 154201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sukhanov, A.A.; Yildiz, E.A.; Kandrashkin, Y.E.; Zhao, J.; Yaglioglu, H.G.; Voronkova, V.K. Radical-Enhanced Intersystem Crossing in a Bay-Substituted Perylene Bisimide−Tempo Dyad and the Electron Spin Polarization Dynamics upon Photoexcitation. ChemPhysChem 2021, 22, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Dyar, S.M.; Margulies, E.A.; Horwitz, N.E.; Brown, K.E.; Krzyaniak, M.D.; Wasielewski, M.R. Photogenerated Quartet State Formation in a Compact Ring-Fused Perylene-Nitroxide. J. Phys. Chem. B 2015, 119, 13560–13569. [Google Scholar] [CrossRef]
- Constantinescu, T.; Ionita, P.; Chiorescu, I.; Ionita, G. Hydrazyl, Nitronyl-, and Imino-Nitroxides: Synthesis, Properties and Reaction with Nitric Oxide and Nitrogen Dioxide. Cent. Eur. J. Chem. 2003, 1, 465–476. [Google Scholar] [CrossRef]
- Latha, A.V.; Ayyappan, M.; Kallar, A.R.; Kakkadavath, R.V.; Victor, S.P.; Selvam, S. Fluorescence Imaging of Nitric Oxide in Living Cells Using O-Phenylenediamine-Rhodamine Based Polymeric Nanosensors. Mater. Sci. Eng. C 2020, 108, 110463. [Google Scholar] [CrossRef]
- Cao, L.Y.; Wu, Q.F.; Li, Q.; Shao, S.J.; Guo, Y. Fluorescence and HPLC Detection of Hydroxyl Radical by a Rhodamine-Nitroxide Probe and its Application in Cell Imaging. J. Fluoresc. 2014, 24, 313–318. [Google Scholar] [CrossRef]
- Yu, H.; Cao, L.; Li, F.; Wu, Q.; Li, Q.; Wang, S.; Guo, Y. The Antioxidant Mechanism of Nitroxide Tempo: Scavenging with Glutathionyl Radicals. RSC Adv. 2015, 5, 63655–63661. [Google Scholar] [CrossRef]
- Cui, X.; Zhao, J.; Lou, Z.; Li, S.; Wu, H.; Han, K.-l. Switching of the Triplet Excited State of Rhodamine/Naphthaleneimide Dyads: An Experimental and Theoretical Study. J. Org. Chem. 2015, 80, 568–581. [Google Scholar] [CrossRef]
- Giacobbe, E.M.; Mi, Q.; Colvin, M.T.; Cohen, B.; Ramanan, C.; Scott, A.M.; Yeganeh, S.; Marks, T.J.; Ratner, M.A.; Wasielewski, M.R. Ultrafast Intersystem Crossing and Spin Dynamics of Photoexcited Perylene-3,4:9,10-Bis(Dicarboximide) Covalently Linked to a Nitroxide Radical at Fixed Distances. J. Am. Chem. Soc. 2009, 131, 3700–3712. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yuan, X.; Kucukoz, B.; Li, S.; Zhang, C.; Majumdar, P.; Karatay, A.; Li, X.; Yaglioglu, H.G.; Elmali, A.; et al. Resonance Energy Transfer-Enhanced Rhodamine–Styryl Bodipy Dyad Triplet Photosensitizers. J. Mater. Chem. C 2014, 2, 3900–3913. [Google Scholar] [CrossRef]
- Kung, J.C.K.; Forman, A.; Jockusch, R.A. The Effect of Methylation on the Intrinsic Photophysical Properties of Simple Rhodamines. Phys. Chem. Chem. Phys. 2019, 21, 10261–10271. [Google Scholar] [CrossRef]
- Beaumont, P.C.; Johnson, D.G.; Parsons, B.J. Excited State and Free Radical Properties of Rhodamine Dyes in Aqueous Solution: A Laser Flash Photolysis and Pulse Radiolysis Study. J. Photochem. Photobiol. A Chem. 1997, 107, 175–183. [Google Scholar] [CrossRef]
- Penzkofer, A.; Beidoun, A. Triplet-Triplet Absorption of Eosin Y in Methanol Determined by Nanosecond Excimer Laser Excitation and Picosecond Light Continuum Probing. Chem. Phys. 1993, 177, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, H.B.; Román, E.S.; Duarte, P.; Machado, I.F.; Vieira Ferreira, L.F. Eosin Y Triplet State as a Probe of Spatial Heterogeneity in Microcrystalline Cellulose. Photochem. Photobiol. 2012, 88, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Richert, S.; Tait, C.E.; Timmel, C.R. Delocalisation of Photoexcited Triplet States Probed by Transient Epr and Hyperfine Spectroscopy. J. Magn. Reson. 2017, 280, 103–116. [Google Scholar] [CrossRef]
- Teki, Y.; Miyamoto, S.; Nakatsuji, M.; Miura, Y. π-Topology and Spin Alignment Utilizing the Excited Molecular Field: Observation of the Excited High-Spin Quartet (S = 3/2) and Quintet (S = 2) States on Purely Organic Π-Conjugated Spin Systems. J. Am. Chem. Soc. 2001, 123, 294–305. [Google Scholar] [CrossRef]
- Teki, Y.; Kimura, M.; Narimatsu, S.; Ohara, K.; Mukai, K. Excited High-Spin Quartet (S = 3/2) State of a Novel Π-Conjugated Organic Spin System, Pyrene–Verdazyl Radical. Bull. Chem. Soc. Jpn. 2004, 77, 95–99. [Google Scholar] [CrossRef]
- Nolden, O.; Fleck, N.; Lorenzo, E.R.; Wasielewski, M.R.; Schiemann, O.; Gilch, P.; Richert, S. Excitation Energy Transfer and Exchange-Mediated Quartet State Formation in Porphyrin-Trityl Systems. Chem. Eur. J. 2021, 27, 2683–2691. [Google Scholar] [CrossRef]
- Montalti, M.; Credi, A.; Prodi, L.; Gandolfi, M.T. Handbook of Photochemistry; CRC Press: Boca Raton, FL, USA, 2006. [Google Scholar]
- Yamashita, M.; Ikeda, H.; Kashiwagi, H. Laser Pumped Magnetophotoselection in Triplet Dyes. J. Chem. Phys. 1975, 63, 1127–1132. [Google Scholar] [CrossRef]
- Takahashi, H.; Iwama, M.; Akai, N.; Shibuya, K.; Kawai, A. Pulsed Epr Study on Large Dynamic Electron Polarisation Created in the Quenching of Photo-Excited Xanthene Dyes by Nitroxide Radicals in Aqueous Solutions. Mol. Phys. 2014, 112, 1012–1020. [Google Scholar] [CrossRef]
- Ghosh, P.; Biswas, S.; Niyogi, U. Photopolymerization of Methyl Methacrylate Using a Combination of Rhodamine 6g and Benzoyl Peroxide as Photoinitiator in Nonaqueous Media. J. Polym. Res. 1986, 24, 1053–1063. [Google Scholar] [CrossRef]
- Kobayashi, K.; Sakai, N.; Matsui, S.; Nakagawa, M. Fluorescent Uv-Curable Resists for Uv Nanoimprint Lithography. Jpn. J. Appl. Phys. 2010, 49, 06GL07. [Google Scholar] [CrossRef]
- Su, H.-L.; Yang, M.-M.; Zhao, L.-M.; Yao, S.-J.; Geng, Q.-N.; Wang, L.-P.; Li, G. Recyclable Magnetic Fe3o4 Supported Photocatalyst for the Metal-Free Atrp. Eur. Polym. J. 2022, 177, 111476. [Google Scholar] [CrossRef]
- Kara, M.; Dadashi-Silab, S.; Yagci, Y. Phenacyl Ethyl Carbazolium as a Long Wavelength Photoinitiator for Free Radical Polymerization. Macromol. Rapid Commun. 2015, 36, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Stoll, S.; Schweiger, A. Easyspin, a Comprehensive Software Package for Spectral Simulation and Analysis in Epr. J. Magn. Reson. 2006, 178, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09W, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Entradas, T.; Waldron, S.; Volk, M. The detection sensitivity of commonly used singlet oxygen probes in aqueous environments. J. Photochem. Photobiol. B Biol. 2020, 204, 111787. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | λabs a/nm b | ε c | λem/nm d | ΦF/% e | τF/ns f | τT/ns g | ΦΔ/% h |
|---|---|---|---|---|---|---|---|
| RB | 554 531 m | 11.55 | 570 560 m | 62.7 | 3.17 i 5.97 m | 94 | 0 k |
| EY | 531 | 10.70 | 548 | 55.3 | 0.95 j (6.9%)/ 3.13 j (93.1%) | 2460 | 23.0 l |
| RB-TEMPO (Open Form) | 555 | 7.99 | 570 | 26.9 | 0.29 i (95.9%)/ 2.37 i (4.1%) | 128 | 40.1 l |
| Sample | D/MHz | E/MHz | Tx | Ty | Tz | g-Factor |
|---|---|---|---|---|---|---|
| RB | −1600 | 530 | 0 | 0 | 1 | 2.005 |
| Eosin Y | −2100 | 618 | 0 | 0 | 1 | 2.005 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, W.; Wu, Y.; Zhang, Y.; Sukhanov, A.A.; Chu, Y.; Zhang, X.; Zhao, J.; Voronkova, V.K. Preparation of Xanthene-TEMPO Dyads: Synthesis and Study of the Radical Enhanced Intersystem Crossing. Int. J. Mol. Sci. 2023, 24, 11220. https://doi.org/10.3390/ijms241311220
Zhu W, Wu Y, Zhang Y, Sukhanov AA, Chu Y, Zhang X, Zhao J, Voronkova VK. Preparation of Xanthene-TEMPO Dyads: Synthesis and Study of the Radical Enhanced Intersystem Crossing. International Journal of Molecular Sciences. 2023; 24(13):11220. https://doi.org/10.3390/ijms241311220
Chicago/Turabian StyleZhu, Wenhui, Yanran Wu, Yiyan Zhang, Andrey A. Sukhanov, Yuqi Chu, Xue Zhang, Jianzhang Zhao, and Violeta K. Voronkova. 2023. "Preparation of Xanthene-TEMPO Dyads: Synthesis and Study of the Radical Enhanced Intersystem Crossing" International Journal of Molecular Sciences 24, no. 13: 11220. https://doi.org/10.3390/ijms241311220
APA StyleZhu, W., Wu, Y., Zhang, Y., Sukhanov, A. A., Chu, Y., Zhang, X., Zhao, J., & Voronkova, V. K. (2023). Preparation of Xanthene-TEMPO Dyads: Synthesis and Study of the Radical Enhanced Intersystem Crossing. International Journal of Molecular Sciences, 24(13), 11220. https://doi.org/10.3390/ijms241311220