
Electrical and Structural Insights into Right Ventricular Outflow Tract Arrhythmogenesis

Abstract
:1. Introduction
2. Predisposing Factors of RV Arrhythmogenesis
2.1. Anatomical and Dynamic Perceptions on RVOT Arrhythmogenesis
2.2. Embryonic Development Relates to RVOT Arrhythmogenesis
2.3. Histopathological Perceptions on RVOT Arrhythmogenesis
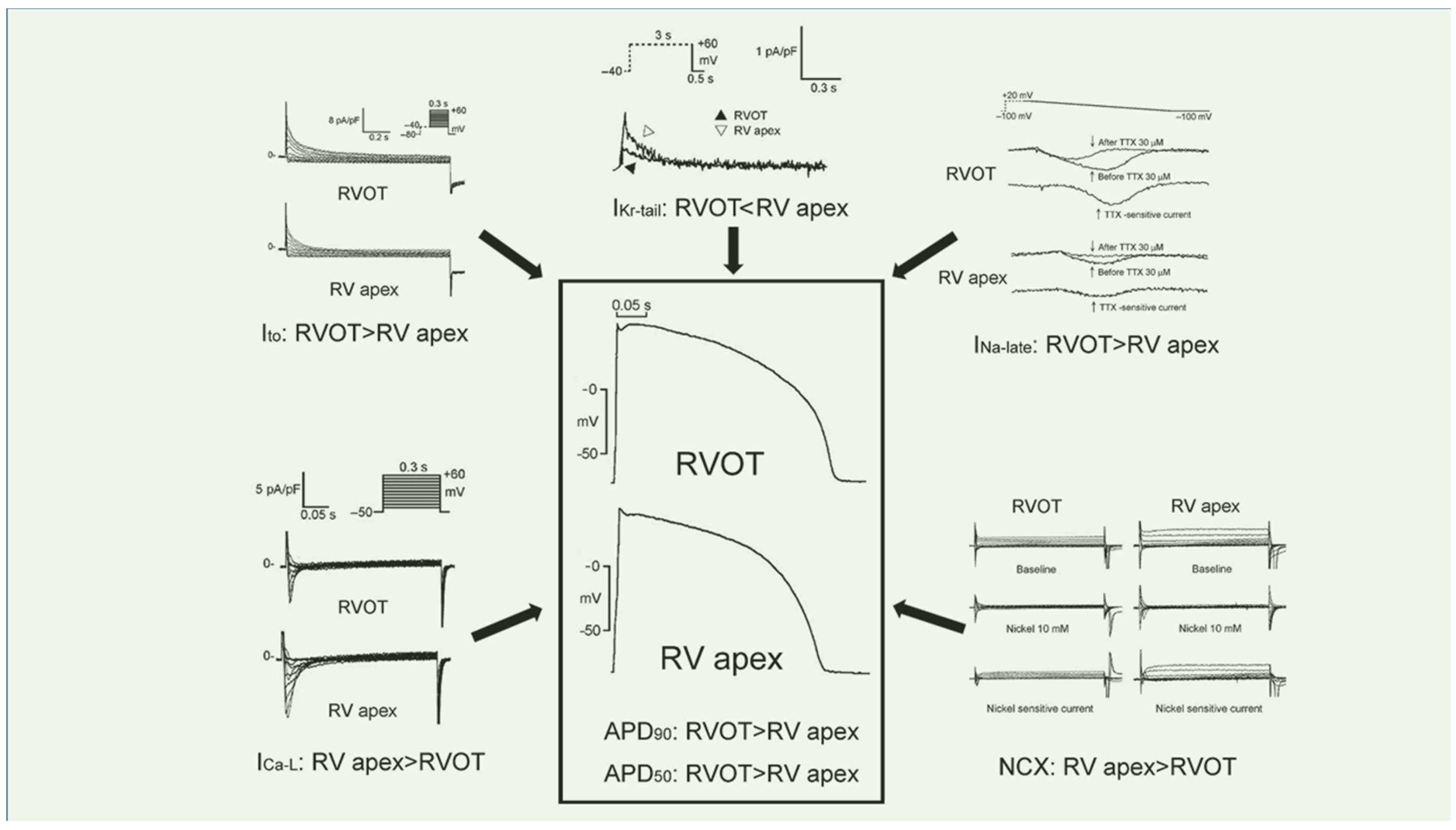
2.4. Cellular Electrophysiology Perceptions on RVOT Arrhythmogenesis
2.5. Ca2+ Homeostasis Perceptions on RVOT Arrhythmogenesis
2.6. Excitation–Contraction Coupling on RVOT Arrhythmogenesis
2.7. Role of Autonomic Nervous Activity in RVOT Arrhythmogenesis
2.8. Sex Hormones
2.9. Mutations in Various Signaling Pathways Leading to RVOT Arrhythmogenesis
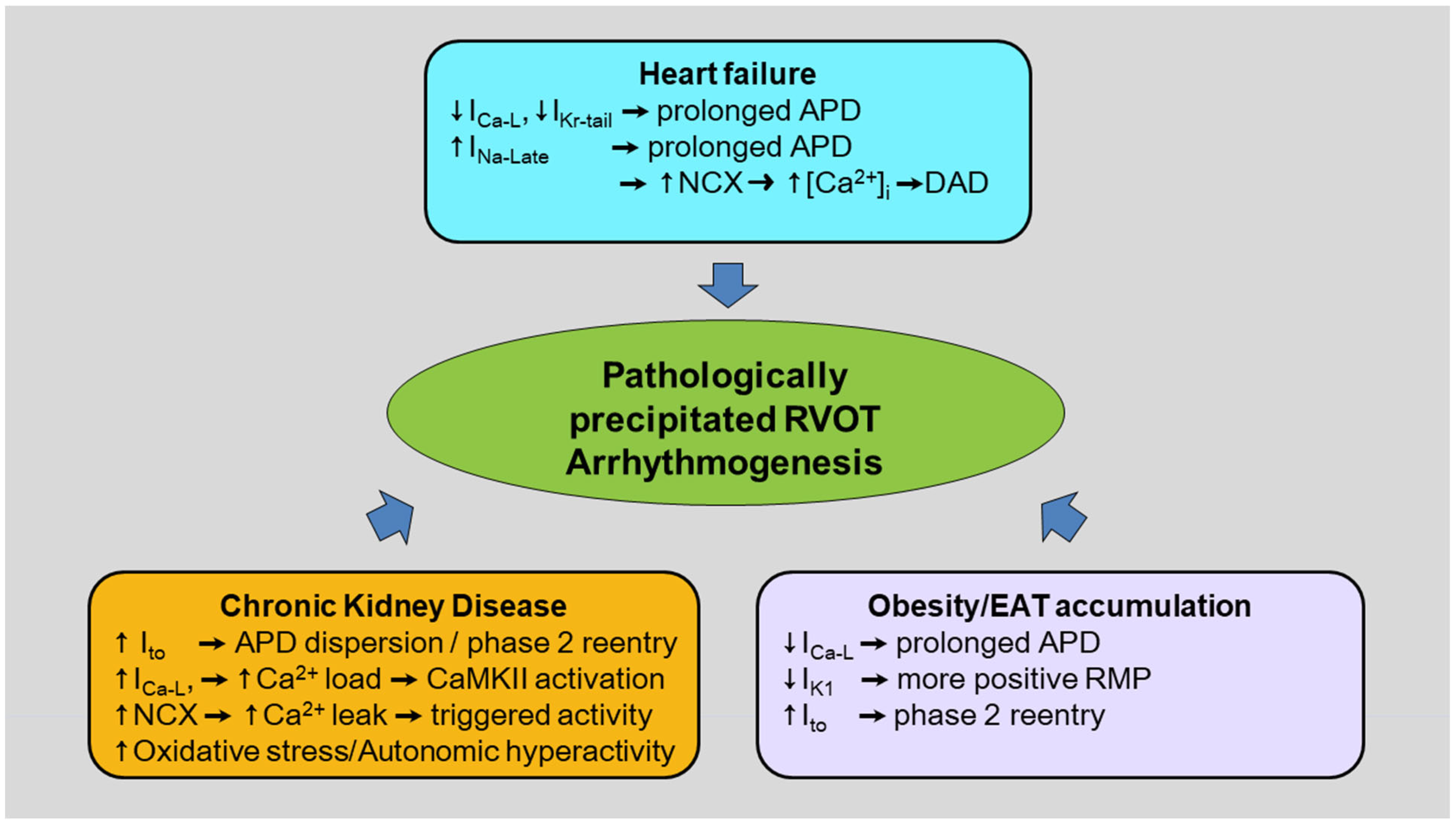
3. Pathological Conditions for RVOT Arrhythmogenesis
3.1. Obesity/Epicardial Adipose Tissue (EAT) Accumulation in the RVOT
3.2. Heart Failure (HF)
3.3. Chronic Kidney Disease (CKD)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kamakura, S.; Shimizu, W.; Matsuo, K.; Taguchi, A.; Suyama, K.; Kurita, T.; Aihara, N.; Ohe, T.; Shimomura, K. Localization of optimal ablation site of idiopathic ventricular tachycardia from right and left ventricular outflow tract by body surface ECG. Circulation 1998, 98, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, H.; Fukushima-Kusano, K.; Nagase, S.; Takenaka-Morita, S.; Nishii, N.; Kakishita, M.; Nakamura, K.; Emori, T.; Matsubara, H.; Ohe, T. Site-specific arrhythmogenesis in patients with Brugada syndrome. J. Cardiovasc. Electrophysiol. 2003, 14, 373–379. [Google Scholar] [CrossRef]
- Tsai, C.F.; Chen, S.A.; Tai, C.T.; Chiang, C.E.; Lee, S.H.; Wen, Z.C.; Huang, J.L.; Ding, Y.A.; Chang, M.S. Idiopathic monomorphic ventricular tachycardia: Clinical outcome, electrophysiologic characteristics and long-term results of catheter ablation. Int. J. Cardiol. 1997, 62, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.J.; Iwai, S.; Markowitz, S.M.; Shah, B.K.; Stein, K.M.; Lerman, B.B. Clinical and electrophysiological spectrum of idiopathic ventricular outflow tract arrhythmias. J. Am. Coll. Cardiol. 2007, 49, 2035–2043. [Google Scholar] [CrossRef] [Green Version]
- Sirichand, S.; Killu, A.M.; Padmanabhan, D.; Hodge, D.O.; Chamberlain, A.M.; Brady, P.A.; Kapa, S.; Noseworthy, P.A.; Packer, D.L.; Munger, T.M.; et al. Incidence of Idiopathic Ventricular Arrhythmias: A Population-Based Study. Circ. Arrhythm. Electrophysiol. 2017, 10, e004662. [Google Scholar] [CrossRef] [Green Version]
- Lerman, B.B.; Stein, K.M.; Markowitz, S.M.; Mittal, S.; Slotwiner, D.J. Ventricular arrhythmias in normal hearts. Cardiol. Clin. 2000, 18, 265–291. [Google Scholar] [CrossRef]
- Dixit, S.; Gerstenfeld, E.P.; Callans, D.J.; Marchlinski, F.E. Electrocardiographic patterns of superior right ventricular outflow tract tachycardias: Distinguishing septal and free-wall sites of origin. J. Cardiovasc. Electrophysiol. 2003, 14, 1–7. [Google Scholar] [CrossRef]
- Anderson, R.D.; Kumar, S.; Parameswaran, R.; Wong, G.; Voskoboinik, A.; Sugumar, H.; Watts, T.; Sparks, P.B.; Morton, J.B.; McLellan, A.; et al. Differentiating Right- and Left-Sided Outflow Tract Ventricular Arrhythmias: Classical ECG Signatures and Prediction Algorithms. Circ. Arrhythm. Electrophysiol. 2019, 12, e007392. [Google Scholar] [CrossRef]
- Mariani, M.V.; Piro, A.; Della Rocca, D.G.; Forleo, G.B.; Pothineni, N.V.; Romero, J.; Di Biase, L.; Fedele, F.; Lavalle, C. Electrocardiographic Criteria for Differentiating Left from Right Idiopathic Outflow Tract Ventricular Arrhythmias. Arrhythm. Electrophysiol. Rev. 2021, 10, 10–16. [Google Scholar] [CrossRef]
- O’Donnell, D.; Cox, D.; Bourke, J.; Mitchell, L.; Furniss, S. Clinical and electrophysiological differences between patients with arrhythmogenic right ventricular dysplasia and right ventricular outflow tract tachycardia. Eur. Heart J. 2003, 24, 801–810. [Google Scholar] [CrossRef] [Green Version]
- Buxton, A.E.; Waxman, H.L.; Marchlinski, F.E.; Simson, M.B.; Cassidy, D.; Josephson, M.E. Right ventricular tachycardia: Clinical and electrophysiologic characteristics. Circulation 1983, 68, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Calvo, N.; Jongbloed, M.; Zeppenfeld, K. Radiofrequency catheter ablation of idiopathic right ventricular outflow tract arrhythmias. Indian Pacing Electrophysiol. J. 2013, 13, 14–33. [Google Scholar] [CrossRef] [Green Version]
- Haissaguerre, M.; Extramiana, F.; Hocini, M.; Cauchemez, B.; Jais, P.; Cabrera, J.A.; Farre, J.; Leenhardt, A.; Sanders, P.; Scavee, C.; et al. Mapping and ablation of ventricular fibrillation associated with long-QT and Brugada syndromes. Circulation 2003, 108, 925–928. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.Y.; Chung, F.P.; Chen, Y.C.; Tsai, C.F.; Kao, Y.H.; Chao, T.F.; Huang, J.H.; Chen, S.A.; Chen, Y.J. Distinctive electrophysiological characteristics of right ventricular out-flow tract cardiomyocytes. J. Cell. Mol. Med. 2014, 18, 1540–1548. [Google Scholar] [CrossRef]
- Ghonim, S.; Voges, I.; Gatehouse, P.D.; Keegan, J.; Gatzoulis, M.A.; Kilner, P.J.; Babu-Narayan, S.V. Myocardial Architecture, Mechanics, and Fibrosis in Congenital Heart Disease. Front. Cardiovasc. Med. 2017, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Tandri, H.; Bluemke, D.A.; Ferrari, V.A.; Bomma, C.; Nasir, K.; Rutberg, J.; Tichnell, C.; James, C.; Lima, J.A.; Calkins, H. Findings on magnetic resonance imaging of idiopathic right ventricular outflow tachycardia. Am. J. Cardiol. 2004, 94, 1441–1445. [Google Scholar] [CrossRef]
- van Riel, A.; Systrom, D.M.; Oliveira, R.K.F.; Landzberg, M.J.; Mulder, B.J.M.; Bouma, B.J.; Maron, B.A.; Shah, A.M.; Waxman, A.B.; Opotowsky, A.R. Hemodynamic and metabolic characteristics associated with development of a right ventricular outflow tract pressure gradient during upright exercise. PLoS ONE 2017, 12, e0179053. [Google Scholar] [CrossRef] [Green Version]
- Gulan, U.; Saguner, A.M.; Akdis, D.; Gotschy, A.; Tanner, F.C.; Kozerke, S.; Manka, R.; Brunckhorst, C.; Holzner, M.; Duru, F. Hemodynamic Changes in the Right Ventricle Induced by Variations of Cardiac Output: A Possible Mechanism for Arrhythmia Occurrence in the Outflow Tract. Sci. Rep. 2019, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Theveniau-Ruissy, M.; Dandonneau, M.; Mesbah, K.; Ghez, O.; Mattei, M.G.; Miquerol, L.; Kelly, R.G. The del22q11.2 candidate gene Tbx1 controls regional outflow tract identity and coronary artery patterning. Circ. Res. 2008, 103, 142–148. [Google Scholar] [CrossRef]
- Rana, M.S.; Theveniau-Ruissy, M.; De Bono, C.; Mesbah, K.; Francou, A.; Rammah, M.; Dominguez, J.N.; Roux, M.; Laforest, B.; Anderson, R.H.; et al. Tbx1 coordinates addition of posterior second heart field progenitor cells to the arterial and venous poles of the heart. Circ. Res. 2014, 115, 790–799. [Google Scholar] [CrossRef]
- Bezzina, C.R.; Barc, J.; Mizusawa, Y.; Remme, C.A.; Gourraud, J.B.; Simonet, F.; Verkerk, A.O.; Schwartz, P.J.; Crotti, L.; Dagradi, F.; et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat. Genet. 2013, 45, 1044–1049. [Google Scholar] [CrossRef] [PubMed]
- Boukens, B.J.; Sylva, M.; de Gier-de Vries, C.; Remme, C.A.; Bezzina, C.R.; Christoffels, V.M.; Coronel, R. Reduced sodium channel function unmasks residual embryonic slow conduction in the adult right ventricular outflow tract. Circ. Res. 2013, 113, 137–141. [Google Scholar] [CrossRef]
- Jongbloed, M.R.; Mahtab, E.A.; Blom, N.A.; Schalij, M.J.; Gittenberger-de Groot, A.C. Development of the cardiac conduction system and the possible relation to predilection sites of arrhythmogenesis. Sci. World J. 2008, 8, 239–269. [Google Scholar] [CrossRef]
- Miles, C.; Westaby, J.; Ster, I.C.; Asimaki, A.; Boardman, P.; Joshi, A.; Papadakis, M.; Sharma, S.; Behr, E.R.; Sheppard, M.N. Morphometric characterization of collagen and fat in normal ventricular myocardium. Cardiovasc. Pathol. 2020, 48, 107224. [Google Scholar] [CrossRef]
- Basso, C.; Thiene, G. Adipositas cordis, fatty infiltration of the right ventricle, and arrhythmogenic right ventricular cardiomyopathy. Just a matter of fat? Cardiovasc. Pathol. 2005, 14, 37–41. [Google Scholar] [CrossRef]
- Pytkowski, M.; Maciag, A.; Sterlinski, M.; Jankowska, A.; Kowalik, I.; Farkowski, M.M.; Kuteszko, R.; Zajac, D.; Firek, B.; Chmielak, Z.; et al. Novel algorithm for arrhythmogenic focus localization in patients with right ventricular outflow tract arrhythmias. Cardiol. J. 2014, 21, 284–292. [Google Scholar] [CrossRef] [Green Version]
- Lerman, B.B. Mechanism, diagnosis, and treatment of outflow tract tachycardia. Nat. Rev. Cardiol. 2015, 12, 597–608. [Google Scholar] [CrossRef]
- Ohkubo, K.; Watanabe, I.; Okumura, Y.; Takagi, Y.; Ashino, S.; Kofune, M.; Sugimura, H.; Nakai, T.; Kasamaki, Y.; Hirayama, A.; et al. Right ventricular histological substrate and conduction delay in patients with Brugada syndrome. Int. Heart J. 2010, 51, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Coronel, R.; Casini, S.; Koopmann, T.T.; Wilms-Schopman, F.J.; Verkerk, A.O.; de Groot, J.R.; Bhuiyan, Z.; Bezzina, C.R.; Veldkamp, M.W.; Linnenbank, A.C.; et al. Right ventricular fibrosis and conduction delay in a patient with clinical signs of Brugada syndrome: A combined electrophysiological, genetic, histopathologic, and computational study. Circulation 2005, 112, 2769–2777. [Google Scholar] [CrossRef] [Green Version]
- Nademanee, K.; Raju, H.; de Noronha, S.V.; Papadakis, M.; Robinson, L.; Rothery, S.; Makita, N.; Kowase, S.; Boonmee, N.; Vitayakritsirikul, V.; et al. Fibrosis, Connexin-43, and Conduction Abnormalities in the Brugada Syndrome. J. Am. Coll. Cardiol. 2015, 66, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Ou, B.; Nakagawa, M.; Kajimoto, M.; Nobe, S.; Ooie, T.; Ichinose, M.; Yonemochi, H.; Ono, N.; Shimada, T.; Saikawa, T. Heterogeneous expression of connexin 43 in the myocardium of rabbit right ventricular outflow tract. Life Sci. 2005, 77, 52–59. [Google Scholar] [CrossRef]
- Kelly, A.; Salerno, S.; Connolly, A.; Bishop, M.; Charpentier, F.; Stolen, T.; Smith, G.L. Normal interventricular differences in tissue architecture underlie right ventricular susceptibility to conduction abnormalities in a mouse model of Brugada syndrome. Cardiovasc. Res. 2018, 114, 724–736. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, M.C.; Stephenson, R.S.; Anderson, R.H.; Benvenuti, L.A.; Loukas, M.; Aiello, V.D. Human subpulmonary infundibulum has an endocardial network of specialized conducting cardiomyocytes. Heart Rhythm 2020, 17, 123–130. [Google Scholar] [CrossRef]
- Aras, K.; Gams, A.; Faye, N.R.; Brennan, J.; Goldrick, K.; Li, J.; Zhong, Y.; Chiang, C.H.; Smith, E.H.; Poston, M.D.; et al. Electrophysiology and Arrhythmogenesis in the Human Right Ventricular Outflow Tract. Circ. Arrhythm. Electrophysiol. 2022, 15, e010630. [Google Scholar] [CrossRef]
- Burke, A.P.; Farb, A.; Tashko, G.; Virmani, R. Arrhythmogenic right ventricular cardiomyopathy and fatty replacement of the right ventricular myocardium: Are they different diseases? Circulation 1998, 97, 1571–1580. [Google Scholar] [CrossRef] [Green Version]
- Lerman, B.B.; Belardinelli, L.; West, G.A.; Berne, R.M.; DiMarco, J.P. Adenosine-sensitive ventricular tachycardia: Evidence suggesting cyclic AMP-mediated triggered activity. Circulation 1986, 74, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Morita, H.; Zipes, D.P.; Morita, S.T.; Wu, J. Differences in arrhythmogenicity between the canine right ventricular outflow tract and anteroinferior right ventricle in a model of Brugada syndrome. Heart Rhythm 2007, 4, 66–74. [Google Scholar] [CrossRef]
- Morita, H.; Zipes, D.P.; Fukushima-Kusano, K.; Nagase, S.; Nakamura, K.; Morita, S.T.; Ohe, T.; Wu, J. Repolarization heterogeneity in the right ventricular outflow tract: Correlation with ventricular arrhythmias in Brugada patients and in an in vitro canine Brugada model. Heart Rhythm 2008, 5, 725–733. [Google Scholar] [CrossRef]
- Rudic, B.; Chaykovskaya, M.; Tsyganov, A.; Kalinin, V.; Tulumen, E.; Papavassiliu, T.; Dosch, C.; Liebe, V.; Kuschyk, J.; Roger, S.; et al. Simultaneous Non-Invasive Epicardial and Endocardial Mapping in Patients With Brugada Syndrome: New Insights Into Arrhythmia Mechanisms. J. Am. Heart Assoc. 2016, 5, e004095. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.; Lin, C.; Li, Y.; Liu, T.; Wang, Y. L-type calcium current in right ventricular outflow tract myocytes of rabbit heart. Sci. China Life Sci. 2012, 55, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Bonatti, V.; Rolli, A.; Botti, G. Recording of monophasic action potentials of the right ventricle in long QT syndromes complicated by severe ventricular arrhythmias. Eur. Heart J. 1983, 4, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Gaborit, N.; Le Bouter, S.; Szuts, V.; Varro, A.; Escande, D.; Nattel, S.; Demolombe, S. Regional and tissue specific transcript signatures of ion channel genes in the non-diseased human heart. J. Physiol. 2007, 582, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Veldkamp, M.W.; Viswanathan, P.C.; Bezzina, C.; Baartscheer, A.; Wilde, A.A.; Balser, J.R. Two distinct congenital arrhythmias evoked by a multidysfunctional Na+ channel. Circ. Res. 2000, 86, E91–E97. [Google Scholar] [CrossRef] [PubMed]
- Kohomoto, O.; Levi, A.J.; Bridge, J.H. Relation between reverse sodium-calcium exchange and sarcoplasmic reticulum calcium release in guinea pig ventricular cells. Circ. Res. 1994, 74, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 2000, 87, 275–281. [Google Scholar] [CrossRef]
- Eisner, D.A.; Caldwell, J.L.; Kistamas, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef]
- Song, Z.; Ko, C.Y.; Nivala, M.; Weiss, J.N.; Qu, Z. Calcium-voltage coupling in the genesis of early and delayed afterdepolarizations in cardiac myocytes. Biophys. J. 2015, 108, 1908–1921. [Google Scholar] [CrossRef] [Green Version]
- Damiano, B.P.; Rosen, M.R. Effects of pacing on triggered activity induced by early afterdepolarizations. Circulation 1984, 69, 1013–1025. [Google Scholar] [CrossRef] [Green Version]
- Patterson, E.; Lazzara, R.; Szabo, B.; Liu, H.; Tang, D.; Li, Y.H.; Scherlag, B.J.; Po, S.S. Sodium-calcium exchange initiated by the Ca2+ transient: An arrhythmia trigger within pulmonary veins. J. Am. Coll. Cardiol. 2006, 47, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Burashnikov, A.; Antzelevitch, C. Late-phase 3 EAD. A unique mechanism contributing to initiation of atrial fibrillation. Pacing Clin. Electrophysiol. 2006, 29, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, T.; Kimura, S.; Catstellanos, A.; Bassett, A.L.; Myerburg, R.J. In vivo induction of “focal” triggered ventricular arrhythmias and responses to overdrive pacing in the canine heart. Circulation 1990, 82, 549–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bers, D.M.; Bassani, J.W.; Bassani, R.A. Na-Ca exchange and Ca fluxes during contraction and relaxation in mammalian ventricular muscle. Ann. N. Y. Acad. Sci. 1996, 779, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Aronson, R.S.; Nordin, C. Arrhythmogenic interaction between low potassium and ouabain in isolated guinea-pig ventricular myocytes. J. Physiol. 1988, 400, 113–134. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.S. A novel mechanism for the treatment of angina, arrhythmias, and diastolic dysfunction: Inhibition of late I(Na) using ranolazine. J. Cardiovasc. Pharmacol. 2009, 54, 279–286. [Google Scholar] [CrossRef] [Green Version]
- Jafri, M.S. Models of excitation-contraction coupling in cardiac ventricular myocytes. Methods Mol. Biol. 2012, 910, 309–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.Y.; Chen, Y.C.; Lin, Y.K.; Chen, S.A.; Chen, Y.J. Mechanoelectrical feedback in pulmonary vein arrhythmogenesis: Clinical challenges and therapeutic opportunities. J. Arrhythm. 2020, 36, 608–614. [Google Scholar] [CrossRef]
- Rios, E. The cell boundary theorem: A simple law of the control of cytosolic calcium concentration. J. Physiol. Sci. 2010, 60, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Antoons, G.; Mubagwa, K.; Nevelsteen, I.; Sipido, K.R. Mechanisms underlying the frequency dependence of contraction and [Ca(2+)](i) transients in mouse ventricular myocytes. J. Physiol. 2002, 543, 889–898. [Google Scholar] [CrossRef]
- Monasky, M.M.; Pappone, C.; Piccoli, M.; Ghiroldi, A.; Micaglio, E.; Anastasia, L. Calcium in Brugada Syndrome: Questions for Future Research. Front. Physiol. 2018, 9, 1088. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.M.; Lin, F.Z.; Chen, Y.C.; Lin, Y.K.; Lu, Y.Y.; Wu, C.I.; Higa, S.; Chen, S.A.; Chen, Y.J. Concurrent increases in post-pacing action potential duration and contractility predict occurrence of ventricular arrhythmia. Pflugers Arch. 2020, 472, 1783–1791. [Google Scholar] [CrossRef]
- Liu, C.M.; Lin, F.J.; Chen, Y.C.; Lin, Y.K.; Lu, Y.Y.; Chan, C.S.; Higa, S.; Chen, S.A.; Chen, Y.J. Modulation of post-pacing action potential duration and contractile responses on ventricular arrhythmogenesis in chloroquine-induced long QT syndrome. Eur. J. Pharmacol. 2023, 941, 175493. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wu, M.; Wang, J.F.; Qiao, Y.J.; Wang, Z. Disruption of the intracellular Ca2+ homeostasis in the cardiac excitation-contraction coupling is a crucial mechanism of arrhythmic toxicity in aconitine-induced cardiomyocytes. Biochem. Biophys. Res. Commun. 2007, 354, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Verberne, H.J.; Blom, M.T.; Bardai, A.; Karemaker, J.M.; Tan, H.L. An inherited sudden cardiac arrest syndrome may be based on primary myocardial and autonomic nervous system abnormalities. Heart Rhythm 2022, 19, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, K.; Kanazawa, H.; Aizawa, Y.; Ardell, J.L.; Shivkumar, K. Cardiac innervation and sudden cardiac death. Circ. Res. 2015, 116, 2005–2019. [Google Scholar] [CrossRef]
- Randall, W.C.; Armour, J.A.; Geis, W.P.; Lippincott, D.B. Regional cardiac distribution of the sympathetic nerves. Fed. Proc. 1972, 31, 1199–1208. [Google Scholar]
- Chang, H.Y.; Lo, L.W.; Chen, Y.R.; Chou, Y.H.; Lin, W.L.; Lin, Y.J.; Yin, W.H.; Feng, A.N.; Chen, S.A. The autonomic neural mechanism of right ventricular outflow tract tachycardia. Auton. Neurosci. 2018, 212, 10–16. [Google Scholar] [CrossRef]
- Schafers, M.; Lerch, H.; Wichter, T.; Rhodes, C.G.; Lammertsma, A.A.; Borggrefe, M.; Hermansen, F.; Schober, O.; Breithardt, G.; Camici, P.G. Cardiac sympathetic innervation in patients with idiopathic right ventricular outflow tract tachycardia. J. Am. Coll. Cardiol. 1998, 32, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Iwai, S.; Cantillon, D.J.; Kim, R.J.; Markowitz, S.M.; Mittal, S.; Stein, K.M.; Shah, B.K.; Yarlagadda, R.K.; Cheung, J.W.; Tan, V.R.; et al. Right and left ventricular outflow tract tachycardias: Evidence for a common electrophysiologic mechanism. J. Cardiovasc. Electrophysiol. 2006, 17, 1052–1058. [Google Scholar] [CrossRef]
- Nakagawa, M.; Takahashi, N.; Nobe, S.; Ichinose, M.; Ooie, T.; Yufu, F.; Shigematsu, S.; Hara, M.; Yonemochi, H.; Saikawa, T. Gender differences in various types of idiopathic ventricular tachycardia. J. Cardiovasc. Electrophysiol. 2002, 13, 633–638. [Google Scholar] [CrossRef]
- Marchlinski, F.E.; Deely, M.P.; Zado, E.S. Sex-specific triggers for right ventricular outflow tract tachycardia. Am. Heart J. 2000, 139, 1009–1013. [Google Scholar] [CrossRef]
- Hodgkinson, K.A.; Parfrey, P.S.; Bassett, A.S.; Kupprion, C.; Drenckhahn, J.; Norman, M.W.; Thierfelder, L.; Stuckless, S.N.; Dicks, E.L.; McKenna, W.J.; et al. The impact of implantable cardioverter-defibrillator therapy on survival in autosomal-dominant arrhythmogenic right ventricular cardiomyopathy (ARVD5). J. Am. Coll. Cardiol. 2005, 45, 400–408. [Google Scholar] [CrossRef] [Green Version]
- Rautaharju, P.M.; Zhou, S.H.; Wong, S.; Calhoun, H.P.; Berenson, G.S.; Prineas, R.; Davignon, A. Sex differences in the evolution of the electrocardiographic QT interval with age. Can. J. Cardiol. 1992, 8, 690–695. [Google Scholar]
- Tsai, W.C.; Yang, L.Y.; Chen, Y.C.; Kao, Y.H.; Lin, Y.K.; Chen, S.A.; Cheng, C.F.; Chen, Y.J. Ablation of the androgen receptor gene modulates atrial electrophysiology and arrhythmogenesis with calcium protein dysregulation. Endocrinology 2013, 154, 2833–2842. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.C.; Lu, Y.Y.; Chen, Y.C.; Chang, C.J.; Kao, Y.H.; Lin, Y.K.; Chen, Y.H.; Chen, S.A.; Yang, L.Y.; Chen, Y.J. Ablation of androgen receptor gene triggers right ventricular outflow tract ventricular tachycardia. Int. J. Cardiol. 2015, 189, 172–181. [Google Scholar] [CrossRef]
- Towbin, J.A.; Vatta, M.; Li, H. Genetics of brugada, long QT, and arrhythmogenic right ventricular dysplasia syndromes. J. Electrocardiol. 2000, 33 (Suppl. S1), 11–22. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Kirsch, G.E.; Zhang, D.; Brugada, R.; Brugada, J.; Brugada, P.; Potenza, D.; Moya, A.; Borggrefe, M.; Breithardt, G.; et al. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 1998, 392, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Malcic, I.; Kniewald, H.; Pivac, V.T.; Jelasic, D.; Buljevic, B. Right Ventricular Arrhytmogenic Cardiomyopathy—Have We Avoided a Family Tragedy by Applying Contemporary Diagnostic and Treatment Approach? Lijec. Vjesn. 2016, 138, 339–344. [Google Scholar]
- Zhou, Y.; Bai, K.; Wang, Y.; Meng, Z.; Zhou, S.; Jiang, S.; Wang, H.; Wang, J.; Yang, M.; Wang, Q.; et al. Identification of Rare Variants in Right Ventricular Outflow Tract Obstruction Congenital Heart Disease by Whole-Exome Sequencing. Front. Cardiovasc. Med. 2021, 8, 811156. [Google Scholar] [CrossRef]
- Hasdemir, C.; Aydin, H.H.; Celik, H.A.; Simsek, E.; Payzin, S.; Kayikcioglu, M.; Aydin, M.; Kultursay, H.; Can, L.H. Transcriptional profiling of septal wall of the right ventricular outflow tract in patients with idiopathic ventricular arrhythmias. Pacing Clin. Electrophysiol. 2010, 33, 159–167. [Google Scholar] [CrossRef]
- Zhang, Y.; Guzadhur, L.; Jeevaratnam, K.; Salvage, S.C.; Matthews, G.D.; Lammers, W.J.; Lei, M.; Huang, C.L.; Fraser, J.A. Arrhythmic substrate, slowed propagation and increased dispersion in conduction direction in the right ventricular outflow tract of murine Scn5a+/- hearts. Acta. Physiol. 2014, 211, 559–573. [Google Scholar] [CrossRef] [Green Version]
- Martin, C.A.; Grace, A.A.; Huang, C.L. Spatial and temporal heterogeneities are localized to the right ventricular outflow tract in a heterozygotic Scn5a mouse model. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H605–H616. [Google Scholar] [CrossRef] [Green Version]
- Pannone, L.; Monaco, C.; Sorgente, A.; Vergara, P.; Gauthey, A.; Calburean, P.A.; Bisignani, A.; Paparella, G.; Ramak, R.; Overeinder, I.; et al. SCN5A mutation in Brugada syndrome is associated with substrate severity detected by electrocardiographic imaging and high-density electroanatomic mapping. Heart Rhythm 2022, 19, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.W.; Ip, J.E.; Yarlagadda, R.K.; Liu, C.F.; Thomas, G.; Xu, L.; Wilkes, D.; Markowitz, S.M.; Lerman, B.B. Adenosine-insensitive right ventricular tachycardia: Novel variant of idiopathic outflow tract tachycardia. Heart Rhythm 2014, 11, 1770–1778. [Google Scholar] [CrossRef]
- Lerman, B.B.; Dong, B.; Stein, K.M.; Markowitz, S.M.; Linden, J.; Catanzaro, D.F. Right ventricular outflow tract tachycardia due to a somatic cell mutation in G protein subunitalphai2. J. Clin. Investig. 1998, 101, 2862–2868. [Google Scholar] [CrossRef] [Green Version]
- Decher, N.; Ortiz-Bonnin, B.; Friedrich, C.; Schewe, M.; Kiper, A.K.; Rinne, S.; Seemann, G.; Peyronnet, R.; Zumhagen, S.; Bustos, D.; et al. Sodium permeable and “hypersensitive” TREK-1 channels cause ventricular tachycardia. EMBO Mol. Med. 2017, 9, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ip, J.E.; Xu, L.; Dai, J.; Steegborn, C.; Jaffre, F.; Evans, T.; Cheung, J.W.; Basson, C.T.; Panaghie, G.; Krogh-Madsen, T.; et al. Constitutively Activating GNAS Somatic Mutation in Right Ventricular Outflow Tract Tachycardia. Circ. Arrhythm. Electrophysiol. 2021, 14, e010082. [Google Scholar] [CrossRef] [PubMed]
- Guh, D.P.; Zhang, W.; Bansback, N.; Amarsi, Z.; Birmingham, C.L.; Anis, A.H. The incidence of co-morbidities related to obesity and overweight: A systematic review and meta-analysis. BMC Public Health 2009, 9, 88. [Google Scholar] [CrossRef] [Green Version]
- Aune, D.; Schlesinger, S.; Norat, T.; Riboli, E. Body mass index, abdominal fatness, and the risk of sudden cardiac death: A systematic review and dose-response meta-analysis of prospective studies. Eur. J. Epidemiol. 2018, 33, 711–722. [Google Scholar] [CrossRef] [Green Version]
- de Divitiis, O.; Fazio, S.; Petitto, M.; Maddalena, G.; Contaldo, F.; Mancini, M. Obesity and cardiac function. Circulation 1981, 64, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Tam, W.C.; Lin, Y.K.; Chan, W.P.; Huang, J.H.; Hsieh, M.H.; Chen, S.A.; Chen, Y.J. Pericardial Fat Is Associated With the Risk of Ventricular Arrhythmia in Asian Patients. Circ. J. 2016, 80, 1726–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messerli, F.H.; Nunez, B.D.; Ventura, H.O.; Snyder, D.W. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch. Intern. Med. 1987, 147, 1725–1728. [Google Scholar] [CrossRef]
- Pan, X.; Chen, X.; Ren, L.; Li, Z.; Chen, S. Correlation between Cardiac Ultrasound Index and Cardiovascular Risk in Healthy Obese and Overweight Populations. Int. J. Clin. Pract. 2022, 2022, 2235994. [Google Scholar] [CrossRef] [PubMed]
- Maury, P. Why is the right ventricular outflow tract so arrhythmogenic? (... or is it really?...). Heart 2011, 97, 1631–1633. [Google Scholar] [CrossRef]
- Lu, Y.Y.; Huang, S.Y.; Lin, Y.K.; Chen, Y.C.; Chen, Y.A.; Chen, S.A.; Chen, Y.J. Epicardial adipose tissue modulates arrhythmogenesis in right ventricle outflow tract cardiomyocytes. Europace 2021, 23, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Doyle, R.; Murphy, D.J.; Hunt, S.A. Right ventricular function in cardiovascular disease, part II: Pathophysiology, clinical importance, and management of right ventricular failure. Circulation 2008, 117, 1717–1731. [Google Scholar] [CrossRef] [Green Version]
- Deveci, B.; Baser, K.; Gul, M.; Sen, F.; Kafes, H.; Avci, S.; Temizer, O.; Ozeke, O.; Tufekcioglu, O.; Golbasi, Z. Right ventricular outflow tract function in chronic heart failure. Indian Heart J. 2016, 68 (Suppl. S1), S10–S14. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.L.; Hsiao, Y.W.; Tsai, Y.N.; Lin, S.F.; Liu, S.H.; Lin, Y.J.; Lo, L.W.; Chung, F.P.; Chao, T.F.; Hu, Y.F.; et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J. Mol. Cell. Cardiol. 2018, 122, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Bayer, J.D.; Lalani, G.G.; Vigmond, E.J.; Narayan, S.M.; Trayanova, N.A. Mechanisms linking electrical alternans and clinical ventricular arrhythmia in human heart failure. Heart Rhythm 2016, 13, 1922–1931. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.Y.; Cheng, C.C.; Tsai, C.F.; Lin, Y.K.; Lee, T.I.; Chen, Y.C.; Chen, S.A.; Chen, Y.J. Discrepant effects of heart failure on electrophysiological property in right ventricular outflow tract and left ventricular outflow tract cardiomyocytes. Clin. Sci. 2017, 131, 1317–1327. [Google Scholar] [CrossRef]
- Reinecke, H.; Studer, R.; Vetter, R.; Holtz, J.; Drexler, H. Cardiac Na+/Ca2+ exchange activity in patients with end-stage heart failure. Cardiovasc. Res. 1996, 31, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Hobai, I.A.; Maack, C.; O’Rourke, B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ. Res. 2004, 95, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Boriani, G.; Savelieva, I.; Dan, G.A.; Deharo, J.C.; Ferro, C.; Israel, C.W.; Lane, D.A.; La Manna, G.; Morton, J.; Mitjans, A.M.; et al. Chronic kidney disease in patients with cardiac rhythm disturbances or implantable electrical devices: Clinical significance and implications for decision making-a position paper of the European Heart Rhythm Association endorsed by the Heart Rhythm Society and the Asia Pacific Heart Rhythm Society. Europace 2015, 17, 1169–1196. [Google Scholar] [CrossRef] [PubMed]
- Chiu, D.Y.; Sinha, S.; Kalra, P.A.; Green, D. Sudden cardiac death in haemodialysis patients: Preventative options. Nephrology 2014, 19, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Bonato, F.O.; Lemos, M.M.; Cassiolato, J.L.; Canziani, M.E. Prevalence of ventricular arrhythmia and its associated factors in nondialyzed chronic kidney disease patients. PLoS ONE 2013, 8, e66036. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.D.; Soliman, E.Z.; Coresh, J.; Matsushita, K.; Chen, L.Y. Two-Week Burden of Arrhythmias across CKD Severity in a Large Community-Based Cohort: The ARIC Study. J. Am. Soc. Nephrol. 2021, 32, 629–638. [Google Scholar] [CrossRef]
- Rautavaara, J.; Kerola, T.; Kaartinen, K.; Vilpakka, M.; Aitkoski, A.; Anttonen, O.; Ahvonen, J.; Koistinen, J.; Vaaraniemi, K.; Miettinen, M.; et al. Asystole episodes and bradycardia in patients with end-stage renal disease. Nephrol. Dial. Transplant. 2022, 37, 575–583. [Google Scholar] [CrossRef]
- Stallworthy, E.J.; Pilmore, H.L.; Webster, M.W.; Sidhu, K.K.; Curry, E.M.; Brown, P.; Scaria, A. Do echocardiographic parameters predict mortality in patients with end-stage renal disease? Transplantation 2013, 95, 1225–1232. [Google Scholar] [CrossRef]
- Yigla, M.; Fruchter, O.; Aharonson, D.; Yanay, N.; Reisner, S.A.; Lewin, M.; Nakhoul, F. Pulmonary hypertension is an independent predictor of mortality in hemodialysis patients. Kidney Int. 2009, 75, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Schleberger, R.; Riess, J.; Brauer, A.; Pinnschmidt, H.O.; Rottner, L.; Moser, F.; Moser, J.; Kany, S.; My, I.; Lemoine, M.D.; et al. Ablation of Outflow Tract Arrhythmias in Patients With and Without Structural Heart Disease-A Comparative Analysis. Front. Cardiovasc. Med. 2022, 9, 910042. [Google Scholar] [CrossRef] [PubMed]
- Sarafidis, P.A.; Kamperidis, V.; Loutradis, C.; Tsilonis, K.; Mpoutsiouki, F.; Saratzis, A.; Giannakoulas, G.; Sianos, G.; Karvounis, H. Haemodialysis acutely deteriorates left and right diastolic function and myocardial performance: An effect related to high ultrafiltration volumes? Nephrol. Dial. Transplant. 2017, 32, 1402–1409. [Google Scholar] [CrossRef] [Green Version]
- Browning, M.C.; Hsu, C.Y.; Wang, P.L.; Tune, B.M. Interaction of ischemic and antibiotic-induced injury in the rabbit kidney. J. Infect. Dis. 1983, 147, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Chen, Y.C.; Kao, Y.H.; Lu, Y.Y.; Lin, Y.K.; Higa, S.; Chen, S.A.; Chen, Y.J. Calcium dysregulation increases right ventricular outflow tract arrhythmogenesis in rabbit model of chronic kidney disease. J. Cell. Mol. Med. 2021, 25, 11264–11277. [Google Scholar] [CrossRef] [PubMed]
- Hsueh, C.H.; Chen, N.X.; Lin, S.F.; Chen, P.S.; Gattone, V.H., 2nd; Allen, M.R.; Fishbein, M.C.; Moe, S.M. Pathogenesis of arrhythmias in a model of CKD. J. Am. Soc. Nephrol. 2014, 25, 2812–2821. [Google Scholar] [CrossRef] [Green Version]
- Waks, J.W.; Tereshchenko, L.G.; Parekh, R.S. Electrocardiographic predictors of mortality and sudden cardiac death in patients with end stage renal disease on hemodialysis. J. Electrocardiol. 2016, 49, 848–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volders, P.G.; Sipido, K.R.; Carmeliet, E.; Spatjens, R.L.; Wellens, H.J.; Vos, M.A. Repolarizing K+ currents ITO1 and IKs are larger in right than left canine ventricular midmyocardium. Circulation 1999, 99, 206–210. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Associated Gene | Inheritance | Mechanism | Model | Reference |
|---|---|---|---|---|
| ATP1A2, CACNA1C, PPP2R2C, PLCD3, GNAO1, Solute Carrier Family 6 (Transporter, Norepinephrine), Member 2(SLC6A2), CAMK2B, PIK3R2 | Down regulation | Myocardial intracellular Ca2+ regulation | Human | Hasdemir C. et al. [79] |
| CAMKK2 and ITPR3 | Up regulation | |||
| TBX3, BMP2, BMPR1B, MYH6, ANKRD23–39 | Down regulation | Cardiovascular functions | Human | Hasdemir C. et al. [79] |
| RGS1 | Up regulation | |||
| Scn5a | Heterozygous missense mutation | Na+ channel dysfunction | Mouse | Boukens BJ. et al. [22] Zhang Y. et al. [80] Martin CA. et al. [81] Pannone L. et al. [82] Chen Q. et al. [76] |
| Human | ||||
| Gja1 | Down regulation | Lower expression of gap junction | Mouse | Boukens BJ. et al. [22] |
| A1 ADO receptor (R296C) | Somatic mutation | Adenosine insensitive | Human | Cheung JW. et al. [83] |
| Inhibitory G protein Gαi2 (F200L) | Somatic mutation | Increase intracellular cAMP concentration Adenosine insensitive | Human | Cheung JW. et al. [83] Lerman BB. et al. [84] |
| TREK-1 (I267T) | Heterozygous point mutation | Stretch-activated K2P K+ channel TREK-1 | Human | Decher N. et al. [85] |
| Androgen receptor | Knockout | Myocardial intracellular Ca2+ regulation | Mouse | Tsai WC. et al. [74] |
| Stimulatory G protein alpha-subunit Gsα (W234R) | Somatic mutation | Increase intracellular cAMP concentration | Human | Ip JE. et al. [86] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.-Y.; Chen, Y.-C.; Lin, Y.-K.; Chen, S.-A.; Chen, Y.-J. Electrical and Structural Insights into Right Ventricular Outflow Tract Arrhythmogenesis. Int. J. Mol. Sci. 2023, 24, 11795. https://doi.org/10.3390/ijms241411795
Lu Y-Y, Chen Y-C, Lin Y-K, Chen S-A, Chen Y-J. Electrical and Structural Insights into Right Ventricular Outflow Tract Arrhythmogenesis. International Journal of Molecular Sciences. 2023; 24(14):11795. https://doi.org/10.3390/ijms241411795
Chicago/Turabian StyleLu, Yen-Yu, Yao-Chang Chen, Yung-Kuo Lin, Shih-Ann Chen, and Yi-Jen Chen. 2023. "Electrical and Structural Insights into Right Ventricular Outflow Tract Arrhythmogenesis" International Journal of Molecular Sciences 24, no. 14: 11795. https://doi.org/10.3390/ijms241411795
APA StyleLu, Y.-Y., Chen, Y.-C., Lin, Y.-K., Chen, S.-A., & Chen, Y.-J. (2023). Electrical and Structural Insights into Right Ventricular Outflow Tract Arrhythmogenesis. International Journal of Molecular Sciences, 24(14), 11795. https://doi.org/10.3390/ijms241411795