
Modulation of Mitochondrial Metabolic Parameters and Antioxidant Enzymes in Healthy and Glaucomatous Trabecular Meshwork Cells with Hybrid Small Molecule SA-2

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. SA-2 Treatment Protects Normal and Glaucomatous TM Cells from TBHP-Induced Cell Death
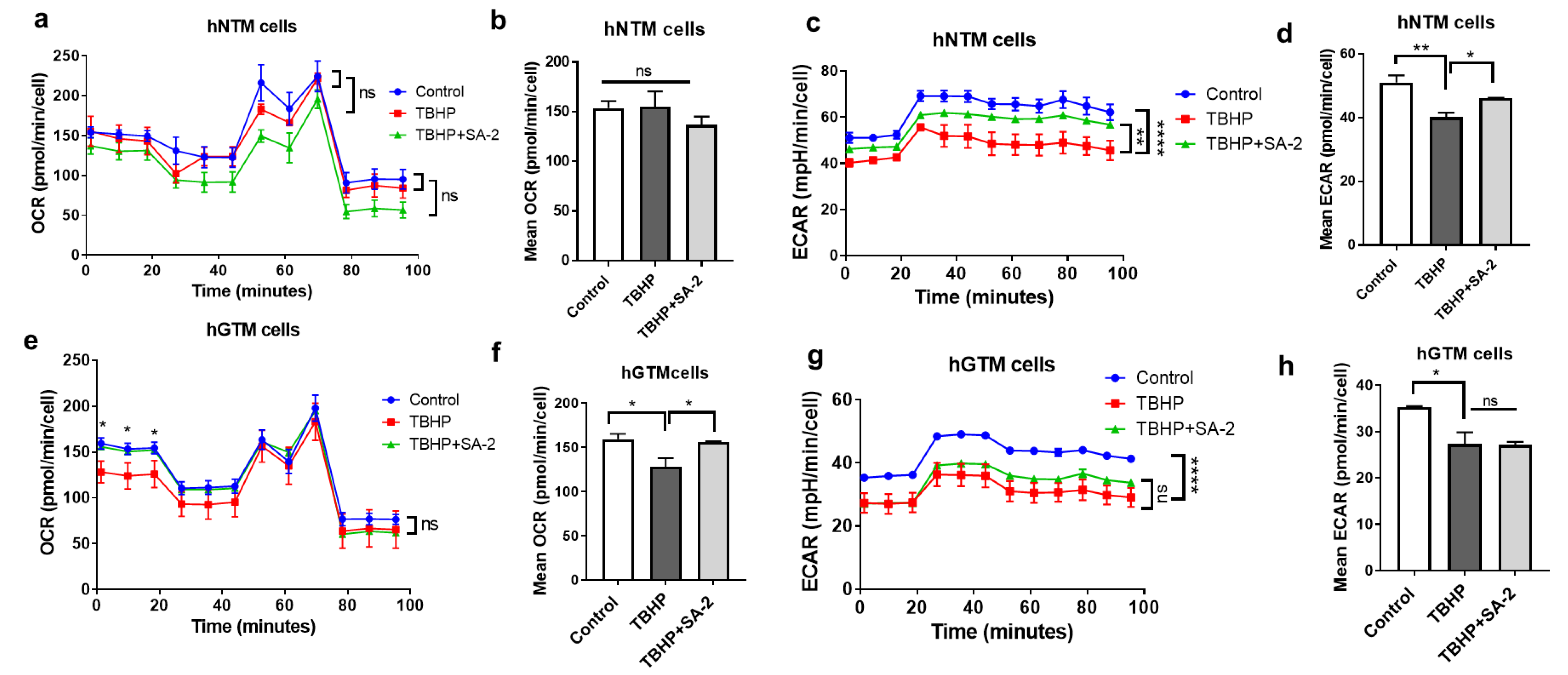
2.2. Effect of SA-2 Treatment on the OCR in Normal and Glaucomatous Human TM Cells
2.2.1. Effect of SA-2 Treatment on ECAR in Normal and Glaucomatous Human TM Cells
2.2.2. Effect of SA-2 Treatment on the Mean OCR and ECAR after TBHP Treatment
2.3. SA-2 Is More Potent Than TEMPOL in Increasing the Maximal Respiration and Spare Capacity in GTM-3 Cells
2.4. SA-2 Treatment Improves Antioxidant Status in the hNTM and hGTM Cells
2.5. SA-2 Treatment Decreases Lipid Peroxidation in hNTM Cells
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Synthesis of SA-2
4.2.1. N-(Cyanomethyl)-N-(1-Hydroxy-2,2,6,6 Tetramethylpiperidin-4-yl) Free Radical 2
4.2.2. N-(Cyanomethyl)-N-(1-Hydroxy-2,2,6,6-Tetramethylpiperidin-4-yl) Nitrous Amide 3
4.2.3. 5-Amino-3-(1-Hydroxy-2,2,6,6-Tetramethylpiperidin-4-yl)-1,2,3-Oxadiazol-3-Ium Chloride (SA-2)
4.3. Culture of Primary hNTM and hGTM Cells
4.4. In Vitro Cell Viability Assay
4.5. In Vitro Cytoprotection Assay
4.6. Measurement of Oxygen Consumption Rate and Extracellular Acidification Rate
4.7. Antioxidant Enzyme Assay
4.7.1. Total Antioxidant Assay
4.7.2. Catalase Assay
4.7.3. Glutathione Peroxidase Assay
4.7.4. Lipid Peroxidation Assay
4.7.5. Immunocytochemistry-Staining for Lipid Peroxidation
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- He, S.; Stankowska, D.L.; Ellis, D.Z.; Krishnamoorthy, R.R.; Yorio, T.J. Targets of Neuroprotection in Glaucoma. J. Ocul. Pharmacol. Ther. 2018, 34, 85–106. [Google Scholar] [PubMed]
- Furlanetto, R.L.; Teixeira, S.H.; Gracitelli, C.P.B.; Lottenberg, C.L.; Emori, F.; Michelan, M.; Amaro, E., Jr.; Paranhos, A., Jr. Structural and functional analyses of the optic nerve and lateral geniculate nucleus in glaucoma. PLoS ONE 2018, 13, e0194038. [Google Scholar]
- Schmidt, M.A.; Knott, M.; Heidemann, R.; Michelson, G.; Kober, T.; Dorfler, A.; Engelhorn, T. Investigation of lateral geniculate nucleus volume and diffusion tensor imaging in patients with normal tension glaucoma using 7 tesla magnetic resonance imaging. PLoS ONE 2018, 13, e0198830. [Google Scholar] [CrossRef] [PubMed]
- Duarte, J.N. Neuroinflammatory Mechanisms of Mitochondrial Dysfunction and Neurodegeneration in Glaucoma. J. Ophthalmol. 2021, 2021, 4581909. [Google Scholar] [CrossRef]
- Garcia-Medina, J.J.; Rubio-Velazquez, E.; Lopez-Bernal, M.D.; Cobo-Martinez, A.; Zanon-Moreno, V.; Pinazo-Duran, M.D.; Del-Rio-Vellosillo, M. Glaucoma and antioxidants: Review and update. Antioxidants 2020, 9, 1031. [Google Scholar] [CrossRef]
- Chrysostomou, V.; Rezania, F.; Trounce, I.A.; Crowston, J.G. Oxidative stress and mitochondrial dysfunction in glaucoma. Curr. Opin. Pharmacol. 2013, 13, 12–15. [Google Scholar] [CrossRef]
- McElnea, E.M.; Quill, B.; Docherty, N.G.; Irnaten, M.; Siah, W.F.; Clark, A.F.; O’Brien, C.J.; Wallace, D.M. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182. [Google Scholar]
- Aydin Yaz, Y.; Yildirim, N.; Yaz, Y.; Tekin, N.; Inal, M.; Sahin, F.M. Role of oxidative stress in pseudoexfoliation syndrome and pseudoexfoliation glaucoma. Turk. J. Ophthalmol. 2019, 49, 61. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Kondkar, A.A.; Mousa, A.; Osman, E.A.; Al-Obeidan, S.A. Decreased total antioxidants status in the plasma of patients with pseudoexfoliation glaucoma. Mol. Vis. 2011, 17, 2769. [Google Scholar]
- Siegfried, C.J.; Shui, Y.B.; Holekamp, N.M.; Bai, F.; Beebe, D.C. Oxygen distribution in the human eye: Relevance to the etiology of open-angle glaucoma after vitrectomy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5731–5738. [Google Scholar] [CrossRef] [Green Version]
- Izzotti, A.; Sacca, S.C.; Longobardi, M.; Cartiglia, C. Sensitivity of ocular anterior chamber tissues to oxidative damage and its relevance to the pathogenesis of glaucoma. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5251–5258. [Google Scholar] [CrossRef] [Green Version]
- Lutjen-Drecoll, E. Morphological changes in glaucomatous eyes and the role of TGFβ2 for the pathogenesis of the disease. Exp. Eye Res. 2005, 81, 1–4. [Google Scholar] [CrossRef]
- Ren, R.; Humphrey, A.A.; Swain, D.L.; Gong, H. Relationships between intraocular pressure, effective filtration area, and morphological changes in the trabecular meshwork of steroid-induced ocular hypertensive mouse eyes. Int. J. Mol. Sci. 2022, 23, 854. [Google Scholar] [CrossRef]
- Abu-Amero, K.K.; Morales, J.; Bosley, T.M. Mitochondrial abnormalities in patients with primary open-angle glaucoma. Investig. Ophthalmol. Vis. Sci. 2006, 47, 2533–2541. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Fan, W.; Procaccio, V. Mitochondrial energetics and therapeutics. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 297–348. [Google Scholar] [CrossRef] [Green Version]
- Stankowska, D.L.; Dibas, A.; Li, L.; Zhang, W.; Krishnamoorthy, V.R.; Chavala, S.H.; Nguyen, T.P.; Yorio, T.; Ellis, D.Z.; Acharya, S. Hybrid compound SA-2 is neuroprotective in animal models of retinal ganglion cell death. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3064–3073. [Google Scholar] [CrossRef] [Green Version]
- Stankowska, D.L.; Millar, J.C.; Kodati, B.; Behera, S.; Chaphalkar, R.M.; Nguyen, T.; Nguyen, K.T.; Krishnamoorthy, R.R.; Ellis, D.Z.; Acharya, S. Nanoencapsulated hybrid compound SA-2 with long-lasting intraocular pressure–lowering activity in rodent eyes. Mol. Vis. 2021, 27, 37. [Google Scholar]
- Amankwa, C.E.; Gondi, S.R.; Dibas, A.; Weston, C.; Funk, A.; Nguyen, T.; Nguyen, K.T.; Ellis, D.Z.; Acharya, S. Novel Thiol Containing Hybrid Antioxidant-Nitric Oxide Donor Small Molecules for Treatment of Glaucoma. Antioxidants 2021, 10, 575. [Google Scholar] [CrossRef]
- Pham, J.H.; Johnson, G.A.; Rangan, R.S.; Amankwa, C.E.; Acharya, S.; Stankowska, D.L. Neuroprotection of rodent and human retinal ganglion cells in vitro/ex vivo by the hybrid small molecule SA-2. Cells 2022, 11, 3741. [Google Scholar] [CrossRef]
- Dogan, S.; Unal, M.; Ozturk, N.; Yargicoglu, P.; Cort, A.; Spasojevic, I.; Aslan, M. Manganese porphyrin reduces retinal injury induced by ocular hypertension in rats. Exp. Eye Res. 2011, 93, 387–396. [Google Scholar] [CrossRef] [Green Version]
- Cort, A.; Ozturk, N.; Akpinar, D.; Unal, M.; Yucel, G.; Ciftcioglu, A.; Aslan, M. Suppressive effect of astaxanthin on retinal injury induced by elevated intraocular pressure. Regul. Toxicol. Pharmacol. 2010, 58, 121–130. [Google Scholar]
- Zhao, J.; Wang, S.; Zhong, W.; Yang, B.; Sun, L.; Zheng, Y. Oxidative stress in the trabecular meshwork. Int. J. Mol. Med. 2016, 38, 995–1002. [Google Scholar]
- Acharya, S.; Rogers, P.; Krishnamoorthy, R.R.; Stankowska, D.L.; Dias, H.V.; Yorio, T. Design and synthesis of novel hybrid sydnonimine and prodrug useful for glaucomatous optic neuropathy. Bioorg. Med. Chem. Lett. 2016, 26, 1490–1494. [Google Scholar] [CrossRef]
- Lindsey, J.D.; Duong-Polk, K.X.; Dai, Y.; Nguyen, D.H.; Leung, C.K.; Weinreb, R.N. Protection by an oral disubstituted hydroxylamine derivative against loss of retinal ganglion cell differentiation following optic nerve crush. PLoS ONE 2013, 8, e65966. [Google Scholar]
- Fan Gaskin, J.C.; Shah, M.H.; Chan, E.C. Oxidative stress and the role of NADPH oxidase in glaucoma. Antioxidants 2021, 10, 238. [Google Scholar] [CrossRef]
- Jassim, A.H.; Fan, Y.; Pappenhagen, N.; Nsiah, N.Y.; Inman, D.M. Oxidative stress and hypoxia modify mitochondrial homeostasis during glaucoma. Antioxid. Redox Signal. 2021, 35, 1341–1357. [Google Scholar] [CrossRef]
- Kamel, K.; Farrell, M.; O’Brien, C. Mitochondrial dysfunction in ocular disease: Focus on glaucoma. Mitochondrion 2017, 35, 44–53. [Google Scholar]
- Lee, S.; Van Bergen, N.J.; Kong, G.Y.; Chrysostomou, V.; Waugh, H.S.; O’Neill, E.C.; Crowston, J.G.; Trounce, I.A. Mitochondrial dysfunction in glaucoma and emerging bioenergetic therapies. Exp. Eye Res. 2011, 93, 204–212. [Google Scholar]
- Kubota, M.; Shui, Y.B.; Liu, M.; Bai, F.; Huang, A.J.; Ma, N.; Beebe, D.C.; Siegfried, C.J. Mitochondrial oxygen metabolism in primary human lens epithelial cells: Association with age, diabetes and glaucoma. Free Radic. Biol. Med. 2016, 97, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Epstein, D.L.; Hashimoto, J.M.; Anderson, P.J.; Grant, W.M. Effect of iodoacetamide perfusion on outflow facility and metabolism of the trabecular meshwork. Investig. Ophthalmol. Vis. Sci. 1981, 20, 625–631. [Google Scholar]
- Reina-Torres, E.; Boussommier-Calleja, A.; Sherwood, J.M.; Overby, D.R. Aqueous humor outflow requires active cellular metabolism in mice. Investig. Ophthalmol. Vis. Sci. 2020, 61, 45. [Google Scholar]
- Hurley, J.B.; Lindsay, K.J.; Du, J.J. Glucose, lactate, and shuttling of metabolites in vertebrate retinas. J. Neurosci. Res. 2015, 93, 1079–1092. [Google Scholar]
- Gil, A.; Martín-Montañez, E.; Valverde, N.; Lara, E.; Boraldi, F.; Claros, S.; Romero-Zerbo, S.-Y.; Fernández, O.; Pavia, J.; Garcia-Fernandez, M. Neuronal metabolism and neuroprotection: Neuroprotective effect of fingolimod on menadione-induced mitochondrial damage. Cells 2020, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Gohil, V.M.; Sheth, S.A.; Nilsson, R.; Wojtovich, A.P.; Lee, J.H.; Perocchi, F.; Chen, W.; Clish, C.B.; Ayata, C.; Brookes, P.S.; et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat. Biotechnol. 2010, 28, 249. [Google Scholar] [CrossRef] [Green Version]
- Jassim, A.H.; Coughlin, L.; Harun-Or-Rashid, M.; Kang, P.T.; Chen, Y.R.; Inman, D.M. Higher reliance on glycolysis limits glycolytic responsiveness in degenerating glaucomatous optic nerve. Mol. Neurobiol. 2019, 56, 7097–7112. [Google Scholar] [CrossRef] [Green Version]
- Brand, M.D.; Nicholls, D.G. Assessing mitochondrial dysfunction in cells. Biochem. J. 2011, 435, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Mariappan, N.; Elks, C.M.; Fink, B.; Francis, J. TNF-induced mitochondrial damage: A link between mitochondrial complex I activity and left ventricular dysfunction. Free Radic. Biol. Med. 2009, 46, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Silva, H.N.M.; Covatti, C.; da Rocha, G.L.; Mizobuti, D.S.; Mancio, R.D.; Hermes, T.A.; Kido, L.A.; Cagnon, V.H.A.; Pereira, E.C.L.; Minatel, E. Oxidative stress, inflammation, and activators of mitochondrial biogenesis: Tempol targets in the diaphragm muscle of exercise trained-mdx mice. Front. Physiol. 2021, 12, 649793. [Google Scholar] [CrossRef]
- Wilcox, C.S. Effects of tempol and redox-cycling nitroxides in models of oxidative stress. Pharmacol. Ther. 2010, 126, 119–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mousa, A.; Kondkar, A.A.; Al-Obeidan, S.A.; Azad, T.A.; Sultan, T.; Osman, E.; Abu-Amero, K.K. Association of total antioxidants level with glaucoma type and severity. Saudi Med. J. 2015, 36, 671. [Google Scholar] [CrossRef]
- Wang, M.; Zheng, Y. Oxidative stress and antioxidants in the trabecular meshwork. Peer J. 2019, 7, e8121. [Google Scholar] [CrossRef] [PubMed]
- Njie-Mbye, Y.F.; Kulkarni-Chitnis, M.; Opere, C.A.; Barrett, A.; Ohia, S.E. Lipid peroxidation: Pathophysiological and pharmacological implications in the eye. Front. Physiol. 2013, 4, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalá, A.; Díaz, M. Impact of lipid peroxidation on the physiology and pathophysiology of cell membranes. Front. Physiol. 2016, 7, 423. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Xu, L.; Porter, N.A. Free radical lipid peroxidation: Mechanisms and analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Leung, K.W.; Zhang, Y.H.; Duan, S.; Zhong, X.F.; Jiang, R.Z.; Peng, Z.; Tombran-Tink, J.; Ge, J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: Protection by antioxidants. Investig. Ophthalmol. Vis. Sci. 2008, 49, 1447–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, K.E.; Bhattacharya, S.K.; Borras, T.; Brunner, T.M.; Chansangpetch, S.; Clark, A.F.; Dismuke, W.M.; Du, Y.; Elliott, M.H.; Ethier, C.R.; et al. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp. Eye Res. 2018, 171, 164–173. [Google Scholar] [CrossRef]
- Clark, A.F.; Steely, H.T.; Dickerson, J.E., Jr.; English-Wright, S.; Stropki, K.; McCartney, M.D.; Jacobson, N.; Shepard, A.R.; Clark, J.I.; Matsushima, H.; et al. Glucocorticoid induction of the glaucoma gene MYOC in human and monkey trabecular meshwork cells and tissues. Investig. Ophthalmol. Vis. Sci. 2001, 42, 1769–1780. [Google Scholar]
- Pang, I.H.; Shade, D.L.; Clark, A.F.; Steely, H.T.; DeSantis, L. Preliminary characterization of a transformed cell strain derived from human trabecular meshwork. Curr. Eye Res. 1994, 13, 51–63. [Google Scholar] [CrossRef]
- Nicholas, D.; Proctor, E.A.; Raval, F.M.; Ip, B.C.; Habib, C.; Ritou, E.; Grammatopoulos, T.N.; Steenkamp, D.; Dooms, H.; Apovian, C.M.; et al. Advances in the quantification of mitochondrial function in primary human immune cells through extracellular flux analysis. PLoS ONE 2017, 12, e0170975. [Google Scholar] [CrossRef] [Green Version]






 Activation,
Activation,  Insult.
Activation, Insult.
Insult.
Activation, Insult.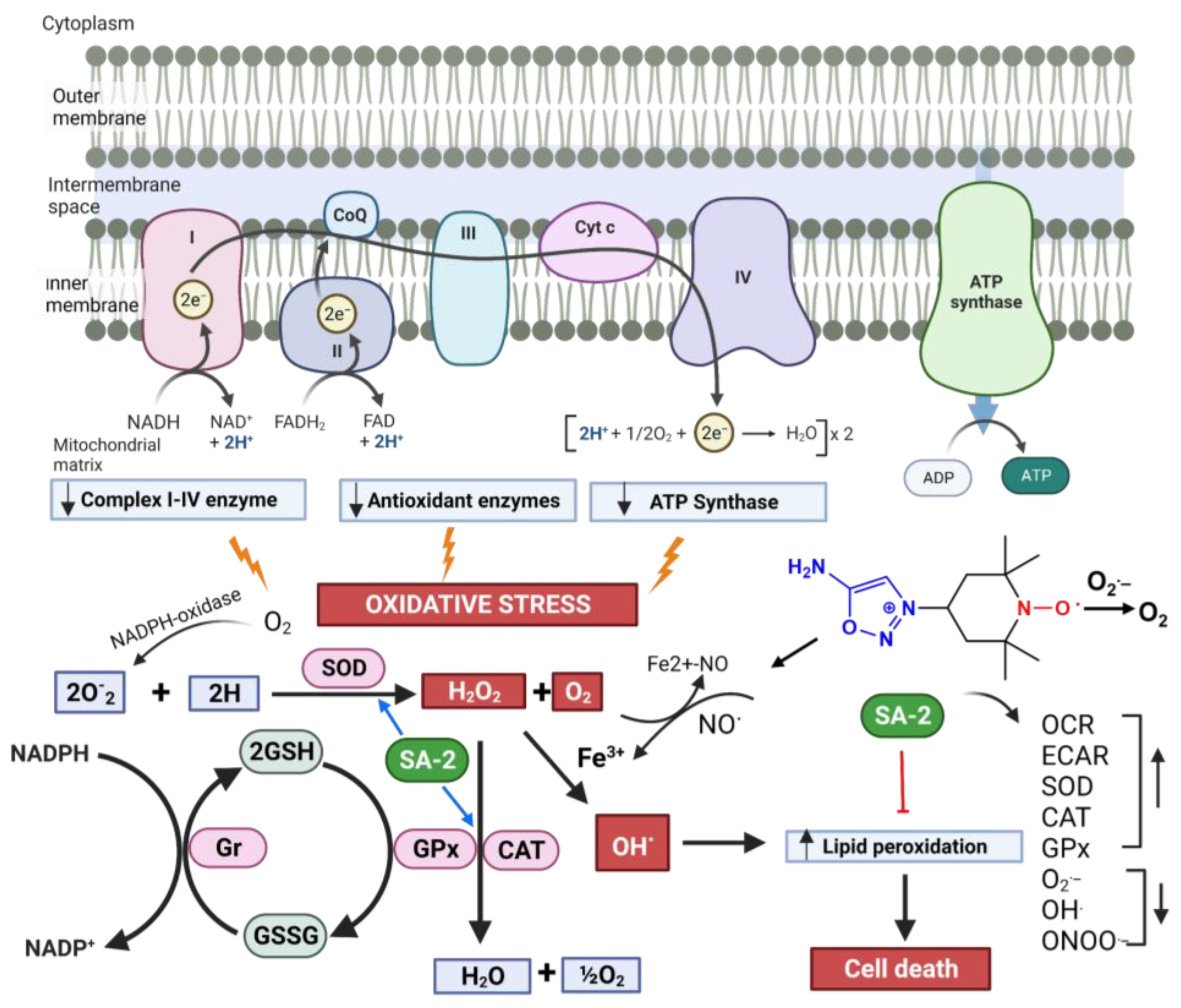

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amankwa, C.E.; Young, O.; DebNath, B.; Gondi, S.R.; Rangan, R.; Ellis, D.Z.; Zode, G.; Stankowska, D.L.; Acharya, S. Modulation of Mitochondrial Metabolic Parameters and Antioxidant Enzymes in Healthy and Glaucomatous Trabecular Meshwork Cells with Hybrid Small Molecule SA-2. Int. J. Mol. Sci. 2023, 24, 11557. https://doi.org/10.3390/ijms241411557
Amankwa CE, Young O, DebNath B, Gondi SR, Rangan R, Ellis DZ, Zode G, Stankowska DL, Acharya S. Modulation of Mitochondrial Metabolic Parameters and Antioxidant Enzymes in Healthy and Glaucomatous Trabecular Meshwork Cells with Hybrid Small Molecule SA-2. International Journal of Molecular Sciences. 2023; 24(14):11557. https://doi.org/10.3390/ijms241411557
Chicago/Turabian StyleAmankwa, Charles E., Olivia Young, Biddut DebNath, Sudershan R. Gondi, Rajiv Rangan, Dorette Z. Ellis, Gulab Zode, Dorota L. Stankowska, and Suchismita Acharya. 2023. "Modulation of Mitochondrial Metabolic Parameters and Antioxidant Enzymes in Healthy and Glaucomatous Trabecular Meshwork Cells with Hybrid Small Molecule SA-2" International Journal of Molecular Sciences 24, no. 14: 11557. https://doi.org/10.3390/ijms241411557