
Impact of Mir196a-2 Genotypes on Colorectal Cancer Risk in Taiwan
 and
and
Abstract
1. Introduction
2. Results
2.1. Characteristics of Study Population
2.2. Mir196a-2 Genotypes, but Not Mir146a Genotypes, Were Associated with CRC Risk
2.3. Associations of Mir196a-2 and Mir146a Alleles with CRC Risk
2.4. Stratified Analyses of Mir196a-2 Genotypes by Age, Gender, Smoking, Alcohol Drinking Behaviors and BMI Status
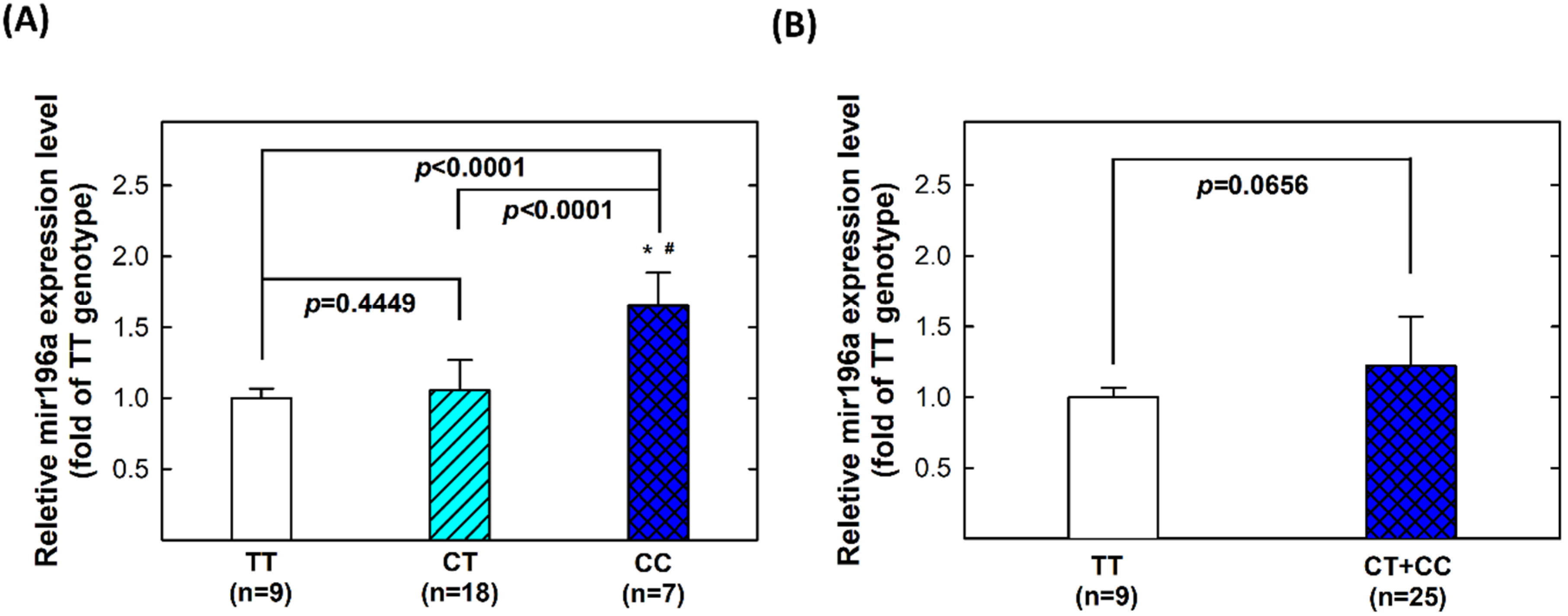
2.5. Genotype-Phenotype Correlation of MiR196a-2 among Controls
3. Discussion
4. Materials and Methods
4.1. Study Population
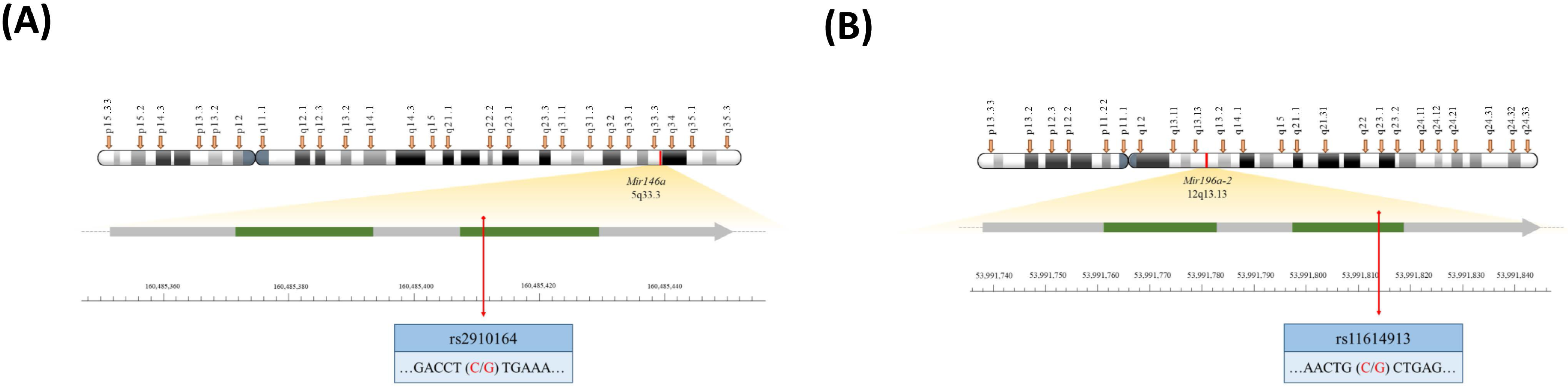
4.2. Genotyping Methodology of Mir146a and Mir196a-2 Polymorphisms
4.3. Quantitative Reverse Transcription Polymerase Chain Reaction for Examining Mir196a-2 Transcriptional Expression
4.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- You, L.; Lv, Z.; Li, C.; Ye, W.; Zhou, Y.; Jin, J.; Han, Q. Worldwide cancer statistics of adolescents and young adults in 2019: A systematic analysis of the Global Burden of Disease Study 2019. ESMO Open 2021, 6, 100255. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Kim, U.; Rose, J.; Hoehn, R.S.; Kucmanic, M.; Eom, K.; Li, S.; Berger, N.A.; Koroukian, S.M. Geographic Variation and Risk Factor Association of Early Versus Late Onset Colorectal Cancer. Cancers 2023, 15, 1006. [Google Scholar] [CrossRef]
- Agache, A.; Mustatea, P.; Mihalache, O.; Bobirca, F.T.; Georgescu, D.E.; Jauca, C.M.; Birligea, A.; Doran, H.; Patrascu, T. Diabetes Mellitus as a Risk-factor for Colorectal Cancer Literature Review—Current Situation and Future Perspectives. Chirurgia 2018, 113, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Lu, B.; Luo, C.; Cai, J.; Lu, M.; Zhang, Y.; Chen, H.; Dai, M. Incidence, mortality, survival, risk factor and screening of colorectal cancer: A comparison among China, Europe, and northern America. Cancer Lett. 2021, 522, 255–268. [Google Scholar] [CrossRef]
- Kao, P.S.; Lin, J.K.; Wang, H.S.; Yang, S.H.; Jiang, J.K.; Chen, W.S.; Lin, T.C.; Li, A.F.; Liang, W.Y.; Chang, S.C. The impact of family history on the outcome of patients with colorectal cancer in a veterans’ hospital. Int. J. Colorectal Dis. 2009, 24, 1249–1254. [Google Scholar] [CrossRef]
- Yeh, Y.L.; Li, M.; Kwok, O.M.; Ma, P.; Chen, L.S. Chinese Americans’ Family History of Colorectal Cancer Communication with Primary Care Physicians. Health Educ. Behav. 2022, 49, 169–178. [Google Scholar] [CrossRef]
- Hung, Y.C.; Chang, W.S.; Chou, A.K.; Pei, J.S.; Yang, M.D.; Yang, H.R.; Yang, T.M.; Wang, Y.C.; Hsiau, Y.C.; Chen, C.P.; et al. Association of Adiponectin Genotypes with Colorectal Cancer Susceptibility in Taiwan. Anticancer Res. 2020, 40, 1297–1306. [Google Scholar] [CrossRef]
- Wu, M.H.; Chen, C.H.; Chen, C.P.; Huang, T.L.; Yueh, T.C.; Wang, Z.H.; Tsai, C.W.; Pei, J.S.; Mong, M.C.; Yang, Y.C.; et al. Contribution of 5-Methyltetrahydrofolate-Homocysteine Methyltransferase Reductase Genotypes to Colorectal Cancer in Taiwan. Anticancer Res. 2022, 42, 2375–2382. [Google Scholar] [CrossRef]
- Wu, M.H.; Hung, Y.W.; Gong, C.L.; Chao, C.C.; Yueh, T.C.; Wang, S.C.; Lai, Y.L.; Hsu, S.W.; Fu, C.K.; Wang, Y.C.; et al. Contribution of Caspase-8 Genotypes to Colorectal Cancer Risk in Taiwan. Anticancer Res. 2019, 39, 2791–2797. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Tzeng, H.E.; Wu, C.N.; Yueh, T.C.; Peng, Y.C.; Tsai, C.H.; Wang, Y.C.; Ke, T.W.; Pei, J.S.; Chang, W.S.; et al. Association of Matrix Metalloproteinase-9 rs3918242 Promoter Genotypes with Colorectal Cancer Risk. Anticancer Res. 2019, 39, 6523–6529. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.H.; Yueh, T.C.; Chang, W.S.; Tsai, C.W.; Fu, C.K.; Yang, M.D.; Yu, C.C.; Bau, D.T. Contribution of Matrix Metalloproteinase-1 Genotypes to Colorectal Cancer in Taiwan. Cancer Genom. Proteom. 2021, 18, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Yueh, T.C.; Hung, Y.C.; Lee, H.T.; Yang, M.D.; Wang, Z.H.; Yang, Y.C.; Ke, T.W.; Pei, J.S.; Tsai, C.W.; Bau, D.T.; et al. Role of Matrix Metallopeptidase-2 Genotypes in Taiwanese Patients with Colorectal Cancer. Anticancer Res. 2022, 42, 5335–5342. [Google Scholar] [CrossRef] [PubMed]
- Devara, D.; Choudhary, Y.; Kumar, S. Role of MicroRNA-502-3p in Human Diseases. Pharmaceuticals 2023, 16, 532. [Google Scholar] [CrossRef]
- Kim, W.R.; Park, E.G.; Lee, D.H.; Lee, Y.J.; Bae, W.H.; Kim, H.S. The Tumorigenic Role of Circular RNA-MicroRNA Axis in Cancer. Int. J. Mol. Sci. 2023, 24, 3050. [Google Scholar] [CrossRef]
- Tajik, F.; Alian, F.; Yousefi, M.; Azadfallah, A.; Hoseini, A.; Mohammadi, F.; Karimi-Dehkordi, M.; Alizadeh-Fanalou, S. MicroRNA-372 acts as a double-edged sword in human cancers. Heliyon 2023, 9, e15991. [Google Scholar] [CrossRef]
- Kontham, S.S.; Walter, C.E.J.; Shankaran, Z.S.; Ramanathan, A.; Karuppasamy, N.; Johnson, T. A microRNA binding site polymorphism in the 3′ UTR region of VEGF-A gene modifies colorectal cancer risk based on ethnicity: A meta-analysis. J. Egypt Natl. Canc. Inst. 2022, 34, 18. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, X.; Zhai, H.; Wang, Y.; Hao, G. Integrated analysis of mRNA-single nucleotide polymorphism-microRNA interaction network to identify biomarkers associated with prostate cancer. Front. Genet. 2022, 13, 922712. [Google Scholar] [CrossRef]
- Sharma, G.; Mo, J.S.; Lamichhane, S.; Chae, S.C. MicroRNA 133A Regulates Cell Proliferation, Cell Migration, and Apoptosis in Colorectal Cancer by Suppressing CDH3 Expression. J. Cancer 2023, 14, 881–894. [Google Scholar] [CrossRef]
- Candiello, E.; Reato, G.; Verginelli, F.; Gambardella, G.; Ambrosio, D.A.; Calandra, N.; Orzan, F.; Iuliano, A.; Albano, R.; Sassi, F.; et al. MicroRNA 483-3p overexpression unleashes invasive growth of metastatic colorectal cancer via NDRG1 downregulation and ensuing activation of the ERBB3/AKT axis. Mol. Oncol. 2023, 17, 1280–1301. [Google Scholar] [CrossRef] [PubMed]
- Pathak, S.; Meng, W.J.; Sriramulu, S.; Jothimani, G.; Jangamreddy, J.R.; Banerjee, A.; Ganesan, A.T.; Adell, G.; Zhang, X.; Sun Zhang, A.; et al. Association of microRNA-652 expression with radiation response of colorectal cancer: A study from rectal cancer patients in a Swedish trial of preoperative radiotherapy. Curr. Gene Ther. 2023. ahead of print. [Google Scholar] [CrossRef]
- Zhao, D.Y.; Yin, T.F.; Sun, X.Z.; Zhou, Y.C.; Wang, Q.Q.; Zhou, G.Y.; Yao, S.K. microRNA-627-5p inhibits colorectal cancer cell proliferation, migration and invasion by targeting Wnt2. World J. Gastrointest. Oncol. 2023, 15, 318–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Y.; Lin, Z.; Cai, L. Association between miRNA-146a polymorphism and lung cancer susceptibility: A meta-analysis involving 6506 cases and 6576 controls. Gene 2020, 757, 144940. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Huang, Z.J.; Li, J.P.; Gao, X.R. rs3735664 Polymorphism Affecting ELFN1-AS1 Adsorption on miR-1231 is Associated with Colorectal Cancer Susceptibility and Tumor Stage. Biomed. Environ. Sci. 2023, 36, 381–383. [Google Scholar]
- Jazdzewski, K.; Murray, E.L.; Franssila, K.; Jarzab, B.; Schoenberg, D.R.; de la Chapelle, A. Common SNP in pre-miR-146a decreases mature miR expression and predisposes to papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 2008, 105, 7269–7274. [Google Scholar] [CrossRef]
- Santos, J.S.D.; Zunta, G.L.; Negrini, A.B.; Ribeiro, M.S.G.; Martinez, C.A.R.; Ribeiro, M.L.; Lourenco, G.J.; Ortega, M.M. The association of a single-nucleotide variant in the microRNA-146a with advanced colorectal cancer prognosis. Tumour Biol. 2020, 42, 1010428320923856. [Google Scholar] [CrossRef]
- Hezova, R.; Kovarikova, A.; Bienertova-Vasku, J.; Sachlova, M.; Redova, M.; Vasku, A.; Svoboda, M.; Radova, L.; Kiss, I.; Vyzula, R.; et al. Evaluation of SNPs in miR-196-a2, miR-27a and miR-146a as risk factors of colorectal cancer. World J. Gastroenterol. 2012, 18, 2827–2831. [Google Scholar] [CrossRef]
- Vinci, S.; Gelmini, S.; Mancini, I.; Malentacchi, F.; Pazzagli, M.; Beltrami, C.; Pinzani, P.; Orlando, C. Genetic and epigenetic factors in regulation of microRNA in colorectal cancers. Methods 2013, 59, 138–146. [Google Scholar] [CrossRef]
- Chayeb, V.; Mahjoub, S.; Zitouni, H.; Jrah-Harzallah, H.; Zouari, K.; Letaief, R.; Mahjoub, T. Contribution of microRNA-149, microRNA-146a, and microRNA-196a2 SNPs in colorectal cancer risk and clinicopathological features in Tunisia. Gene 2018, 666, 100–107. [Google Scholar] [CrossRef]
- Parlayan, C.; Ikeda, S.; Sato, N.; Sawabe, M.; Muramatsu, M.; Arai, T. Association analysis of single nucleotide polymorphisms in miR-146a and miR-196a2 on the prevalence of cancer in elderly Japanese: A case-control study. Asian Pac. J. Cancer Prev. 2014, 15, 2101–2107. [Google Scholar] [CrossRef]
- Ying, H.Q.; Peng, H.X.; He, B.S.; Pan, Y.Q.; Wang, F.; Sun, H.L.; Liu, X.; Chen, J.; Lin, K.; Wang, S.K. MiR-608, pre-miR-124-1 and pre-miR26a-1 polymorphisms modify susceptibility and recurrence-free survival in surgically resected CRC individuals. Oncotarget 2016, 7, 75865–75873. [Google Scholar] [CrossRef]
- Lindor, N.M.; Larson, M.C.; DeRycke, M.S.; McDonnell, S.K.; Baheti, S.; Fogarty, Z.C.; Win, A.K.; Potter, J.D.; Buchanan, D.D.; Clendenning, M.; et al. Germline miRNA DNA variants and the risk of colorectal cancer by subtype. Genes Chromosomes Cancer 2017, 56, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, S.; Tang, W.; Qiu, Z. Lack of association between miR-146a rs2910164 C/G locus and colorectal cancer: From a case-control study to a meta-analysis. Biosci. Rep. 2021, 41, BSR20191729. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Li, Y.; Jing, F.; Cai, S.; Zhang, Z.; Li, Q.; Ma, X.; Wang, J.; Jin, M.; Chen, K. Association of a genetic variant in microRNA-146a with risk of colorectal cancer: A population-based case-control study. Tumour Biol. 2014, 35, 6961–6967. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhu, Z.; Zhang, S. miR-146a rs2910164 polymorphism and the risk of colorectal cancer in Chinese population. J. Cancer Res. Ther. 2018, 14, S97–S99. [Google Scholar] [CrossRef] [PubMed]
- Min, K.T.; Kim, J.W.; Jeon, Y.J.; Jang, M.J.; Chong, S.Y.; Oh, D.; Kim, N.K. Association of the miR-146aC>G, 149C>T, 196a2C>T, and 499A>G polymorphisms with colorectal cancer in the Korean population. Mol. Carcinog. 2012, 51 (Suppl. S1), E65–E73. [Google Scholar] [CrossRef]
- Dikaiakos, P.; Gazouli, M.; Rizos, S.; Zografos, G.; Theodoropoulos, G.E. Evaluation of genetic variants in miRNAs in patients with colorectal cancer. Cancer Biomark. 2015, 15, 157–162. [Google Scholar] [CrossRef]
- Chae, Y.S.; Kim, J.G.; Lee, S.J.; Kang, B.W.; Lee, Y.J.; Park, J.Y.; Jeon, H.S.; Park, J.S.; Choi, G.S. A miR-146a polymorphism (rs2910164) predicts risk of and survival from colorectal cancer. Anticancer Res. 2013, 33, 3233–3239. [Google Scholar]
- Kupcinskas, J.; Bruzaite, I.; Juzenas, S.; Gyvyte, U.; Jonaitis, L.; Kiudelis, G.; Skieceviciene, J.; Leja, M.; Pauzas, H.; Tamelis, A.; et al. Lack of association between miR-27a, miR-146a, miR-196a-2, miR-492 and miR-608 gene polymorphisms and colorectal cancer. Sci. Rep. 2014, 4, 5993. [Google Scholar] [CrossRef]
- Ma, L.; Zhu, L.; Gu, D.; Chu, H.; Tong, N.; Chen, J.; Zhang, Z.; Wang, M. A genetic variant in miR-146a modifies colorectal cancer susceptibility in a Chinese population. Arch. Toxicol. 2013, 87, 825–833. [Google Scholar] [CrossRef]
- Lv, M.; Dong, W.; Li, L.; Zhang, L.; Su, X.; Wang, L.; Gao, L.; Zhang, L. Association between genetic variants in pre-miRNA and colorectal cancer risk in a Chinese population. J. Cancer Res. Clin. Oncol. 2013, 139, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.F.; Chen, L.H.; Chen, Z.X.; Yuan, Y.W.; Xie, G.Z.; Sun, A.M.; Liu, Y. A functional variant in microRNA-196a2 is associated with susceptibility of colorectal cancer in a Chinese population. Arch. Med. Res. 2011, 42, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Chu, H.; Gu, D.; Ma, L.; Shi, D.; Zhong, D.; Tong, N.; Zhang, Z.; Wang, M. A functional polymorphism in miRNA-196a2 is associated with colorectal cancer risk in a Chinese population. DNA Cell Biol. 2012, 31, 350–354. [Google Scholar] [CrossRef]
- Haerian, M.S.; Haerian, B.S.; Molanaei, S.; Kosari, F.; Sabeti, S.; Bidari-Zerehpoosh, F.; Abdolali, E. MIR196A2 rs11614913 contributes to susceptibility to colorectal cancer in Iranian population: A multi-center case-control study and meta-analysis. Gene 2018, 669, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Fawzy, M.S.; Mohammed, E.A.; Hussein, M.H.; El-Labban, M.M. MicroRNA-196a2 Biomarker and Targetome Network Analysis in Solid Tumors. Mol. Diagn. Ther. 2016, 20, 559–577. [Google Scholar] [CrossRef]
- Chen, H.; Sun, L.Y.; Chen, L.L.; Zheng, H.Q.; Zhang, Q.F. A variant in microRNA-196a2 is not associated with susceptibility to and progression of colorectal cancer in Chinese. Intern. Med. J. 2012, 42, e115–e119. [Google Scholar] [CrossRef]
- Soltanian, A.R.; Hosseini, B.; Mahjub, H.; Bahreini, F.; Nazemalhosseini Mojarad, E.; Ghaffari, M.E. Association between rs11614913 Polymorphism of The MiR-196-a2 Gene and Colorectal Cancer in The Presence of Departure from Hardy-Weinberg Equilibrium. Cell J. 2021, 23, 313–318. [Google Scholar]
- Pao, J.B.; Lu, T.L.; Ting, W.C.; Chen, L.M.; Bao, B.Y. Association of Genetic Variants of Small Non-Coding RNAs with Survival in Colorectal Cancer. Int. J. Med. Sci. 2018, 15, 217–222. [Google Scholar] [CrossRef]
- Jang, M.J.; Kim, J.W.; Min, K.T.; Jeon, Y.J.; Oh, D.; Kim, N.K. Prognostic significance of microRNA gene polymorphisms in patients with surgically resected colorectal cancer. Exp. Ther. Med. 2011, 2, 1127–1132. [Google Scholar] [CrossRef]
- Yang, M.D.; Lin, K.C.; Lu, M.C.; Jeng, L.B.; Hsiao, C.L.; Yueh, T.C.; Fu, C.K.; Li, H.T.; Yen, S.T.; Lin, C.W.; et al. Contribution of matrix metalloproteinases-1 genotypes to gastric cancer susceptibility in Taiwan. Biomedicine 2017, 7, 10. [Google Scholar] [CrossRef]
- Tsai, C.W.; Chang, W.S.; Xu, Y.; Huang, M.; Bau, D.T.; Gu, J. Associations of genetically predicted circulating insulin-like growth factor-1 and insulin-like growth factor binding protein-3 with bladder cancer risk. Mol. Carcinog. 2021, 60, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.S.; Chang, W.S.; Hsu, P.C.; Chen, C.C.; Chin, Y.T.; Huang, T.L.; Hsu, Y.N.; Kuo, C.C.; Wang, Y.C.; Tsai, C.W.; et al. Significant Association Between the MiR146a Genotypes and Susceptibility to Childhood Acute Lymphoblastic Leukemia in Taiwan. Cancer Genom. Proteom. 2020, 17, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.; Hsu, P.C.; Shih, L.C.; Hsu, Y.N.; Kuo, C.C.; Chao, C.Y.; Chang, W.S.; Tsai, C.W.; Bau, D.T.; Pei, J.S. MiR-196a-2 Genotypes Determine the Susceptibility and Early Onset of Childhood Acute Lymphoblastic Leukemia. Anticancer Res. 2020, 40, 4465–4469. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Characteristic | Controls, n = 362 | Cases, n = 362 | p-Value a | ||
|---|---|---|---|---|---|
| n | % | n | % | ||
| Age (years) | |||||
| ≤60 | 95 | 26.2% | 95 | 26.2% | 1.0000 |
| >60 | 267 | 73.8% | 267 | 73.8% | |
| Gender | |||||
| Male | 203 | 56.1% | 203 | 56.1% | 1.0000 |
| Female | 159 | 43.6% | 159 | 43.9% | |
| Smoking | |||||
| Yes | 84 | 23.2% | 91 | 25.1% | 0.5434 |
| No | 278 | 76.8% | 271 | 74.9% | |
| Alcohol drinking | |||||
| Yes | 51 | 14.1% | 44 | 12.2% | 0.4410 |
| No | 311 | 85.9% | 318 | 87.8% | |
| BMI | |||||
| <24 | 175 | 48.3% | 193 | 53.3% | 0.1809 |
| ≥24 | 187 | 51.7% | 169 | 46.7% | |
| Tumor size (cm) | |||||
| <5 | 195 | 53.9% | |||
| ≥5 | 167 | 46.1% | |||
| Location | |||||
| Colon | 257 | 71.0% | |||
| Rectum | 105 | 29.0% | |||
| Lymph node involvement | |||||
| Negative | 210 | 58.0% | |||
| Positive | 152 | 42.0% | |||
| Stage | |||||
| Stage 1 | 94 | 26.0% | |||
| Stage 2 | 72 | 19.9% | |||
| Stage 3 | 134 | 37.0% | |||
| Stage 4 | 62 | 17.1% | |||
| SNP | Genotype | Cases | Controls | p-Value | OR (95%CI) |
|---|---|---|---|---|---|
| rs11614913 | TT | 89 (24.6%) | 117 (32.3%) | 1.00 (Ref) | |
| CT | 163 (45.0%) | 174 (48.1%) | 0.2792 | 1.23 (0.87–1.75) | |
| CC | 110 (30.4%) | 71 (19.6%) | 0.0008 * | 2.04 (1.36–3.06) | |
| Ptrend | 0.0019 * | ||||
| CT + CC | 273 (75.4%) | 245 (67.7%) | 0.0261 * | 1.46 (1.06–2.03) | |
| PHWE | 0.6631 |
| SNP | Genotype | Cases | Controls | p-Value | OR (95%CI) |
|---|---|---|---|---|---|
| rs2910164 | CC | 124 (34.3%) | 119 (32.9%) | 1.00 (Ref) | |
| CG | 160 (44.2%) | 164 (45.3%) | 0.7618 | 0.94 (0.67–1.31) | |
| GG | 78 (21.5%) | 79 (21.8%) | 0.8723 | 0.95 (0.63–1.42) | |
| Ptrend | 0.9237 | ||||
| CG + GG | 238 (65.7%) | 243 (67.1%) | 0.7529 | 0.94 (0.69–1.28) | |
| PHWE | 0.1155 |
| Allele | Cases | Controls | p-Value | OR (95%CI) |
|---|---|---|---|---|
| mir146a rs2910164 | ||||
| C | 408 (56.4%) | 402 (55.5%) | 1.00 (Ref) | |
| G | 316 (43.6%) | 322 (45.5%) | 0.7913 | 0.97 (0.79–1.19) |
| mir196a-2 rs11614913 | ||||
| T | 341 (47.1%) | 408 (56.4%) | 1.00 (Ref) | |
| C | 383 (52.9%) | 316 (43.6%) | 0.0005 * | 1.45 (1.18–1.78) |
| Characteristics | Controls | Cases | OR (95% CI) a | aOR (95% CI) b | p-Value c |
|---|---|---|---|---|---|
| Age | |||||
| ≤60 years old | |||||
| TT | 32 | 23 | 1.00 (ref) | 1.00 (ref) | |
| CT | 48 | 43 | 1.25 (0.63–2.45) | 1.21 (0.67–2.39) | 0.6400 |
| CC | 15 | 29 | 2.69 (1.18–6.12) | 2.77 (1.23–4.98) | 0.0291 * |
| >60 years old | |||||
| TT | 85 | 66 | 1.00 (ref) | 1.00 (ref) | |
| CT | 126 | 120 | 1.23 (0.82–1.84) | 1.33 (0.79–2.11) | 0.3791 |
| CC | 56 | 81 | 1.86 (1.17–2.98) | 2.04 (1.24–3.58) | 0.0126 * |
| Gender | |||||
| Males | |||||
| TT | 69 | 52 | 1.00 (ref) | 1.00 (ref) | |
| CT | 94 | 89 | 1.26 (0.79–1.99) | 1.19 (0.81–1.86) | 0.3948 |
| CC | 40 | 62 | 2.06 (1.20–3.52) | 2.28 (1.19–3.23) | 0.0119 * |
| Females | |||||
| TT | 48 | 37 | 1.00 (ref) | 1.00 (ref) | |
| CT | 80 | 74 | 1.20 (0.70–2.04) | 1.19 (0.74–2.17) | 0.5922 |
| CC | 31 | 48 | 2.01 (1.08–3.74) | 2.04 (1.12–3.88) | 0.0404 * |
| Smoking behaviors | |||||
| Non-smokers | |||||
| TT | 91 | 68 | 1.00 (ref) | 1.00 (ref) | |
| CT | 133 | 122 | 1.23 (0.82–1.83) | 1.31 (0.92–2.53) | 0.3646 |
| CC | 54 | 81 | 2.01 (1.26–3.20) | 2.27 (1.23–2.97) | 0.0047 * |
| Smokers | |||||
| TT | 26 | 21 | 1.00 (ref) | 1.00 (ref) | |
| CT | 41 | 41 | 1.24 (0.60–2.54) | 1.27 (0.58–2.49) | 0.6900 |
| CC | 17 | 29 | 2.11 (0.92–4.84) | 2.16 (0.97–4.38) | 0.1169 |
| Alcohol drinking behaviors | |||||
| Non-drinkers | |||||
| TT | 99 | 78 | 1.00 (ref) | 1.00 (ref) | |
| CT | 150 | 143 | 1.21 (0.83–1.76) | 1.28 (0.85–2.04) | 0.3672 |
| CC | 62 | 97 | 1.99 (1.28–3.07) | 2.04 (1.17–2.89) | 0.0028 * |
| Drinkers | |||||
| TT | 18 | 11 | 1.00 (ref) | 1.00 (ref) | |
| CT | 24 | 20 | 1.36 (0.52–3.55) | 1.29 (0.48–3.42) | 0.6933 |
| CC | 9 | 13 | 2.36 (0.76–7.34) | 2.24 (0.74–6.93) | 0.2239 |
| BMI | |||||
| <24 | |||||
| TT | 57 | 49 | 1.00 (ref) | 1.00 (ref) | |
| CT | 81 | 86 | 1.24 (0.76–2.01) | 1.22 (0.73–1.98) | 0.4686 |
| CC | 37 | 58 | 1.82 (1.04–3.20) | 1.84 (1.09–3.35) | 0.0498 * |
| ≥24 | |||||
| TT | 60 | 40 | 1.00 (ref) | 1.00 (ref) | |
| CT | 93 | 77 | 1.24 (0.75–2.05) | 1.21 (0.72–2.34) | 0.4712 |
| CC | 34 | 52 | 2.29 (1.27–4.13) | 2.38 (1.34–3.97) | 0.0084 * |
| First Author | Year | Ethnicity | TT, CT, CC Genotype # of the Controls | TT, CT, CC Genotype # of the Cases | OR (95%CI) a | p-Value b | Ref # |
|---|---|---|---|---|---|---|---|
| Chen | 2011 | Chinese | 107:206:94 | 35:64:27 | 0.94 (0.71–1.25) | 0.715 | [47] |
| Zhan | 2011 | Chinese | 163:267:113 | 56:128:68 | 1.32 (1.07–1.64) | 0.011 | [43] |
| Min | 2012 | Korean | 148:254:100 | 125:201:120 | 1.19 (0.99–1.42) | 0.073 | [37] |
| Hezova | 2012 | European | 22:103:87 | 26:89:82 | 0.95 (0.71–1.27) | 0.811 | [28] |
| Vinci | 2013 | European | 11:84:83 | 12:86:62 | 0.81 (0.59–1.12) | 0.231 | [29] |
| Lv | 2013 | Chinese | 91:331:109 | 114:223:10 | 0.50 (0.41–0.61) | <0.001 | [42] |
| Kupcinskas | 2014 | European | 54:174:199 | 27:87:79 | 0.86 (0.67–1.10) | 0.254 | [40] |
| Parlayan | 2014 | Japanese | 390:679:282 | 34:59:23 | 0.97 (0.74–1.27) | 0.881 | [31] |
| Dikaiakos | 2015 | European | 117:149:33 | 69:69:19 | 0.92 (0.69–1.23) | 0.624 | [38] |
| Chayeb | 2018 | Tunisian | 29:85:47 | 31:82:39 | 0.89 (0.65–1.22) | 0.508 | [30] |
| Haerian | 2018 | Iranian | 187:551:505 | 262:196:449 | 0.90 (0.80–1.02) | 0.105 | [45] |
| Soltanian | 2021 | Iranian | 56:122:108 | 29:91:74 | 1.11 (0.85–1.45) | 0.477 | [48] |
| Yueh | 2023 | Taiwanese | 117:174:71 | 89:163:110 | 1.45 (1.18–1.78) | 0.001 | current |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yueh, T.-C.; Wang, Y.-C.; Chin, Y.-T.; Hung, Y.-C.; Mong, M.-C.; Yang, Y.-C.; Pei, J.-S.; Gu, J.; Tsai, C.-W.; Bau, D.-T.; et al. Impact of Mir196a-2 Genotypes on Colorectal Cancer Risk in Taiwan. Int. J. Mol. Sci. 2023, 24, 11613. https://doi.org/10.3390/ijms241411613
Yueh T-C, Wang Y-C, Chin Y-T, Hung Y-C, Mong M-C, Yang Y-C, Pei J-S, Gu J, Tsai C-W, Bau D-T, et al. Impact of Mir196a-2 Genotypes on Colorectal Cancer Risk in Taiwan. International Journal of Molecular Sciences. 2023; 24(14):11613. https://doi.org/10.3390/ijms241411613
Chicago/Turabian StyleYueh, Te-Cheng, Yun-Chi Wang, Yu-Ting Chin, Yi-Chih Hung, Mei-Chin Mong, Ya-Chen Yang, Jen-Sheng Pei, Jian Gu, Chia-Wen Tsai, Da-Tian Bau, and et al. 2023. "Impact of Mir196a-2 Genotypes on Colorectal Cancer Risk in Taiwan" International Journal of Molecular Sciences 24, no. 14: 11613. https://doi.org/10.3390/ijms241411613
APA StyleYueh, T.-C., Wang, Y.-C., Chin, Y.-T., Hung, Y.-C., Mong, M.-C., Yang, Y.-C., Pei, J.-S., Gu, J., Tsai, C.-W., Bau, D.-T., & Chang, W.-S. (2023). Impact of Mir196a-2 Genotypes on Colorectal Cancer Risk in Taiwan. International Journal of Molecular Sciences, 24(14), 11613. https://doi.org/10.3390/ijms241411613