
Mechanism of Cell Death by Combined Treatment with an xCT Inhibitor and Paclitaxel: An Alternative Therapeutic Strategy for Patients with Ovarian Clear Cell Carcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of SAS on GSH Levels and ROS Accumulation in OCCC Cells
2.2. Effect of SAS on Cytotoxicity of Cisplatin or Paclitaxel in OCCC Cells
2.3. ROS Generation in OCCC Cells Treated with SAS and/or PTX
2.4. Apoptosis or Ferroptosis Underlying Cell Death Induced by Combined Treatment with PTX and SAS in OCCC Cells
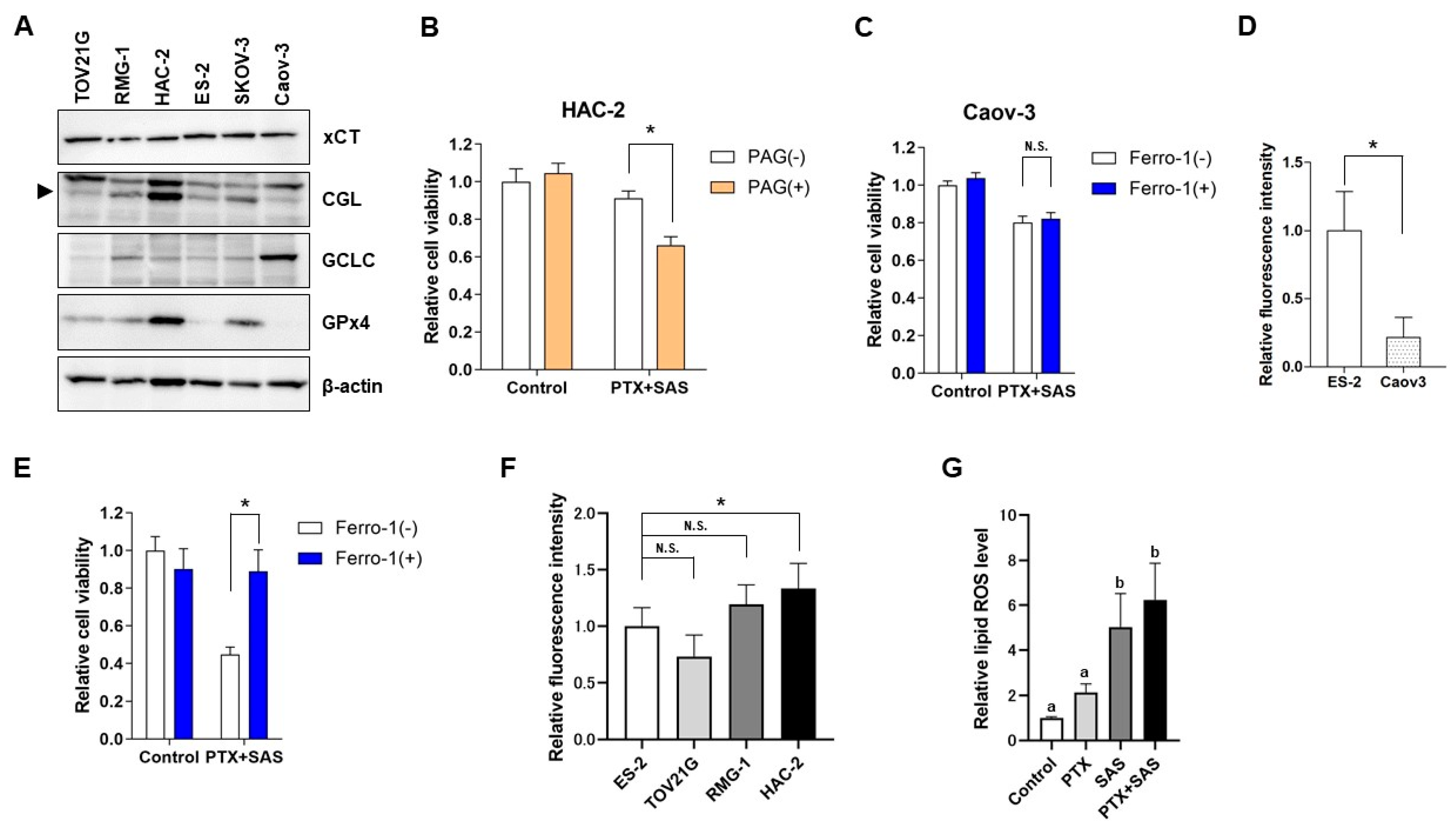
2.5. The Effect of SAS Depends on the GSH Synthesis Pathway-Related Proteins in OCCC Cells and the Association between the Intercellular Iron Concentration and Ferroptosis Induced by Combined Treatment with PTX and SAS
2.6. The Effect of Treatment with PTX and SAS on Tumor Growth Inhibition
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Antibodies and Reagents
4.3. Cell Viability Assays
4.4. Glutathione Analysis
4.5. ROS and Lipid ROS Measurements
4.6. Immunoblotting Analysis
4.7. Detection of Intracellular Fe2+
4.8. Animal Experiments
4.9. Immunofluorescent Staining
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aure, J.C.; Hoeg, K.; Kolstad, K. Clinical and histologic studies of ovarian carcinoma. Long-term follow-up of 990 cases. Obstet. Gynecol. 1971, 37, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nagase, S.; Ohta, T.; Takahashi, F.; Yamagami, W.; Yaegashi, N. Board Members of the 2020 Committee on Gynecologic Oncology of the Japan Society of Obstetrics and Gynecology. Annual report of the Committee on Gynecologic Oncology, the Japan Society of Obstetrics and Gynecology: Annual Patient Report for 2018 and Annual Treatment Report for 2013. J. Obstet. Gynaecol. Res. 2022, 48, 541–552. [Google Scholar] [PubMed]
- Sugiyama, T.; Kamura, T.; Kigawa, J.; Kikuchi, Y.; Kita, T.; Suzuki, M.; Sato, I.; Taguchi, K. Clinical characteristics of clear cell carcinoma of the ovary: A distinct histologic type with poor prognosis and resistance to platinum-based chemotherapy. Cancer 2020, 88, 2484–2589. [Google Scholar] [CrossRef]
- Chan, J.K.; Teoh, D.; Hu, J.M.; Shin, J.Y.; Osann, K.; Kapp, D.S. Do clear cell ovarian carcinomas have poorer prognosis com-pared to other epithelial cell types? A study of 1411 clear cell ovarian cancers. Gynecol. Oncol. 2008, 109, 370–376. [Google Scholar] [PubMed]
- Itamochi, H.; Kigawa, J.; Sugiyama, T.; Kikuchi, Y.; Suzuki, M.; Terakawa, N. Low proliferation activity may be associated with chemoresistance in clear cell carcinoma of the ovary. Obstet. Gynecol. 2002, 100, 281–287. [Google Scholar]
- Jones, S.; Li, M.; Parsons, D.W.; Zhang, X.; Wesseling, J.; Kristel, P.; Schmidt, M.K.; Markowitz, S.; Yan, H.; Bigner, D.; et al. Somatic mutations in the chromatin remodeling gene ARID1A occur in several tumor types. Hum. Mutat. 2012, 33, 100–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell. 2019, 35, 177–190. [Google Scholar] [CrossRef] [Green Version]
- Seino, M.; Ohta, T.; Sugiyama, A.; Sakaki, H.; Sudo, T.; Tsutsumi, S.; Shigeta, S.; Tokunaga, H.; Toyoshima, M.; Yaegashi, N.; et al. Metabolomic analysis of uterine serous carcinoma with acquired resistance to paclitaxel. Oncotarget 2018, 9, 31985–31998. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, H.; Sato, H.; Kuriyama-Matsumura, K.; Sato, K.; Maebara, K.; Wang, H.; Tamba, M.; Itoh, K.; Yamamoto, M.; Bannai, S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J. Biol. Chem. 2002, 277, 44765–44771. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, M.; Tsuchihashi, K.; Ishimoto, T.; Yae, T.; Motohara, T.; Sugihara, E.; Onishi, N.; Masuko, T.; Yoshizawa, K.; Kawashiri, S.; et al. xCT inhibition depletes CD44v-expressing tumor cells that are resistant to EGFR-targeted therapy in head and neck squamous cell carcinoma. Cancer Res. 2013, 73, 1855–1866. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.J.; Zhang, Y.; Zeng, Z.L.; Li, X.B.; Hu, K.S.; Luo, H.Y.; Yang, J.; Huang, P.; Xu, R.H. β-phenylethyl isothiocyanate re-verses platinum resistance by a GSH-dependent mechanism in cancer cells with epithelial-mesenchymal transition pheno-type. Biochem. Pharmacol. 2013, 85, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.Z.; Chen, G.; Wang, P.; Lu, W.H.; Zhu, C.F.; Song, M.; Yang, J.; Wen, S.; Xu, R.H.; Hu, Y.; et al. Xc- inhibitor sulfasala-zine sensitizes colorectal cancer to cisplatin by a GSH-dependent mechanism. Cancer Lett. 2015, 368, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Zenlea, T.; Peppercorn, M.A. Immunosuppressive therapies for inflammatory bowel disease. World J. Gastroenterol. 2014, 20, 3146–3152. [Google Scholar] [CrossRef] [PubMed]
- Lo, M.; Wang, Y.Z.; Gout, P.W. The x(c)- cystine/glutamate antiporter: A potential target for therapy of cancer and other dis-eases. J. Cell. Physiol. 2018, 215, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Sendo, K.; Seino, M.; Ohta, T.; Nagase, S. Impact of the glutathione synthesis pathway on sulfasalazine-treated endometrial cancer. Oncotarget 2022, 13, 224–236. [Google Scholar] [CrossRef]
- Sehm, T.; Rauh, M.; Wiendieck, K.; Buchfelder, M.; Eyupoglu, I.Y.; Savaskan, N.E. Temozolomide toxicity operates in a xCT/SLC7a11 dependent manner and is fostered by ferroptosis. Oncotarget 2016, 7, 74630–74647. [Google Scholar] [CrossRef]
- Roh, J.L.; Kim, E.H.; Jang, H.J.; Park, J.Y.; Shin, D. Induction of ferroptotic cell death for overcoming cisplatin resistance of head and neck cancer. Cancer Lett. 2016, 381, 96–103. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, R.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, H.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–370. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, A.; Ohta, T.; Obata, M.; Takahashi, K.; Seino, M.; Nagase, S. xCT inhibitor sulfasalazine depletes paclitaxel resistant tumor cells through ferroptosis in uterine serous carcinoma. Oncol. Lett. 2020, 20, 2689–2700. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic func-tions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [Green Version]
- Weaver, B.A. How Taxol/paclitaxel kills cancer cells. Mol. Biol. Cell. 2014, 25, 2677–2681. [Google Scholar] [CrossRef]
- Rosado, J.O.; Salvador, M.; Bonatto, D. Importance of the trans-sulfuration pathway in cancer prevention and promotion. Mol. Cell. Biochem. 2007, 301, 1–12. [Google Scholar] [CrossRef]
- Kandil, S.; Brennan, L.; McBean, G.J. Glutathione depletion causes a JNK and p38MAPK-mediated increase in expression of cystathionine-gamma-lyase and upregulation of the transsulfuration pathway in C6 glioma cells. Neurochem. Int. 2010, 56, 611–619. [Google Scholar] [CrossRef]
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2016, 23, 270–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, S.; Liang, B.; Huang, Q.; Dong, S.; Wu, Z.; He, W.; Shi, M. Metabolic networks in ferroptosis. Oncol. Lett. 2018, 15, 5405–5411. [Google Scholar]
- Kim, J.; Hong, S.J.; Park, J.H.; Park, S.Y.; Kim, S.W.; Cho, E.Y.; Do, I.G.; Joh, J.W.; Kim, D.S. Expression of cystathionine be-ta-synthase is downregulated in hepatocellular carcinoma and associated with poor prognosis. Oncol. Rep. 2009, 21, 1449–1454. [Google Scholar] [CrossRef]
- Zhao, H.; Li, Q.; Wang, J.; Su, X.; Ng, K.M.; Qju, T.; Shan, L.; Ling, Y.; Wang, L.; Cai, J.; et al. Frequent epigenetic silencing of the folate-metabolising gene cystathionine-beta-synthase in gastrointestinal cancer. PLoS ONE 2012, 7, e49683. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhao, Y.; Zhang, Z.; Tan, N.; Zhao, F.; Ge, C.; Liang, L.; Jia, D.; Chen, T.; Yao, M.; et al. Disruption of xCT inhibits cell growth via the ROS/autophagy pathway in hepatocellular carcinoma. Cancer Lett. 2011, 312, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Verschoor, M.L.; Singh, G. Ets-1 regulated intracellular glutathione levels: Key target for resistant ovarian cancer. Mol. Cancer 2013, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Fan, X.; Zhao, M.; Zhu, P. The Relationship between Ferroptosis and Tumors: A Novel Landscape for Therapeutic Approach. Curr. Gene Ther. 2019, 19, 117–124. [Google Scholar] [CrossRef]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, K.; Mandai, M.; Toyokuni, S.; Hamanishi, J.; Higuchi, T.; Takakura, K.; Fujii, S. Contents of endometriotic cysts, especially the high concentration of free iron, are a possible cause of carcinogenesis in the cysts through the iron-induced persistent oxidative stress. Clin. Cancer Res. 2008, 14, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Robe, P.A.; Martin, D.H.; Nguyen-Khac, M.T.; Artesi, M.; Deprez, M.; Albert, A.; Vanbelle, S.; Califice, S.; Bredel, M.; Bours, V. Early termination of ISRCTN45828668, a phase 1/2 prospective, randomized study of sulfasalazine for the treatment of pro-gressing malignant gliomas in adults. BMC Cancer 2009, 9, 372. [Google Scholar] [CrossRef] [Green Version]
- Otsubo, K.; Nosaki, K.; Imamura, C.K.; Ogata, H.; Fujita, A.; Sakata, S.; Hirai, F.; Toyokawa, G.; Iwama, E.; Harada, T.; et al. Phase I study of salazosulfapyridine in combination with cisplatin and pemetrexed for advanced non-small-cell lung cancer. Cancer Sci. 2017, 108, 1843–1849. [Google Scholar] [CrossRef] [Green Version]
- Sandborn, W.J.; Feagan, B.G. Review article: Mild to moderate Crohn's disease defining the basis for a new treatment algorithm. Aliment. Pharmacol. Ther. 2003, 18, 263–277. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idei, U.; Ohta, T.; Yamatani, H.; Seino, M.; Nagase, S. Mechanism of Cell Death by Combined Treatment with an xCT Inhibitor and Paclitaxel: An Alternative Therapeutic Strategy for Patients with Ovarian Clear Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 11781. https://doi.org/10.3390/ijms241411781
Idei U, Ohta T, Yamatani H, Seino M, Nagase S. Mechanism of Cell Death by Combined Treatment with an xCT Inhibitor and Paclitaxel: An Alternative Therapeutic Strategy for Patients with Ovarian Clear Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(14):11781. https://doi.org/10.3390/ijms241411781
Chicago/Turabian StyleIdei, Urara, Tsuyoshi Ohta, Hizuru Yamatani, Manabu Seino, and Satoru Nagase. 2023. "Mechanism of Cell Death by Combined Treatment with an xCT Inhibitor and Paclitaxel: An Alternative Therapeutic Strategy for Patients with Ovarian Clear Cell Carcinoma" International Journal of Molecular Sciences 24, no. 14: 11781. https://doi.org/10.3390/ijms241411781