

Distinct Molecular Signatures of Amyloid-Beta and Tau in Alzheimer’s Disease Associated with Down Syndrome
 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Clinical Profiles and Demographics
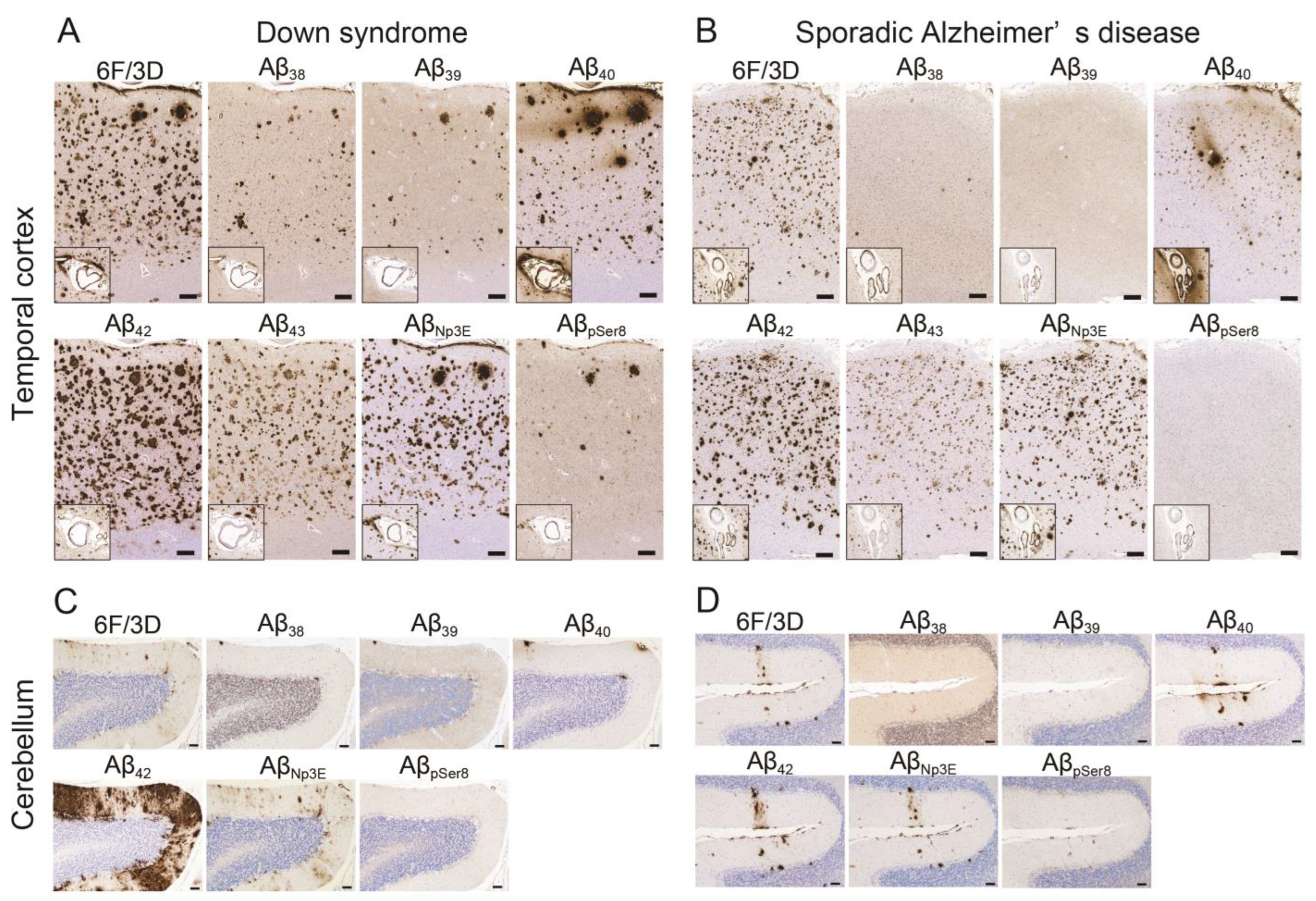
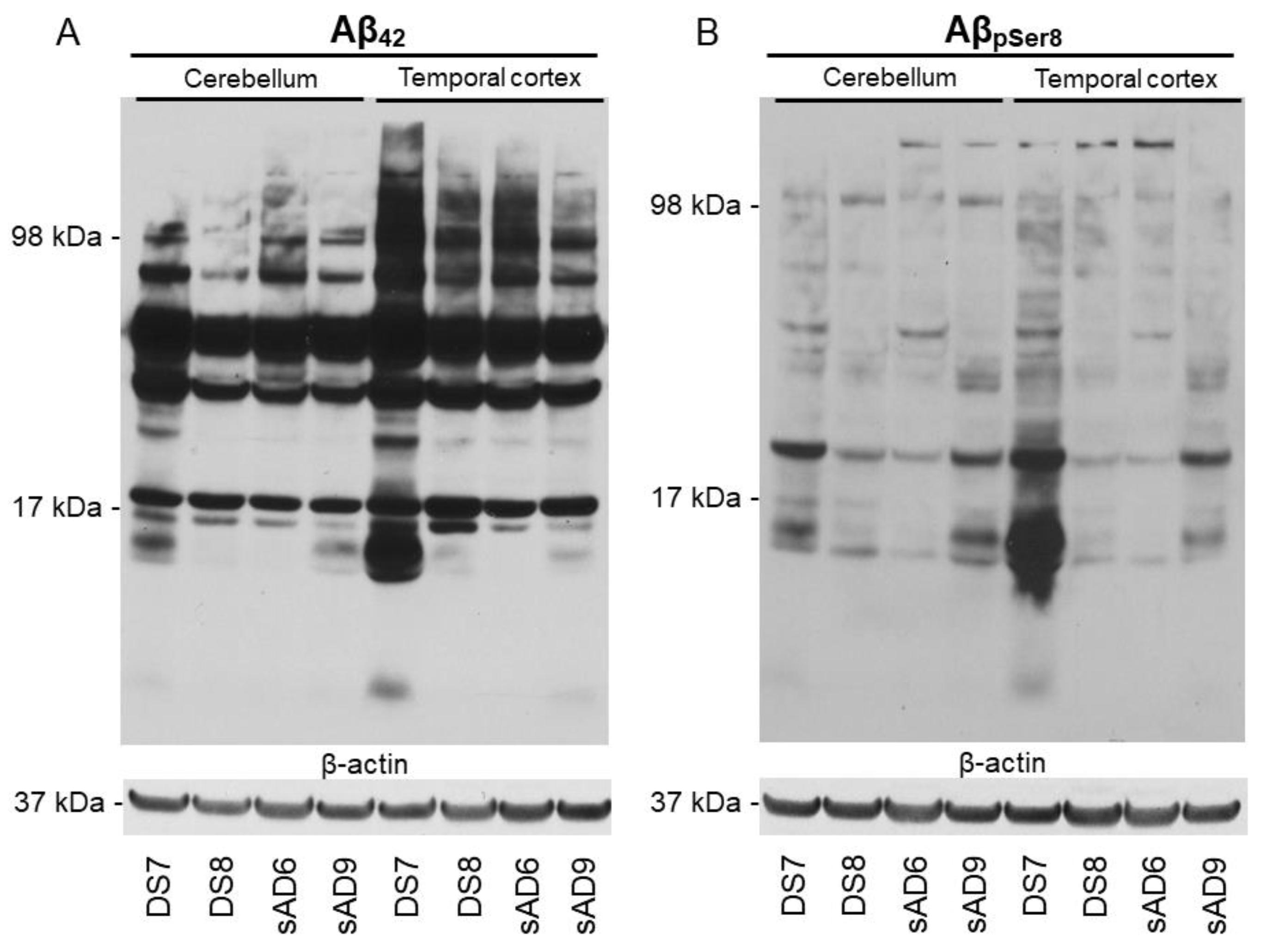
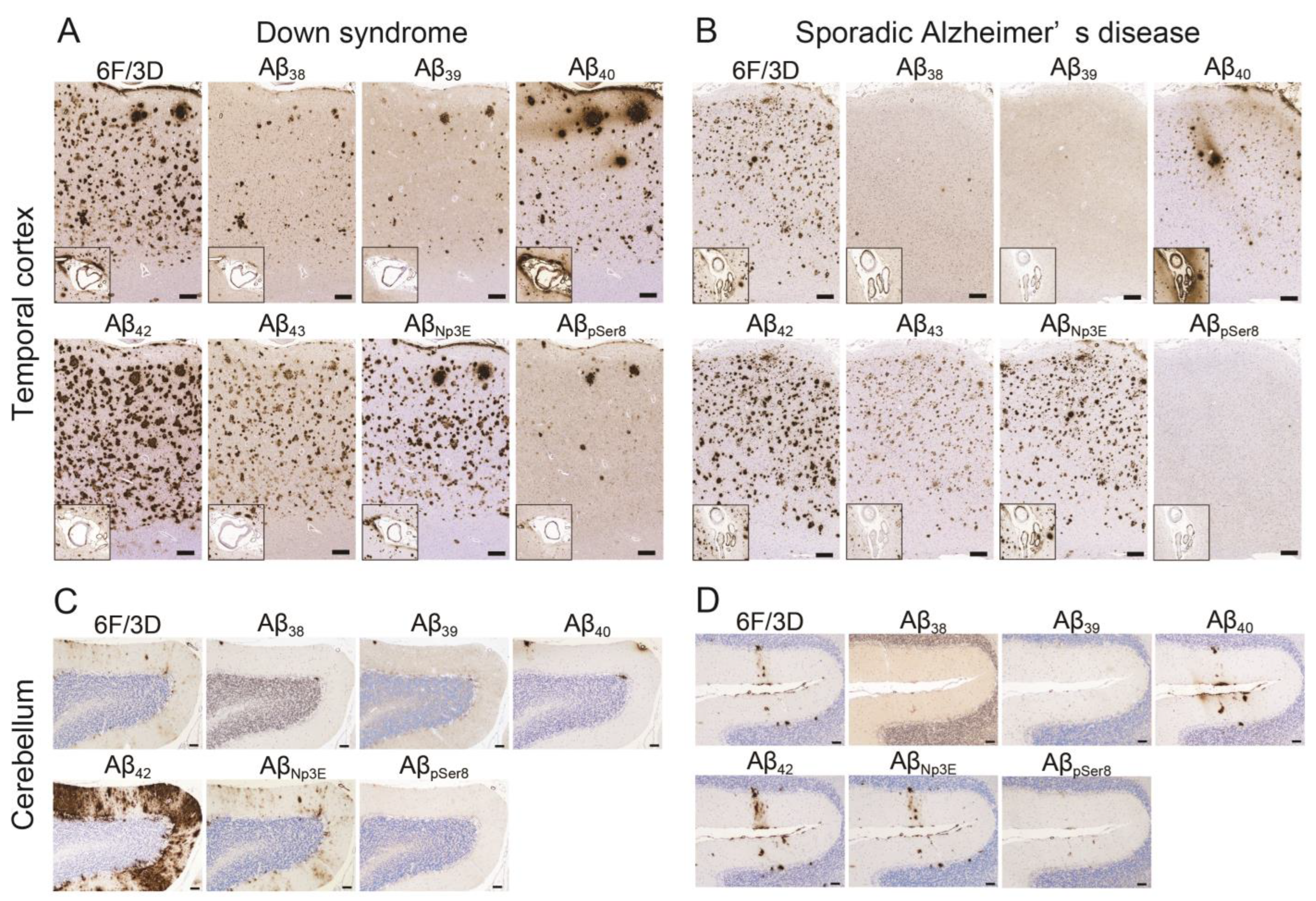
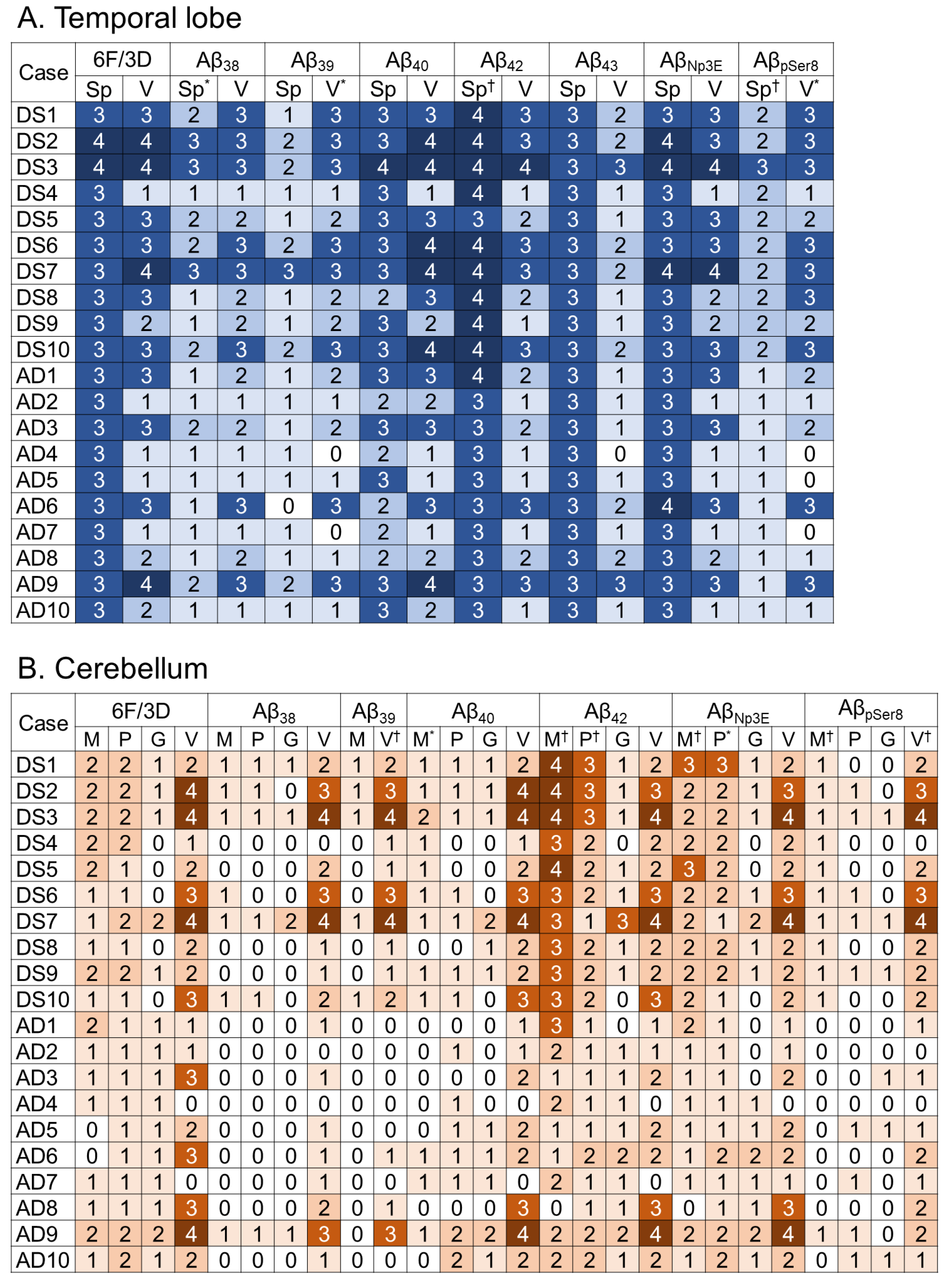
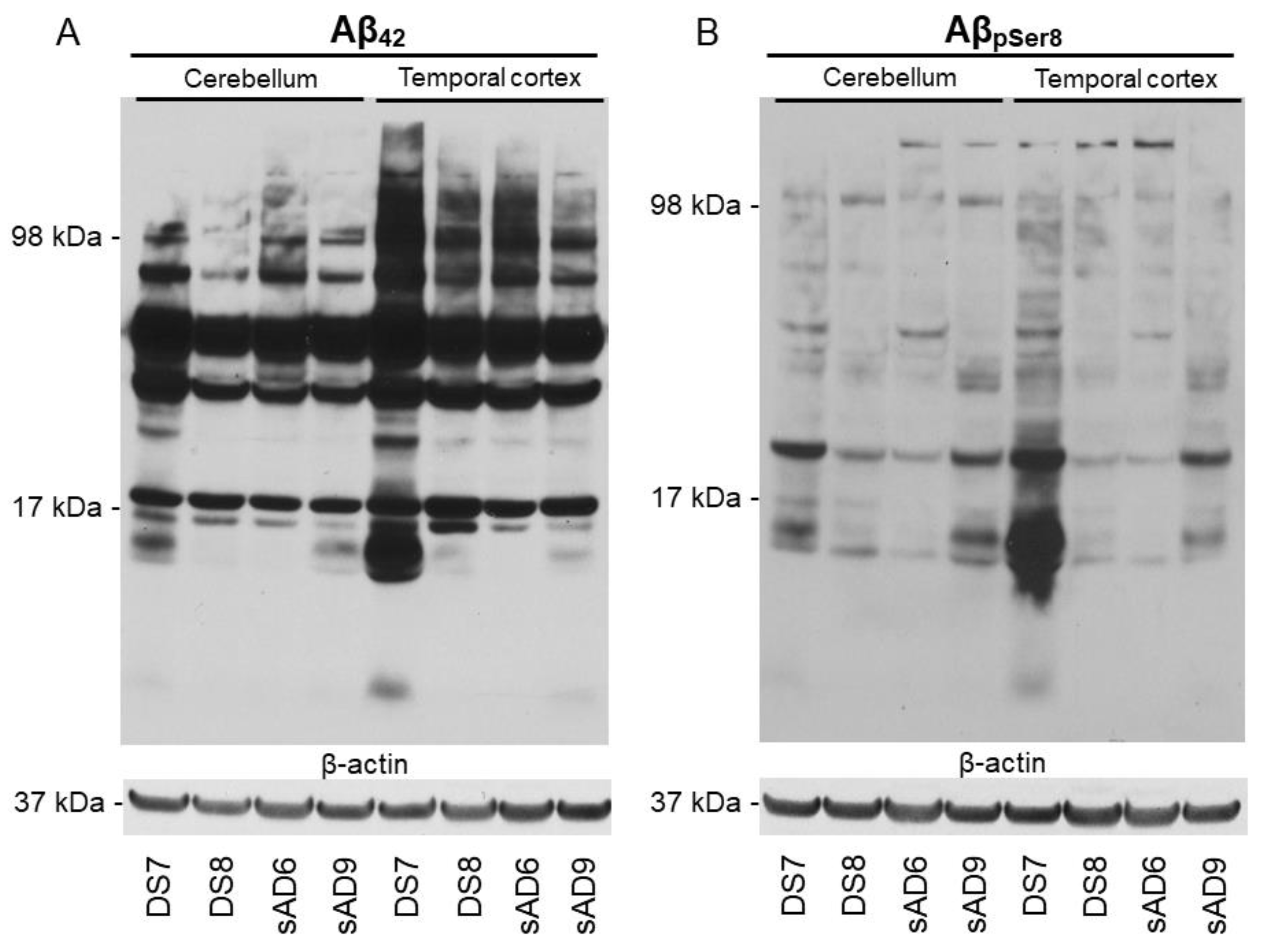
2.2. Aβ Pathology
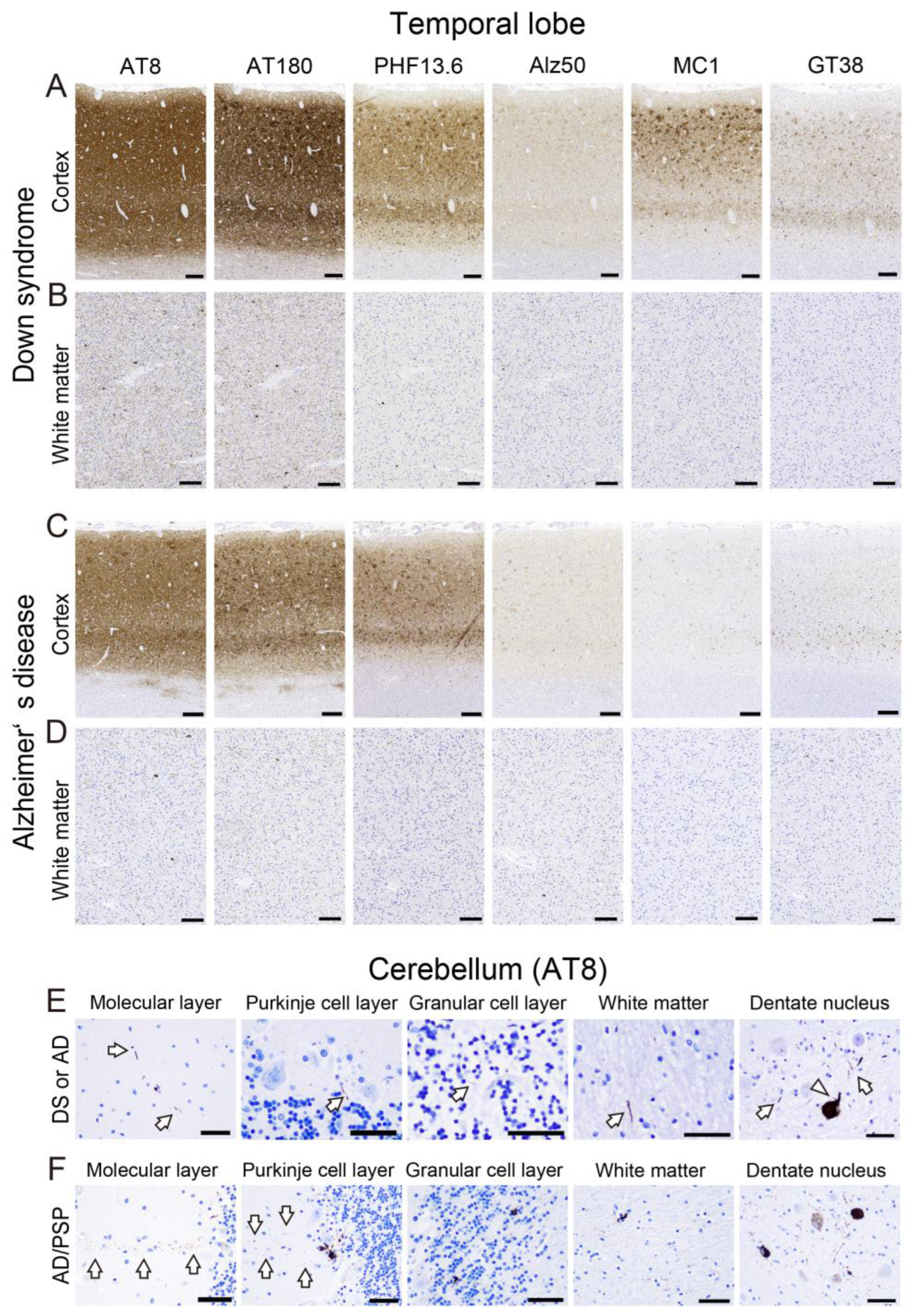
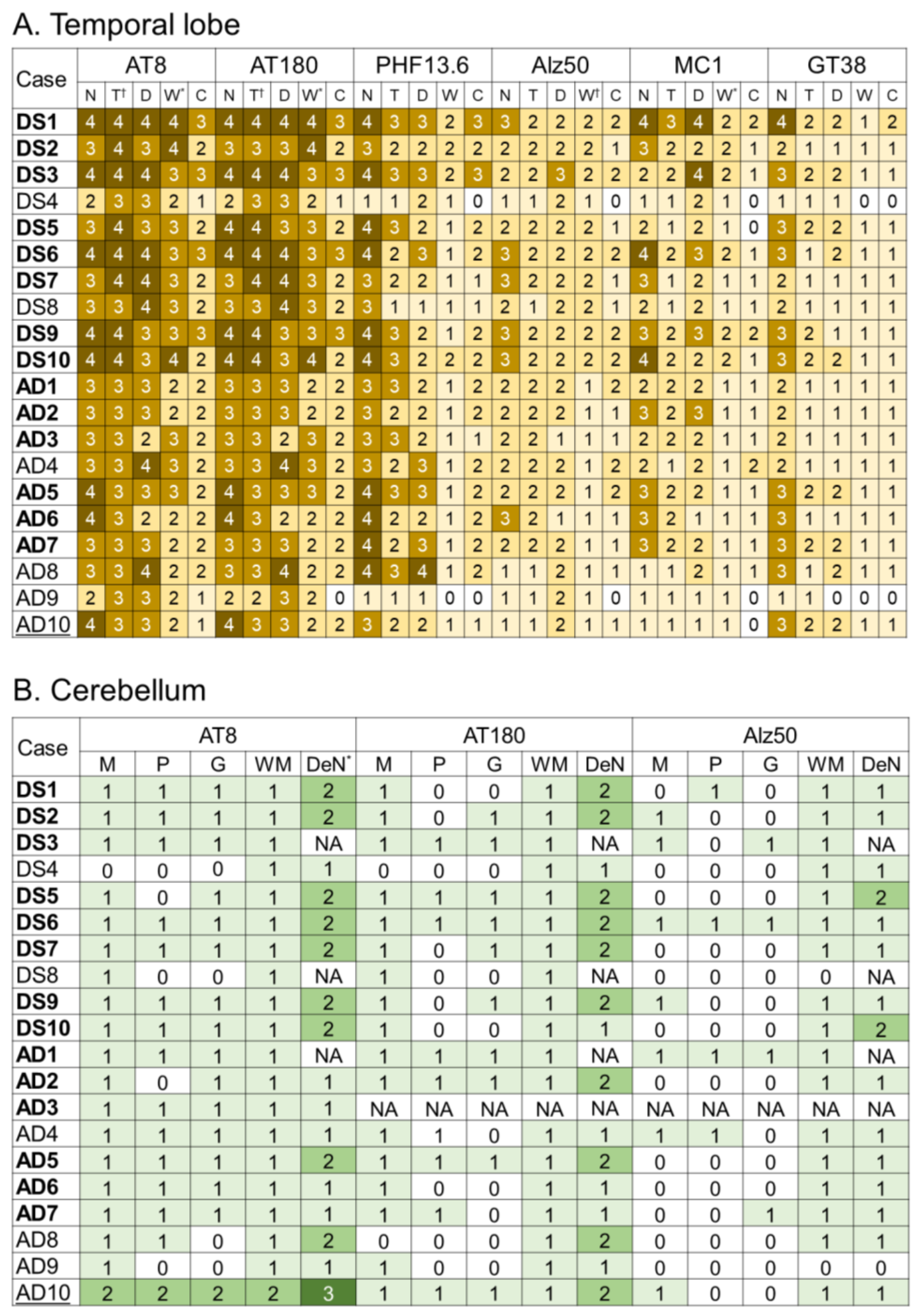
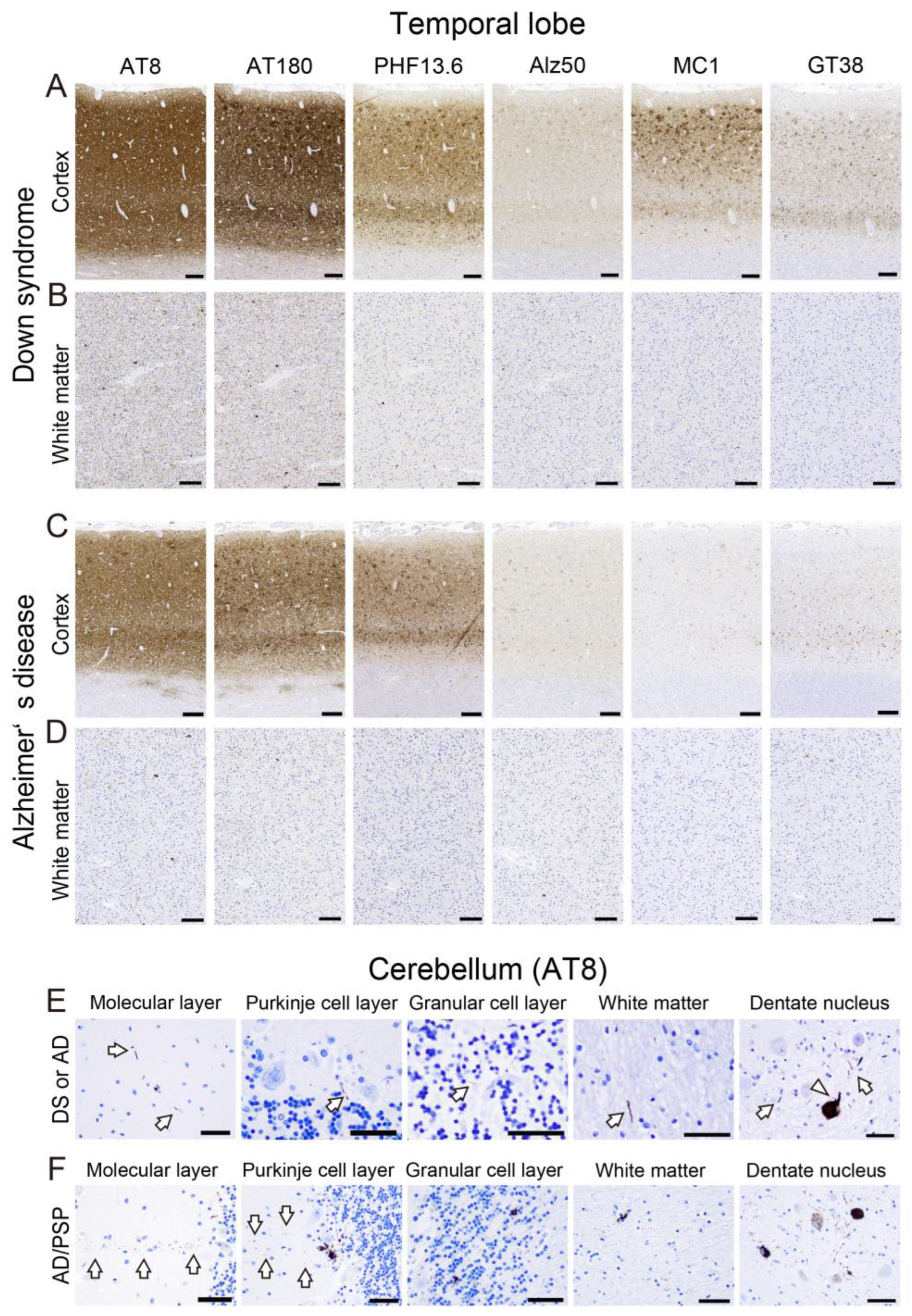
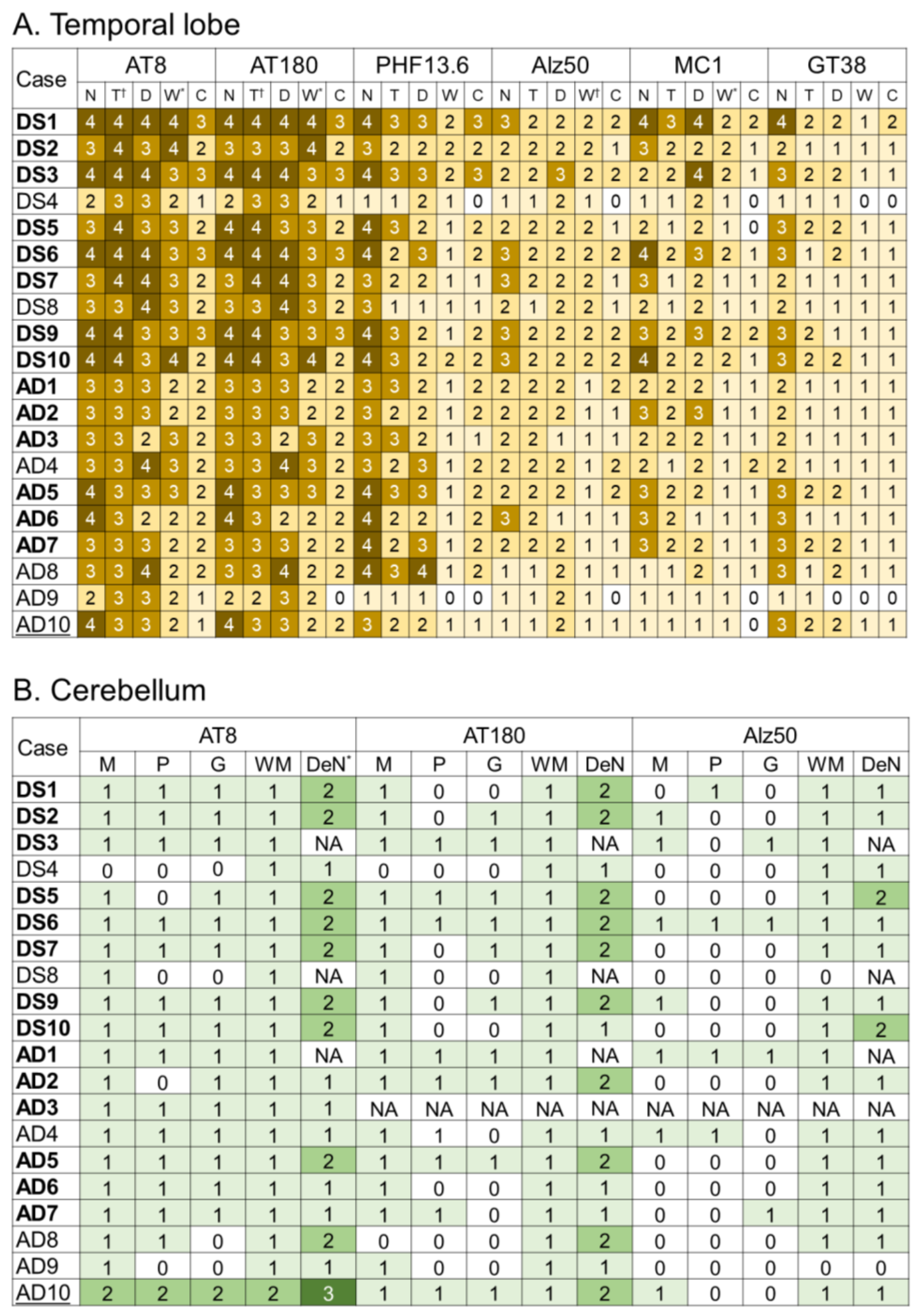
2.3. Tau Pathology
2.4. Correlations between Various Aβ and Tau Deposition Burdens
3. Discussion
4. Materials and Methods
4.1. Case Selection
4.2. Immunohistochemistry
4.3. Protein Extraction and Western Blotting
4.4. Pathological Assessment and Semiquantitative Grading System of Aβ and Tau Pathology
4.5. Image Analysis
4.6. Tau Seeding Assay
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bull, M.J. Down Syndrome. N. Engl. J. Med. 2020, 382, 2344–2352. [Google Scholar] [CrossRef] [PubMed]
- Wegiel, J.; Kaczmarski, W.; Barua, M.; Kuchna, I.; Nowicki, K.; Wang, K.C.; Wegiel, J.; Yang, S.M.; Frackowiak, J.; Mazur-Kolecka, B.; et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J. Neuropathol. Exp. Neurol. 2011, 70, 36–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, J.; Dowjat, K.; Kaczmarski, W.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Mazur Kolecka, B.; Wegiel, J.; Silverman, W.P.; Reisberg, B.; et al. The role of overexpressed DYRK1A protein in the early onset of neurofibrillary degeneration in Down syndrome. Acta Neuropathol. 2008, 116, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Snyder, H.M.; Bain, L.J.; Brickman, A.M.; Carrillo, M.C.; Esbensen, A.J.; Espinosa, J.M.; Fernandez, F.; Fortea, J.; Hartley, S.L.; Head, E.; et al. Further understanding the connection between Alzheimer’s disease and Down syndrome. Alzheimers Dement. 2020, 16, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Davidson, Y.S.; Robinson, A.; Prasher, V.P.; Mann, D.M.A. The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol. Commun. 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Hof, P.R.; Bouras, C.; Perl, D.P.; Sparks, D.L.; Mehta, N.; Morrison, J.H. Age-related distribution of neuropathologic changes in the cerebral cortex of patients with Down’s syndrome. Quantitative regional analysis and comparison with Alzheimer’s disease. Arch. Neurol. 1995, 52, 379–391. [Google Scholar] [CrossRef]
- Fortea, J.; Zaman, S.H.; Hartley, S.; Rafii, M.S.; Head, E.; Carmona-Iragui, M. Alzheimer’s disease associated with Down syndrome: A genetic form of dementia. Lancet Neurol. 2021, 20, 930–942. [Google Scholar] [CrossRef]
- Ichimata, S.; Yoshida, K.; Visanji, N.P.; Lang, A.E.; Nishida, N.; Kovacs, G.G. Patterns of Mixed Pathologies in Down Syndrome. J. Alzheimers Dis. 2022, 87, 595–607. [Google Scholar] [CrossRef]
- Ichimata, S.; Martinez-Valbuena, I.; Forrest, S.L.; Kovacs, G.G. Expanding the spectrum of amyloid-β plaque pathology: The Down syndrome associated ‘bird-nest plaque’. Acta Neuropathol. 2022, 144, 1171–1174. [Google Scholar] [CrossRef]
- Drummond, E.; Kavanagh, T.; Pires, G.; Marta-Ariza, M.; Kanshin, E.; Nayak, S.; Faustin, A.; Berdah, V.; Ueberheide, B.; Wisniewski, T. The amyloid plaque proteome in early onset Alzheimer’s disease and Down syndrome. Acta Neuropathol. Commun. 2022, 10, 53. [Google Scholar] [CrossRef]
- Maxwell, A.M.; Yuan, P.; Rivera, B.M.; Schaaf, W.; Mladinov, M.; Prasher, V.P.; Robinson, A.C.; DeGrado, W.F.; Condello, C. Emergence of distinct and heterogeneous strains of amyloid beta with advanced Alzheimer’s disease pathology in Down syndrome. Acta Neuropathol. Commun. 2021, 9, 201. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, I.; Jarc, J.; Dassler, E.; Aronica, E.; Iyer, A.; Adle-Biassette, H.; Scharrer, A.; Reischer, T.; Hainfellner, J.A.; Kovacs, G.G. The physiological phosphorylation of tau is critically changed in fetal brains of individuals with Down syndrome. Neuropathol. Appl. Neurobiol. 2018, 44, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Mondragon-Rodriguez, S.; Perry, G.; Luna-Munoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein at sites Ser(396–404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef]
- Condello, C.; Maxwell, A.M.; Castillo, E.; Aoyagi, A.; Graff, C.; Ingelsson, M.; Lannfelt, L.; Bird, T.D.; Keene, C.D.; Seeley, W.W.; et al. Abeta and tau prions feature in the neuropathogenesis of Down syndrome. Proc. Natl. Acad. Sci. USA 2022, 119, e2212954119. [Google Scholar] [CrossRef]
- Thal, D.R.; Rüb, U.; Orantes, M.; Braak, H. Phases of A beta-deposition in the human brain and its relevance for the development of AD. Neurology 2002, 58, 1791–1800. [Google Scholar] [CrossRef]
- Mann, D.M.; Iwatsubo, T.; Snowden, J.S. Atypical amyloid (A beta) deposition in the cerebellum in Alzheimer’s disease: An immunohistochemical study using end-specific A beta monoclonal antibodies. Acta Neuropathol. 1996, 91, 647–653. [Google Scholar] [CrossRef]
- Mann, D.M.; Iwatsubo, T. Diffuse plaques in the cerebellum and corpus striatum in Down’s syndrome contain amyloid beta protein (A beta) only in the form of A beta 42(43). Neurodegeneration 1996, 5, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.T.; Woodruff-Pak, D.S.; Trojanowski, J.Q. Amyloid plaques in cerebellar cortex and the integrity of Purkinje cell dendrites. Neurobiol. Aging 1994, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Cole, G.; Neal, J.W.; Singhrao, S.K.; Jasani, B.; Newman, G.R. The distribution of amyloid plaques in the cerebellum and brain stem in Down’s syndrome and Alzheimer’s disease: A light microscopical analysis. Acta Neuropathol. 1993, 85, 542–552. [Google Scholar] [CrossRef]
- Mann, D.M.; Jones, D.; Prinja, D.; Purkiss, M.S. The prevalence of amyloid (A4) protein deposits within the cerebral and cerebellar cortex in Down’s syndrome and Alzheimer’s disease. Acta Neuropathol. 1990, 80, 318–327. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hirai, S.; Morimatsu, M.; Shoji, M.; Nakazato, Y. Diffuse type of senile plaques in the cerebellum of Alzheimer-type dementia demonstrated by beta protein immunostain. Acta Neuropathol. 1989, 77, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Joachim, C.L.; Morris, J.H.; Selkoe, D.J. Diffuse senile plaques occur commonly in the cerebellum in Alzheimer’s disease. Am. J. Pathol. 1989, 135, 309–319. [Google Scholar] [PubMed]
- Miguel, J.C.; Perez, S.E.; Malek-Ahmadi, M.; Mufson, E.J. Cerebellar Calcium-Binding Protein and Neurotrophin Receptor Defects in Down Syndrome and Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 645334. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.A.; Davidson, Y.S.; Robinson, A.C.; Allen, N.; Hashimoto, T.; Richardson, A.; Jones, M.; Snowden, J.S.; Pendleton, N.; Potier, M.C.; et al. Patterns and severity of vascular amyloid in Alzheimer’s disease associated with duplications and missense mutations in APP gene, Down syndrome and sporadic Alzheimer’s disease. Acta Neuropathol. 2018, 136, 569–587. [Google Scholar] [CrossRef] [PubMed]
- Head, E.; Phelan, M.J.; Doran, E.; Kim, R.C.; Poon, W.W.; Schmitt, F.A.; Lott, I.T. Cerebrovascular pathology in Down syndrome and Alzheimer disease. Acta Neuropathol. Commun. 2017, 5, 93. [Google Scholar] [CrossRef] [Green Version]
- Reinert, J.; Richard, B.C.; Klafki, H.W.; Friedrich, B.; Bayer, T.A.; Wiltfang, J.; Kovacs, G.G.; Ingelsson, M.; Lannfelt, L.; Paetau, A.; et al. Deposition of C-terminally truncated Abeta species Abeta37 and Abeta39 in Alzheimer’s disease and transgenic mouse models. Acta Neuropathol. Commun. 2016, 4, 24. [Google Scholar] [CrossRef] [Green Version]
- van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Banerjee, D. A Primer on the Evolution of Aducanumab: The First Antibody Approved for Treatment of Alzheimer’s Disease. J. Alzheimers Dis. 2021, 83, 1537–1552. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Robinson, J.L.; Xie, S.X.; Lee, E.B.; Grossman, M.; Wolk, D.A.; Irwin, D.J.; Weintraub, D.; Kim, C.F.; Schuck, T.; et al. Evaluating the Patterns of Aging-Related Tau Astrogliopathy Unravels Novel Insights Into Brain Aging and Neurodegenerative Diseases. J. Neuropathol. Exp. Neurol. 2017, 76, 270–288. [Google Scholar] [CrossRef] [Green Version]
- Roemer, S.F.; Grinberg, L.T.; Crary, J.F.; Seeley, W.W.; McKee, A.C.; Kovacs, G.G.; Beach, T.G.; Duyckaerts, C.; Ferrer, I.A.; Gelpi, E.; et al. Rainwater Charitable Foundation criteria for the neuropathologic diagnosis progressive supranuclear palsy. Acta Neuropathol. 2022, 144, 603–614. [Google Scholar] [CrossRef]
- Kovacs, G.G. Astroglia and Tau: New Perspectives. Front. Aging Neurosci. 2020, 12, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengoa-Vergniory, N.; Velentza-Almpani, E.; Silva, A.M.; Scott, C.; Vargas-Caballero, M.; Sastre, M.; Wade-Martins, R.; Alegre-Abarrategui, J. Tau-proximity ligation assay reveals extensive previously undetected pathology prior to neurofibrillary tangles in preclinical Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 18. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondragon-Rodriguez, S.; Basurto-Islas, G.; Santa-Maria, I.; Mena, R.; Binder, L.I.; Avila, J.; Smith, M.A.; Perry, G.; Garcia-Sierra, F. Cleavage and conformational changes of tau protein follow phosphorylation during Alzheimer’s disease. Int. J. Exp. Pathol. 2008, 89, 81–90. [Google Scholar] [CrossRef]
- Hirayama, A.; Horikoshi, Y.; Maeda, M.; Ito, M.; Takashima, S. Characteristic developmental expression of amyloid β40, 42 and 43 in patients with Down syndrome. Brain Dev. 2003, 25, 180–185. [Google Scholar] [CrossRef]
- Lemere, C.A.; Blusztajn, J.K.; Yamaguchi, H.; Wisniewski, T.; Saido, T.C.; Selkoe, D.J. Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: Implications for initial events in amyloid plaque formation. Neurobiol. Dis. 1996, 3, 16–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwatsubo, T.; Mann, D.M.; Odaka, A.; Suzuki, N.; Ihara, Y. Amyloid beta protein (A beta) deposition: A beta 42(43) precedes A beta 40 in Down syndrome. Ann. Neurol. 1995, 37, 294–299. [Google Scholar] [CrossRef]
- Saido, T.C.; Iwatsubo, T.; Mann, D.M.; Shimada, H.; Ihara, Y.; Kawashima, S. Dominant and differential deposition of distinct beta-amyloid peptide species, A beta N3(pE), in senile plaques. Neuron 1995, 14, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Lemere, C.A.; Walter, J. Phosphorylated Abeta peptides in human Down syndrome brain and different Alzheimer’s-like mouse models. Acta Neuropathol. Commun. 2020, 8, 118. [Google Scholar] [CrossRef]
- Braun, G.A.; Dear, A.J.; Sanagavarapu, K.; Zetterberg, H.; Linse, S. Amyloid-beta peptide 37, 38 and 40 individually and cooperatively inhibit amyloid-beta 42 aggregation. Chem. Sci. 2022, 13, 2423–2439. [Google Scholar] [CrossRef]
- Sepulveda-Falla, D.; Matschke, J.; Bernreuther, C.; Hagel, C.; Puig, B.; Villegas, A.; Garcia, G.; Zea, J.; Gomez-Mancilla, B.; Ferrer, I.; et al. Deposition of hyperphosphorylated tau in cerebellum of PS1 E280A Alzheimer’s disease. Brain Pathol. 2011, 21, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Rijal Upadhaya, A.; Kosterin, I.; Kumar, S.; von Arnim, C.A.; Yamaguchi, H.; Fandrich, M.; Walter, J.; Thal, D.R. Biochemical stages of amyloid-beta peptide aggregation and accumulation in the human brain and their association with symptomatic and pathologically preclinical Alzheimer’s disease. Brain 2014, 137, 887–903. [Google Scholar] [CrossRef] [PubMed]
- Hunter, S.; Brayne, C. Do anti-amyloid beta protein antibody cross reactivities confound Alzheimer disease research? J. Negat. Results Biomed. 2017, 16, 1. [Google Scholar] [CrossRef] [Green Version]
- Lalowski, M.; Golabek, A.; Lemere, C.A.; Selkoe, D.J.; Wisniewski, H.M.; Beavis, R.C.; Frangione, B.; Wisniewski, T. The “nonamyloidogenic” p3 fragment (amyloid beta17–42) is a major constituent of Down’s syndrome cerebellar preamyloid. J. Biol. Chem. 1996, 271, 33623–33631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teller, J.K.; Russo, C.; DeBusk, L.M.; Angelini, G.; Zaccheo, D.; Dagna-Bricarelli, F.; Scartezzini, P.; Bertolini, S.; Mann, D.M.; Tabaton, M.; et al. Presence of soluble amyloid beta-peptide precedes amyloid plaque formation in Down’s syndrome. Nat. Med. 1996, 2, 93–95. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, A.J.; Abrams, B.S.; Knowlton, S.; Raskatov, J.A. Alzheimer’s Disease “Non-amyloidogenic” p3 Peptide Revisited: A Case for Amyloid-alpha. ACS Chem. Neurosci. 2020, 11, 1539–1544. [Google Scholar] [CrossRef]
- Wei, W.; Norton, D.D.; Wang, X.; Kusiak, J.W. Abeta 17–42 in Alzheimer’s disease activates JNK and caspase-8 leading to neuronal apoptosis. Brain 2002, 125, 2036–2043. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, A.J.; Raskatov, J. Is the p3 Peptide (Abeta17–40, Abeta17–42) Relevant to the Pathology of Alzheimer’s Disease? J. Alzheimers Dis. 2020, 74, 43–53. [Google Scholar] [CrossRef]
- Leverenz, J.B.; Raskind, M.A. Early amyloid deposition in the medial temporal lobe of young Down syndrome patients: A regional quantitative analysis. Exp. Neurol. 1998, 150, 296–304. [Google Scholar] [CrossRef]
- Guidi, S.; Ciani, E.; Bonasoni, P.; Santini, D.; Bartesaghi, R. Widespread proliferation impairment and hypocellularity in the cerebellum of fetuses with down syndrome. Brain Pathol. 2011, 21, 361–373. [Google Scholar] [CrossRef]
- Kanaumi, T.; Milenkovic, I.; Adle-Biassette, H.; Aronica, E.; Kovacs, G.G. Non-neuronal cell responses differ between normal and Down syndrome developing brains. Int. J. Dev. Neurosci. 2013, 31, 796–803. [Google Scholar] [CrossRef] [PubMed]
- McAleese, K.E.; Walker, L.; Graham, S.; Moya, E.L.J.; Johnson, M.; Erskine, D.; Colloby, S.J.; Dey, M.; Martin-Ruiz, C.; Taylor, J.P.; et al. Parietal white matter lesions in Alzheimer’s disease are associated with cortical neurodegenerative pathology, but not with small vessel disease. Acta Neuropathol. 2017, 134, 459–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAleese, K.E.; Miah, M.; Graham, S.; Hadfield, G.M.; Walker, L.; Johnson, M.; Colloby, S.J.; Thomas, A.J.; DeCarli, C.; Koss, D.; et al. Frontal white matter lesions in Alzheimer’s disease are associated with both small vessel disease and AD-associated cortical pathology. Acta Neuropathol. 2021, 142, 937–950. [Google Scholar] [CrossRef]
- Forrest, S.L.; Wagner, S.; Kim, A.; Kovacs, G.G. Association of glial tau pathology and LATE-NC in the ageing brain. Neurobiol. Aging 2022, 119, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.; Vincze, A.; Veszpremi, B.; Kravjak, A.; Gomori, E.; Kovacs, G.G.; Seress, L. Impaired myelination of the human hippocampal formation in Down syndrome. Int. J. Dev. Neurosci. 2012, 30, 147–158. [Google Scholar] [CrossRef]
- Jin, N.; Gu, J.; Wu, R.; Chu, D.; Tung, Y.C.; Wegiel, J.; Wisniewski, T.; Gong, C.X.; Iqbal, K.; Liu, F. Tau seeding activity in various regions of down syndrome brain assessed by two novel assays. Acta Neuropathol. Commun. 2022, 10, 132. [Google Scholar] [CrossRef]
- Piao, Y.S.; Hayashi, S.; Wakabayashi, K.; Kakita, A.; Aida, I.; Yamada, M.; Takahashi, H. Cerebellar cortical tau pathology in progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol. 2002, 103, 469–474. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-beta and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Tudorascu, D.L.; Laymon, C.M.; Zammit, M.; Minhas, D.S.; Anderson, S.J.; Ellison, P.A.; Zaman, S.; Ances, B.M.; Sabbagh, M.; Johnson, S.C.; et al. Relationship of amyloid beta and neurofibrillary tau deposition in Neurodegeneration in Aging Down Syndrome (NiAD) study at baseline. Alzheimers Dement. 2020, 6, e12096. [Google Scholar] [CrossRef]
- Grigorova, M.; Mak, E.; Brown, S.S.G.; Beresford-Webb, J.; Hong, Y.T.; Fryer, T.D.; Coles, J.P.; Aigbirhio, F.I.; Tudorascu, D.; Cohen, A.; et al. Amyloid- beta and tau deposition influences cognitive and functional decline in Down syndrome. Neurobiol. Aging 2022, 119, 36–45. [Google Scholar] [CrossRef]
- Cline, E.N.; Bicca, M.A.; Viola, K.L.; Klein, W.L. The Amyloid-beta Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimers Dis. 2018, 64, S567–S610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnsten, A.F.T.; Datta, D.; Del Tredici, K.; Braak, H. Hypothesis: Tau pathology is an initiating factor in sporadic Alzheimer’s disease. Alzheimers Dement. 2021, 17, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G.; Ghetti, B.; Goedert, M. Classification of diseases with accumulation of Tau protein. Neuropathol. Appl. Neurobiol. 2022, 48, e12792. [Google Scholar] [CrossRef] [PubMed]
- Hithersay, R.; Startin, C.M.; Hamburg, S.; Mok, K.Y.; Hardy, J.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Nizetic, D.; Strydom, A. Association of Dementia With Mortality Among Adults With Down Syndrome Older Than 35 Years. JAMA Neurol. 2019, 76, 152–160. [Google Scholar] [CrossRef]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Swinkin, E.; Santamaria, E.; Fernandez-Irigoyen, J.; Sackmann, V.; Kim, A.; Li, J.; Gonzalez-Latapi, P.; Kuhlman, G.; Bhowmick, S.S.; et al. α-Synuclein molecular behavior and nigral proteomic profiling distinguish subtypes of Lewy body disorders. Acta Neuropathol. 2022, 144, 167–185. [Google Scholar] [CrossRef]
- Martinez-Valbuena, I.; Visanji, N.P.; Kim, A.; Lau, H.H.C.; So, R.W.L.; Alshimemeri, S.; Gao, A.; Seidman, M.A.; Luquin, M.R.; Watts, J.C.; et al. α-synuclein seeding shows a wide heterogeneity in multiple system atrophy. Transl. Neurodegener. 2022, 11, 7. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Montine, T.J.; Phelps, C.H.; Beach, T.G.; Bigio, E.H.; Cairns, N.J.; Dickson, D.W.; Duyckaerts, C.; Frosch, M.P.; Masliah, E.; Mirra, S.S.; et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012, 123, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Thal, D.R.; Ghebremedhin, E.; Rüb, U.; Yamaguchi, H.; Del Tredici, K.; Braak, H. Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol. 2002, 61, 282–293. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimata, S.; Kim, A.; Nishida, N.; Kovacs, G.G. Lack of difference between amyloid-beta burden at gyral crests and sulcal depths in diverse neurodegenerative diseases. Neuropathol. Appl. Neurobiol. 2023, 49, e12869. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Furman, J.L.; Mahan, T.E.; Yamasaki, T.R.; Mirbaha, H.; Eades, W.C.; Belaygorod, L.; Cairns, N.J.; Holtzman, D.M.; Diamond, M.I. Proteopathic tau seeding predicts tauopathy in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, E4376–E4385. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age ** | Gender | BW (g) * | Braak NFT Stage | Thal Phase | CERAD Score | NIA-AA AD Level | CAA Type |
|---|---|---|---|---|---|---|---|---|
| DS1 | 42 | F | 900 | 6 | 5 | C | High | 2 |
| DS2 | 49 | M | 1100 | 6 | 5 | C | High | 2 |
| DS3 | 51 | F | 750 | 6 | 5 | C | High | 1 |
| DS4 | 54 | M | 1130 | 3 | 5 | C | Int | 2 |
| DS5 | 57 | F | 756 | 6 | 5 | C | High | 2 |
| DS6 | 58 | M | 1100 | 6 | 5 | C | High | 1 |
| DS7 | 61 | M | 905 | 6 | 5 | C | High | 1 |
| DS8 | 61 | F | 900 | 5 | 5 | C | High | 2 |
| DS9 | 65 | F | 880 | 6 | 5 | C | High | 1 |
| DS10 | 66 | F | 760 | 6 | 5 | C | High | 1 |
| sAD1 | 77 | M | 1200 | 6 | 5 | C | High | 2 |
| sAD2 | 83 | F | 1000 | 6 | 5 | C | High | 2 |
| sAD3 | 78 | M | 1220 | 6 | 5 | C | High | 2 |
| sAD4 | 77 | F | 1200 | 5 | 5 | C | High | 2 |
| sAD5 | 79 | M | 1200 | 6 | 5 | C | High | 2 |
| sAD6 | 78 | M | N/A | 6 | 5 | C | High | 2 |
| sAD7 | 94 | F | 830 | 6 | 5 | C | High | 2 |
| sAD8 | 82 | F | N/A | 5 | 5 | C | High | 2 |
| sAD9 | 75 | M | 1240 | 4 | 5 | C | Int | 1 |
| sAD10 | 85 | M | N/A | 4 | 5 | C | Int | 2 |
| Plaque | Antibodies | DS | sAD | p-Value * |
|---|---|---|---|---|
| Cerebral Aβ burden in all cases (N = 10 cases, each) | ||||
| Size (range) | 6F/3D | 59.4 ± 13.3 (42.0–77.3) | 36.8 ± 12.7 (15.6–55.0) | <0.01 |
| Aβ42 | 83.2 ± 21.5 (38.4–111.4) | 50.6 ± 17.0 (28.2–75.5) | <0.01 | |
| AβNp3E | 97.5 ± 38.4 (46.1–173.2) | 57.1 ± 28.3 (25.5–124.8) | 0.02 | |
| Load (range) | 6F/3D | 7.6 ± 4.6 (1.8–16.2) | 4.7 ± 1.8 (2.1–8.2) | 0.22 |
| Aβ42 | 18.3 ± 6.7 (10.2–28.0) | 8.3 ± 2.6 (4.5–12.0) | <0.01 | |
| AβNp3E | 8.9 ± 4.8 (1.8–18.2) | 6.1 ± 2.0 (3.7–10.1) | 0.19 | |
| Cerebral Aβ burden in cases with Braak NFT stage VI (N = 8 and 6 cases, respectively) | ||||
| Size (range) | 6F/3D | 60.0 ± 13.8 (42.0–77.3) | 38.7 ± 10.2 (25.0–52.1) | 0.04 |
| Aβ42 | 78.1 ± 21.1 (38.4–111.4) | 49.3 ± 17.0 (28.2–75.5) | 0.03 | |
| AβNp3E | 105.7 ± 37.7 (56.8–173.2) | 52.3 ± 19.5 (25.5–84.0) | <0.01 | |
| Load (range) | 6F/3D | 9.0 ± 4.0 (4.4–16.2) | 5.8 ± 1.3 (4.3–8.2) | 0.18 |
| Aβ42 | 20.2 ± 6.2 (10.7–28.0) | 9.0 ± 2.6 (5.0–12.0) | <0.01 | |
| AβNp3E | 10.3 ± 4.2 (5.2–18.2) | 7.0 ± 2.1 (3.7–10.1) | 0.18 | |
| Cerebellar molecular layer Aβ burden (N = 10 and 7 cases, respectively) | ||||
| Load (range) | 6F/3D | 2.0 ± 1.6 (0.2–5.1) | 0.9 ± 0.8 (0.1–3.4) | 0.16 |
| Aβ42 | 35.3 ± 11.8 (20.7–55.6) | 8.7 ± 8.4 (1.6–28.4) | <0.01 | |
| AβNp3E | 4.9 ± 2.2 (2.0–8.2) | 2.2 ± 1.3 (0.1–5.5) | 0.06 | |
| Antibody | DS-Cx | sAD-Cx | DS-WM | sAD-WM | p-Value (Cx/WM) * |
|---|---|---|---|---|---|
| Mean total deposition burden in all cases (range) [N = 10 and 9 cases, respectively] | |||||
| AT8 | 56.9 ± 19.1 (16.9–77.0) | 46.2 ± 13.9 (15.3–65.2) | 6.0 ± 2.8 (1.0–10.9) | 2.4 ± 1.2 (0.5–4.6) | 0.18/<0.01 |
| AT180 | 53.4 ± 18.9 (12.4–73.9) | 39.5 ± 13.0 (13.7–56.2) | 5.8 ± 2.6 (0.9–10.1) | 2.2 ± 1.1 (0.6–4.0) | 0.07/<0.01 |
| PHF13.6 | 21.7 ± 16.0 (0.9–45.6) | 18.9 ± 9.9 (0.2–38.7) | NA ** | NA ** | 0.60/NE |
| Alz50 | 5.7 ± 3.6 (1.3–12.6) | 3.9 ± 2.2 (1.0–7.3) | 1.2 ± 0.6 (0.3–2.6) | 0.6 ± 0.3 (0.1–1.2) | 0.40/0.02 |
| MC1 | 6.3 ± 8.6 (0.4–30.6) | 2.2 ± 1.9 (0.2–6.3) | 0.5 ± 0.4 (0.2–1.6) | 0.4 ± 0.2 (0.2–0.8) | 0.24/0.28 |
| GT38 | 3.1 ± 3.1 (0.0–8.6) | 0.9 ± 0.8 (0.0–2.4) | 0.2 ± 0.2 (0.0–0.9) | 0.2 ± 0.2 (0.0–0.7) | 0.32/0.97 |
| Mean total deposition burden in Braak NFT stage VI cases (range) [N = 8, 6 cases, respectively] | |||||
| AT8 | 64.2 ± 12.5 (44.0–77.0) | 50.9 ± 10.4 (32.7–65.2) | 6.8 ± 2.4 (3.6–10.9) | 2.8 ± 1.1 (1.0–4.6) | 0.11/<0.01 |
| AT180 | 61.1 ± 11.0 (38.3–73.9) | 43.5 ± 11.2 (22.7–56.2) | 6.6 ± 2.1 (4.1–10.1) | 2.6 ± 1.0 (0.9–4.0) | 0.01/<0.01 |
| PHF13.6 | 26.8 ± 13.6 (2.9–45.6) | 22.8 ± 7.9 (12.5–38.7) | NA ** | NA ** | 0.49/NE |
| Alz50 | 6.7 ± 3.4 (2.8–12.6) | 5.1 ± 1.5 (3.3–7.3) | 1.3 ± 0.6 (0.9–2.6) | 0.7 ± 0.3 (0.4–1.2) | 0.76/<0.01 |
| MC1 | 7.7 ± 9.0 (1.0–30.6) | 3.1 ± 1.7 (1.4–6.3) | 0.6 ± 0.4 (0.2–1.6) | 0.4 ± 0.2 (0.2–0.8) | 0.41/0.57 |
| GT38 | 3.9 ± 3.0 (0.1–8.6) | 1.1 ± 0.9 (0.1–2.4) | 0.1 ± 0.1 (0.0–0.3) | 0.2 ± 0.2 (0.0–0.7) | 0.18/1.00 |
| 6F/3D (r/p-Value) | Aβ42 (r/p-Value) | AβNp3E (r/p-Value) | ||||
|---|---|---|---|---|---|---|
| DS | sAD | DS | sAD | DS | sAD | |
| AT8 | 0.42/0.23 | 0.50/0.17 | 0.50/0.14 | 0.65/0.06 | 0.42/0.23 | 0.02/0.97 |
| AT180 | 0.36/0.31 | 0.47/0.21 | 0.38/0.28 | 0.63/0.07 | 0.33/0.35 | 0.00/1.00 |
| PHF13.6 | 0.33/0.35 | 0.50/0.17 | 0.43/0.21 | 0.68/0.04 | 0.33/0.35 | 0.27/0.49 |
| Alz50 | 0.30/0.41 | 0.62/0.08 | 0.37/0.29 | 0.12/0.77 | 0.35/0.33 | 0.27/0.49 |
| MC1 | 0.37/0.29 | 0.55/0.13 | 0.49/0.15 | 0.20/0.61 | 0.43/0.21 | 0.22/0.72 |
| GT38 | 0.19/0.60 | 0.25/0.52 | 0.24/0.51 | 0.30/0.43 | 0.16/0.65 | 0.02/0.97 |
| Antibody | Source | Clone | Dilution | 1st Antigen Retrieval | 2nd Antigen Retrieval |
|---|---|---|---|---|---|
| Aβaa8–17 | Dako (Santa Clara, CA, USA) | 6F/3D | 1:50 | 80% FA 60 min | None |
| Aβ38 | Synaptic Systems (Göttingen, Germany) | Polyclonal | 1:1000 | Heat | 88% FA 3 min |
| Aβ39 | Cell Signaling (Danvers, MA, USA) | D5Y9L | 1:500 | Heat | 88% FA 3 min |
| Aβ40 | BioLegend (San Diego, CA, USA) | QA18A67 | 1:2000 | 70% FA 10 min | None |
| Aβ42 | BioLegend | 1-11-13 | 1:500 | 70% FA 10 min | None |
| Aβ43 | IBL (Fujioka, Japan) | Polyclonal | 1:100 | 88% FA 5 min | None |
| AβNp3E | BioLegend | 337.48 | 1:800 | Heat | 88% FA 3 min |
| AβpSer8 | Sigma-Aldrich (St. Louis, MO, USA) | 1E4E11 | 1:200 | Heat | 88% FA 3 min |
| p-tau (Ser202, Thr205) | Thermo Fischer (Waltham, MA, USA) | AT8 | 1:1000 | Heat | None |
| p-tau (Thr 231) | Thermo Fischer | AT180 | 1:1000 | Heat | None |
| p-tau (Ser396) | Thermo Fischer | PHF13.6 | 1:500 | Heat | None |
| Anti-tau (Alz50) | Gifted * | Alz50 | 1:100 | Heat | None |
| Anti-tau (MC1) | Gifted * | MC1 | 1:500 | Heat | None |
| Anti-tau AD antibody | Abcam (Cambridge, UK) | GT38 | 1:1000 | Heat * | None |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ichimata, S.; Martinez-Valbuena, I.; Lee, S.; Li, J.; Karakani, A.M.; Kovacs, G.G. Distinct Molecular Signatures of Amyloid-Beta and Tau in Alzheimer’s Disease Associated with Down Syndrome. Int. J. Mol. Sci. 2023, 24, 11596. https://doi.org/10.3390/ijms241411596
Ichimata S, Martinez-Valbuena I, Lee S, Li J, Karakani AM, Kovacs GG. Distinct Molecular Signatures of Amyloid-Beta and Tau in Alzheimer’s Disease Associated with Down Syndrome. International Journal of Molecular Sciences. 2023; 24(14):11596. https://doi.org/10.3390/ijms241411596
Chicago/Turabian StyleIchimata, Shojiro, Ivan Martinez-Valbuena, Seojin Lee, Jun Li, Ali M. Karakani, and Gabor G. Kovacs. 2023. "Distinct Molecular Signatures of Amyloid-Beta and Tau in Alzheimer’s Disease Associated with Down Syndrome" International Journal of Molecular Sciences 24, no. 14: 11596. https://doi.org/10.3390/ijms241411596
APA StyleIchimata, S., Martinez-Valbuena, I., Lee, S., Li, J., Karakani, A. M., & Kovacs, G. G. (2023). Distinct Molecular Signatures of Amyloid-Beta and Tau in Alzheimer’s Disease Associated with Down Syndrome. International Journal of Molecular Sciences, 24(14), 11596. https://doi.org/10.3390/ijms241411596