
Kinase Suppressor of RAS 1 (KSR1) Maintains the Transformed Phenotype of BRAFV600E Mutant Human Melanoma Cells
 , , , , and
, , , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Knocking Out KSR1 in BRAFV600E-Mutated SK-MEL-239 Cells Does Not Impact Bulk RAF-ERK Signalling
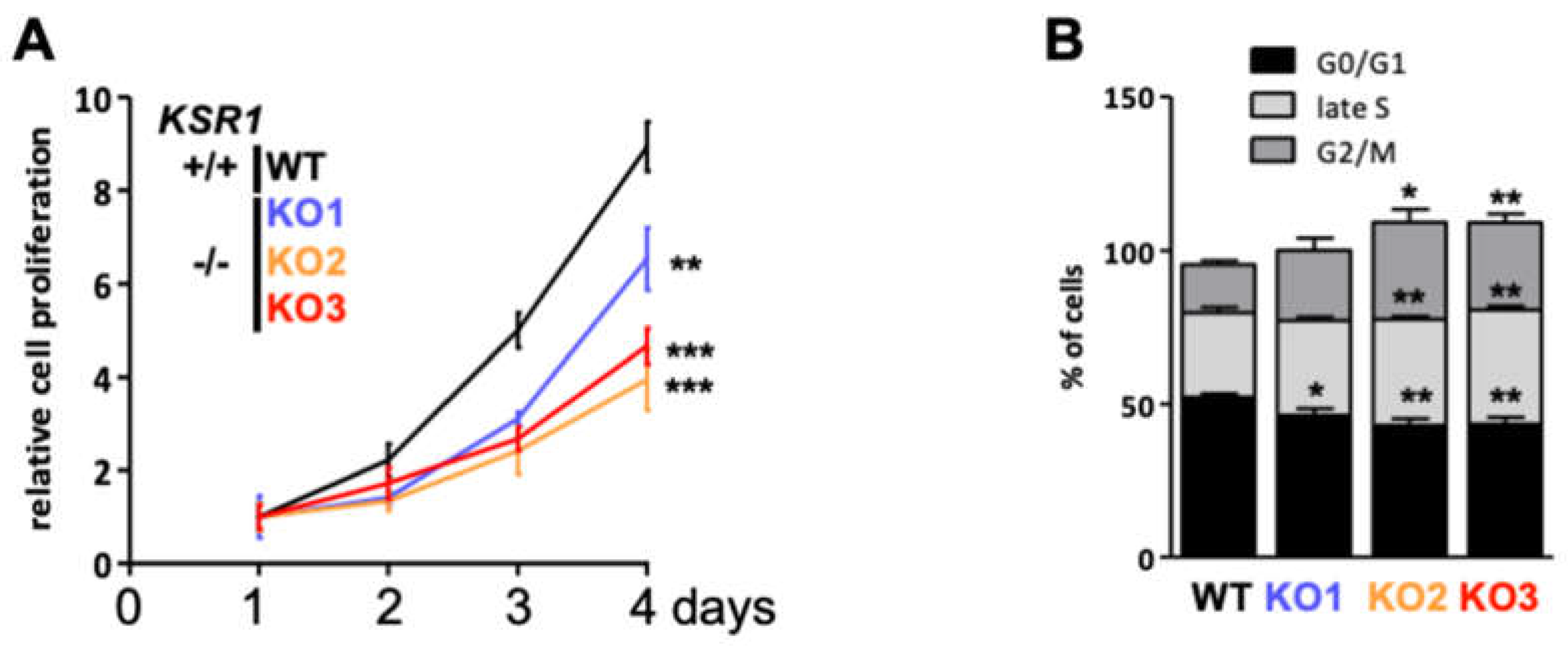
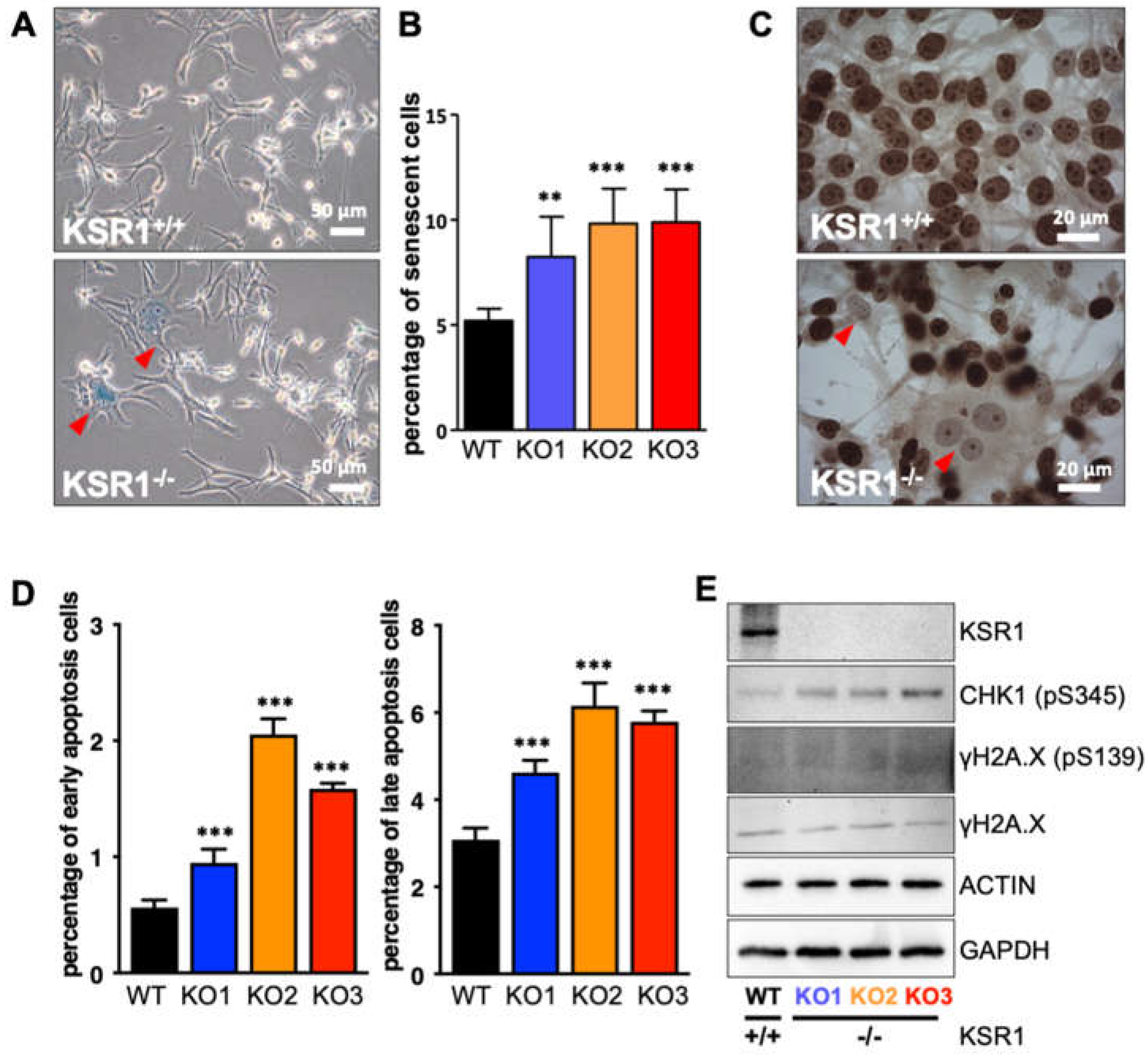
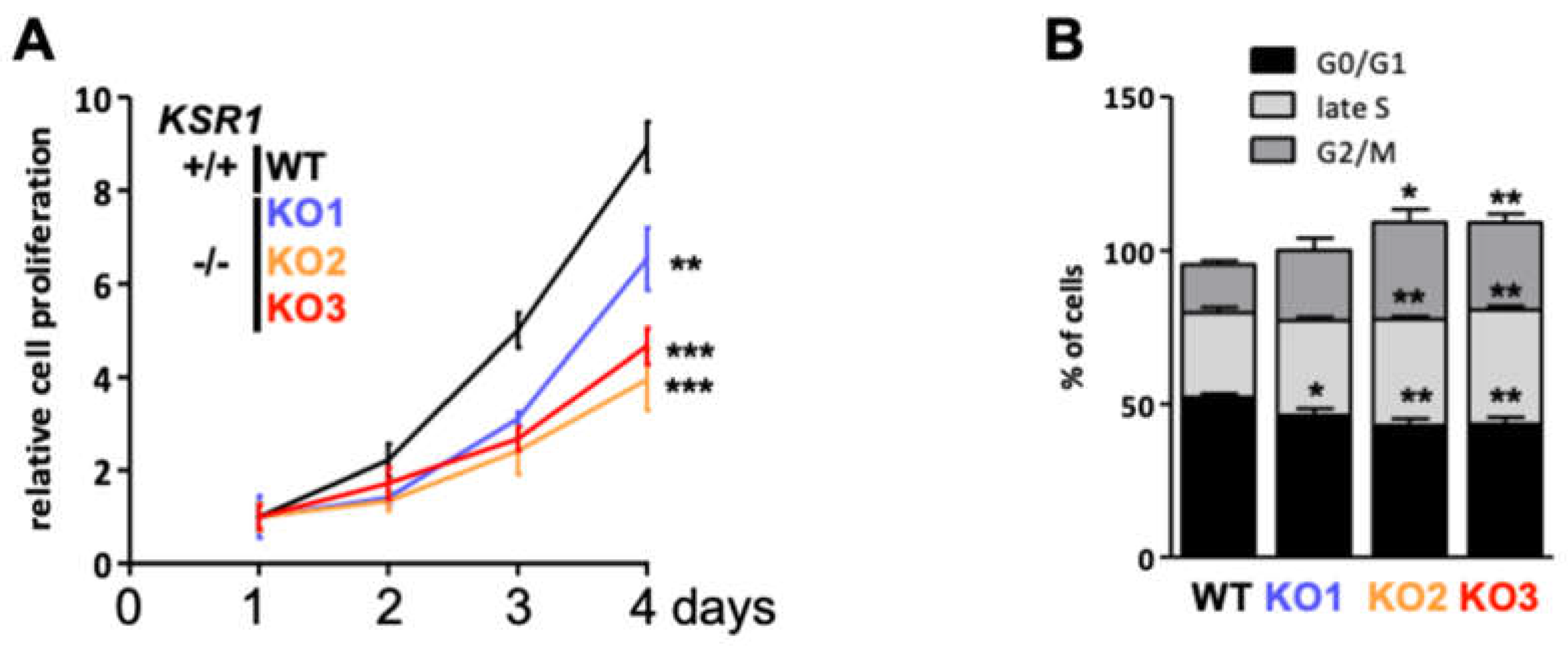
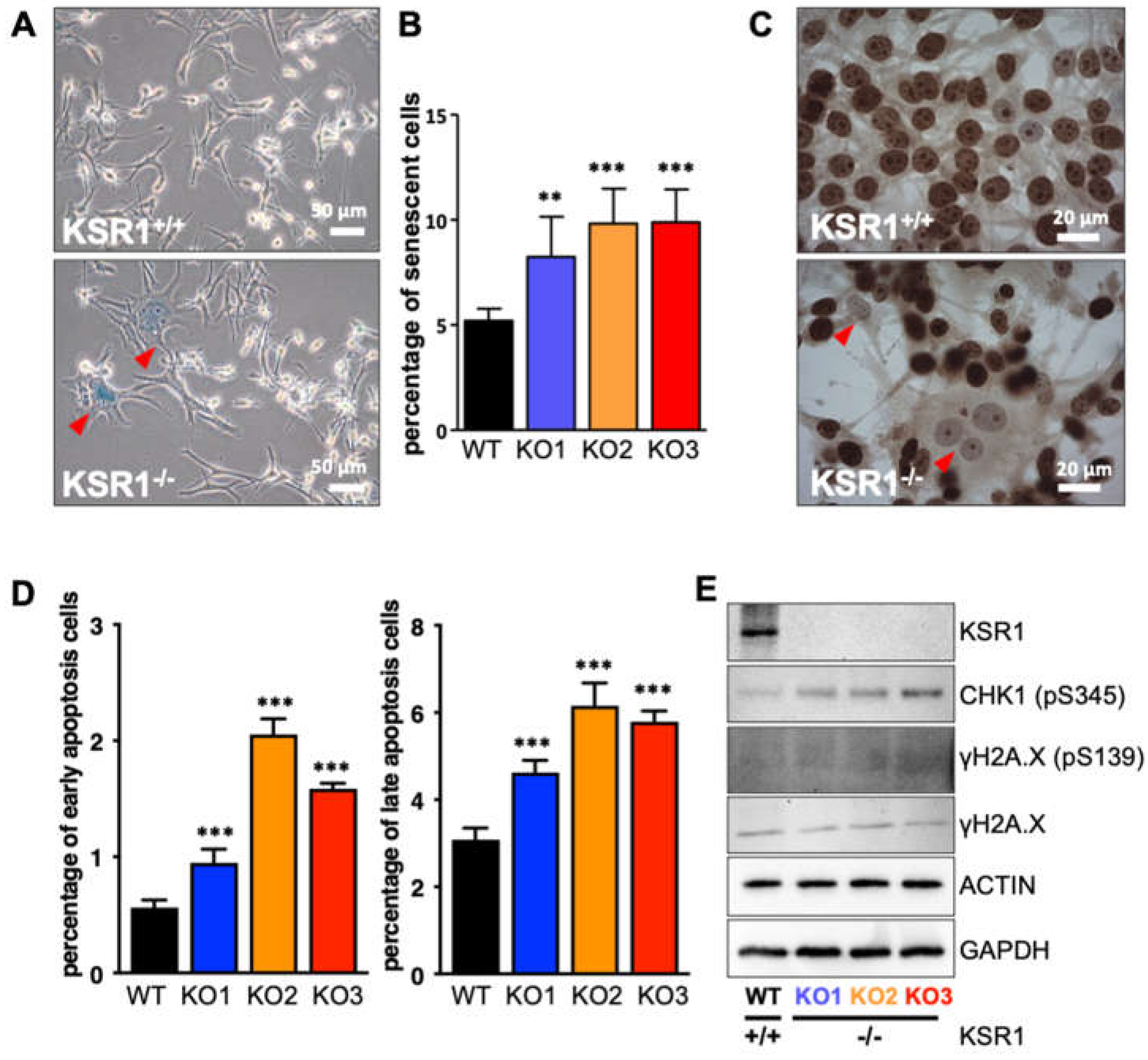
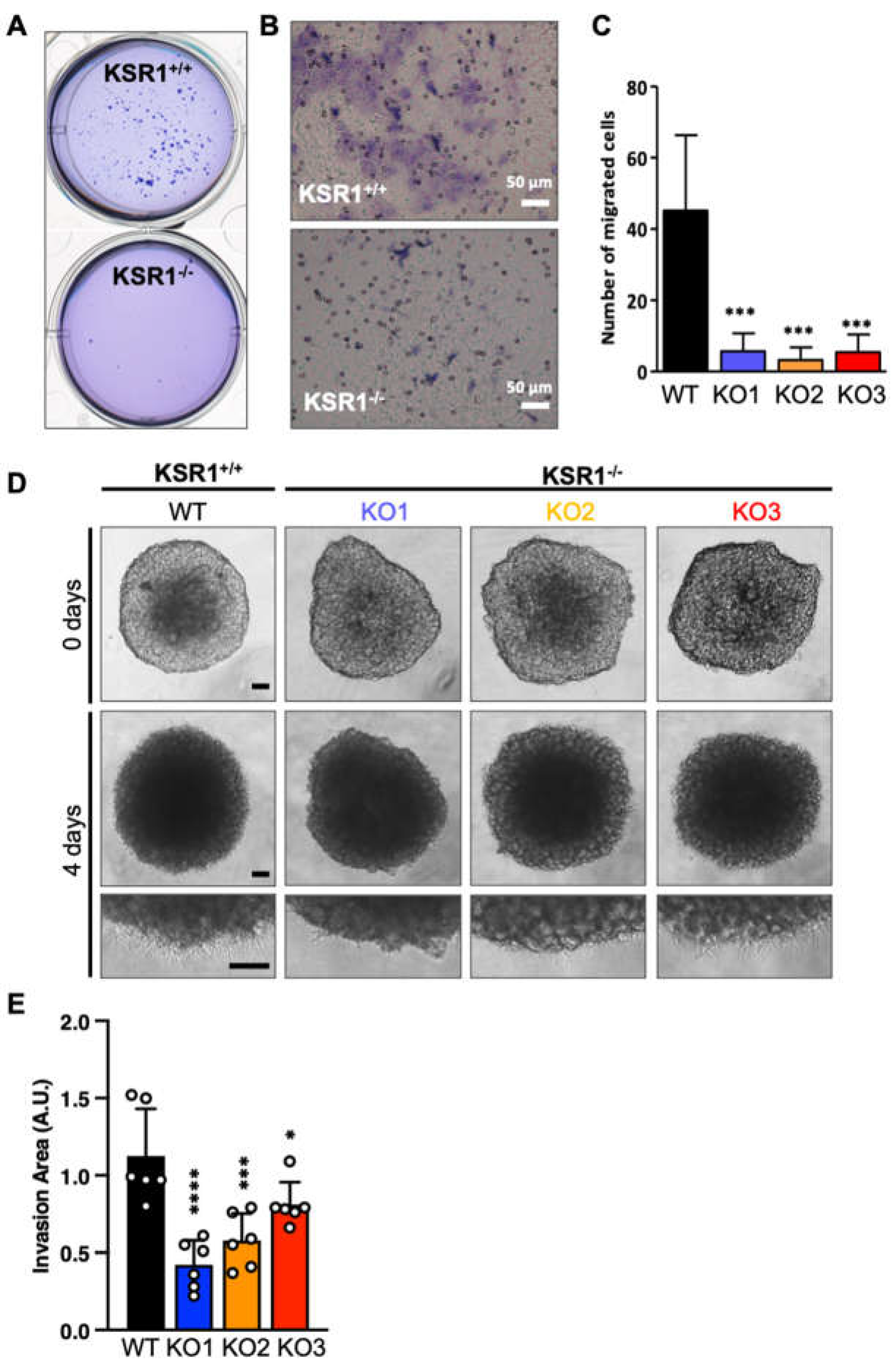
2.2. The Biological Phenotype of KSR1 Loss
2.3. ERK Substrateomics
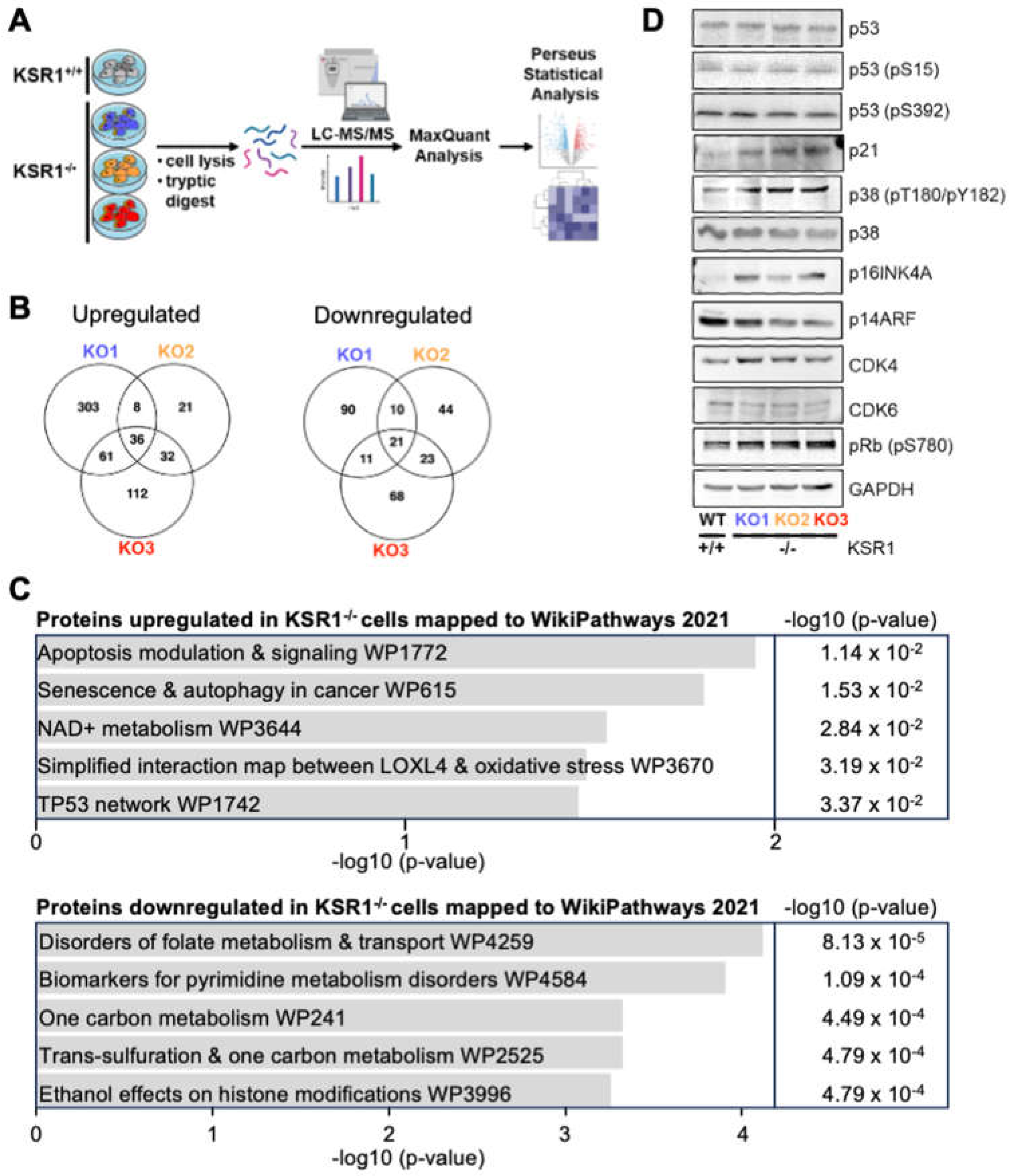
2.4. KSR1-Dependent Global Changes in Protein Expression
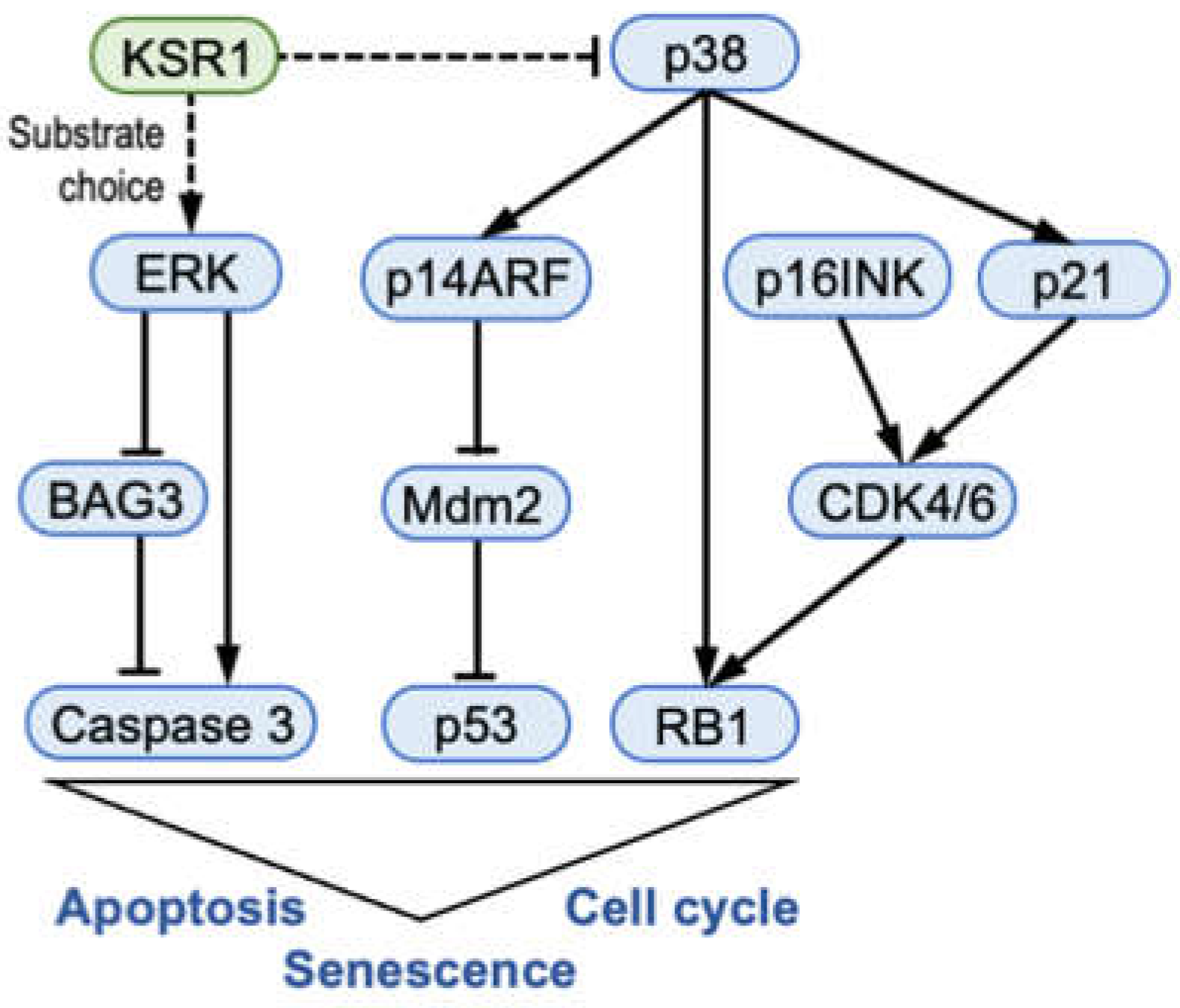
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, S.; Rauch, J.; Kolch, W. Targeting MAPK Signaling in Cancer: Mechanisms of Drug Resistance and Sensitivity. Int. J. Mol. Sci. 2020, 21, 1102. [Google Scholar] [CrossRef] [Green Version]
- Roskoski, R. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol. Res. 2019, 14, 151–168. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Frodyma, D.; Neilsen, B.; Costanzo-Garvey, D.; Fisher, K.; Lewis, R. Coordinating ERK signaling via the molecular scaffold Kinase Suppressor of Ras. F1000Research 2017, 6, 1621. [Google Scholar] [CrossRef] [Green Version]
- Lavoie, H.; Sahmi, M.; Maisonneuve, P.; Marullo, S.A.; Thevakumaran, N.; Jin, T.; Kurinov, I.; Sicheri, F.; Therrien, M. MEK drives BRAF activation through allosteric control of KSR proteins. Nature 2018, 554, 549–553. [Google Scholar] [CrossRef]
- Casar, B.; Arozarena, I.; Sanz-Moreno, V.; Pinto, A.; Agudo-Ibáñez, L.; Marais, R.; Lewis, R.E.; Berciano, M.T.; Crespo, P. Ras subcellular localization defines extracellular signal-regulated kinase 1 and 2 substrate specificity through distinct utilization of scaffold proteins. Mol. Cell. Biol. 2009, 29, 1338–1353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano, J.; Xing, R.; Cai, Z.; Jensen, H.L.; Trempus, C.; Mark, W.; Cannon, R.; Kolesnick, R. Deficiency of kinase suppressor of Ras1 prevents oncogenic ras signaling in mice. Cancer Res. 2003, 63, 4232–4238. [Google Scholar]
- Germino, E.A.; Miller, J.P.; Diehl, L.; Swanson, C.J.; Durinck, S.; Modrusan, Z.; Miner, J.H.; Shaw, A.S. Homozygous KSR1 deletion attenuates morbidity but does not prevent tumor development in a mouse model of RAS-driven pancreatic cancer. PLoS ONE 2018, 13, e0194998. [Google Scholar] [CrossRef] [Green Version]
- Kortum, R.L.; Lewis, R.E. The molecular scaffold KSR1 regulates the proliferative and oncogenic potential of cells. Mol. Cell. Biol. 2004, 24, 4407–4416. [Google Scholar] [CrossRef] [Green Version]
- Yan, F.; John, S.K.; Wilson, G.; Jones, D.S.; Washington, M.K.; Polk, D.B. Kinase suppressor of Ras-1 protects intestinal epithelium from cytokine-mediated apoptosis during inflammation. J. Clin. Investig. 2004, 114, 1272–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortum, R.L.; Johnson, H.J.; Costanzo, D.L.; Volle, D.J.; Razidlo, G.L.; Fusello, A.M.; Shaw, A.S.; Lewis, R.E. The molecular scaffold kinase suppressor of Ras 1 is a modifier of RasV12-induced and replicative senescence. Mol. Cell. Biol. 2006, 26, 2202–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortum, R.L.; Fernandez, M.R.; Costanzo-Garvey, D.L.; Johnson, H.J.; Fisher, K.W.; Volle, D.J.; Lewis, R.E. Caveolin-1 is required for kinase suppressor of Ras 1 (KSR1)-mediated extracellular signal-regulated kinase 1/2 activation, H-RasV12-induced senescence, and transformation. Mol. Cell. Biol. 2014, 34, 3461–3472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, C.; Frodyma, D.E.; Southekal, S.; Svoboda, R.A.; Black, A.R.; Guda, C.; Mizutani, T.; Clevers, H.; Johnson, K.R.; Fisher, K.W.; et al. KSR1- and ERK-dependent translational regulation of the epithelial-to-mesenchymal transition. eLife 2021, 10, e66608. [Google Scholar] [CrossRef] [PubMed]
- Neilsen, B.K.; Frodyma, D.E.; Lewis, R.E.; Fisher, K.W. KSR as a therapeutic target for Ras-dependent cancers. Expert Opin. Ther. Targets 2017, 21, 499–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53. [Google Scholar] [CrossRef] [Green Version]
- Topham, C.H.; Taylor, S.S. Mitosis and apoptosis: How is the balance set? Curr. Opin. Cell Biol. 2013, 25, 780–785. [Google Scholar] [CrossRef]
- Lemmens, M.; Fischer, B.; Zogg, M.; Rodrigues, L.; Kerr, G.; Del Rio-Espinola, A.; Schaeffer, F.; Maddalo, D.; Dubost, V.; Piaia, A.; et al. Evaluation of two in vitro assays for tumorigenicity assessment of CRISPR-Cas9 genome-edited cells. Mol. Ther. Methods Clin. Dev. 2021, 23, 241–253. [Google Scholar] [CrossRef]
- Yang, L.; Zheng, L.; Chng, W.J.; Ding, J.L. Comprehensive Analysis of ERK1/2 Substrates for Potential Combination Immunotherapies. Trends Pharmacol. Sci. 2019, 40, 897–910. [Google Scholar] [CrossRef] [Green Version]
- Ünal, E.B.; Uhlitz, F.; Blüthgen, N. A compendium of ERK targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef] [Green Version]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Zhuang, S.; Yan, Y.; Daubert, R.A.; Han, J.; Schnellmann, R.G. ERK promotes hydrogen peroxide-induced apoptosis through caspase-3 activation and inhibition of Akt in renal epithelial cells. Am. J. Physiol. Ren. Physiol 2007, 292, F440–F447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturner, E.; Behl, C. The Role of the Multifunctional BAG3 Protein in Cellular Protein Quality Control and in Disease. Front. Mol. Neurosci. 2017, 10, 177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; Kim, Y.S.; Yun, H.H.; Im, C.N.; Ko, J.H.; Lee, J.H. ERK-mediated phosphorylation of BIS regulates nuclear translocation of HSF1 under oxidative stress. Exp. Mol. Med. 2016, 48, e260. [Google Scholar] [CrossRef]
- Wang, S.; Bellen, H.J. The retromer complex in development and disease. Development 2015, 142, 2392–2396. [Google Scholar] [CrossRef] [Green Version]
- Weir, M.L.; Xie, H.; Klip, A.; Trimble, W.S. VAP-A binds promiscuously to both v- and tSNAREs. Biochem. Biophys. Res. Commun. 2001, 286, 616–621. [Google Scholar] [CrossRef]
- Lee, J.E.; Silhavy, J.L.; Zaki, M.S.; Schroth, J.; Bielas, S.L.; Marsh, S.E.; Olvera, J.; Brancati, F.; Iannicelli, M.; Ikegami, K.; et al. CEP41 is mutated in Joubert syndrome and is required for tubulin glutamylation at the cilium. Nat. Genet. 2012, 44, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Mannava, S.; Grachtchouk, V.; Wheeler, L.J.; Im, M.; Zhuang, D.; Slavina, E.G.; Mathews, C.K.; Shewach, D.S.; Nikiforov, M.A. Direct role of nucleotide metabolism in C-MYC-dependent proliferation of melanoma cells. Cell Cycle 2008, 7, 2392–2400. [Google Scholar] [CrossRef] [PubMed]
- Remmerie, M.; Janssens, V. PP2A: A Promising Biomarker and Therapeutic Target in Endometrial Cancer. Front. Oncol. 2019, 9, 462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birmpilis, A.I.; Karachaliou, C.E.; Samara, P.; Ioannou, K.; Selemenakis, P.; Kostopoulos, I.V.; Kavrochorianou, N.; Kalbacher, H.; Livaniou, E.; Haralambous, S.; et al. Antitumor Reactive T-Cell Responses Are Enhanced In Vivo by DAMP Prothymosin Alpha and Its C-Terminal Decapeptide. Cancers 2019, 11, 1764. [Google Scholar] [CrossRef] [Green Version]
- Fortis, S.P.; Anastasopoulou, E.A.; Voutsas, I.F.; Baxevanis, C.N.; Perez, S.A.; Mahaira, L.G. Potential Prognostic Molecular Signatures in a Preclinical Model of Melanoma. Anticancer. Res. 2017, 37, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Kito, Y.; Bai, J.; Goto, N.; Okubo, H.; Adachi, Y.; Nagayama, T.; Takeuchi, T. Pathobiological properties of the ubiquitin ligase Nedd4L in melanoma. Int. J. Exp. Pathol. 2014, 95, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Vella, L.J.; Xavier, D.; Amirkhani, A.; Parker, J.; Cebon, J.; Molloy, M.P. Phosphoproteomic Analysis of Cell-Based Resistance to BRAF Inhibitor Therapy in Melanoma. Front. Oncol. 2015, 5, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Lee, D.S.; Kim, M.J.; Kang, T.C. PLPP/CIN-mediated NEDD4-2 S448 dephosphorylation regulates neuronal excitability via GluA1 ubiquitination. Cell Death Dis. 2019, 10, 545. [Google Scholar] [CrossRef] [Green Version]
- Willems, P.; Wanschers, B.F.; Esseling, J.; Szklarczyk, R.; Kudla, U.; Duarte, I.; Forkink, M.; Nooteboom, M.; Swarts, H.; Gloerich, J.; et al. BOLA1 is an aerobic protein that prevents mitochondrial morphology changes induced by glutathione depletion. Antioxid. Redox Signal. 2013, 18, 129–138. [Google Scholar] [CrossRef] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129. [Google Scholar] [CrossRef]
- Lafarga, V.; Cuadrado, A.; Lopez de Silanes, I.; Bengoechea, R.; Fernandez-Capetillo, O.; Nebreda, A.R. p38 Mitogen-activated protein kinase- and HuR-dependent stabilization of p21(Cip1) mRNA mediates the G(1)/S checkpoint. Mol. Cell. Biol. 2009, 29, 4341–4351. [Google Scholar] [CrossRef] [Green Version]
- Iwasa, H.; Han, J.; Ishikawa, F. Mitogen-activated protein kinase p38 defines the common senescence-signalling pathway. Genes Cells 2003, 8, 131–144. [Google Scholar] [CrossRef]
- Spallarossa, P.; Altieri, P.; Barisione, C.; Passalacqua, M.; Aloi, C.; Fugazza, G.; Frassoni, F.; Podestà, M.; Canepa, M.; Ghigliotti, G.; et al. p38 MAPK and JNK antagonistically control senescence and cytoplasmic p16INK4A expression in doxorubicin-treated endothelial progenitor cells. PLoS ONE 2010, 5, e15583. [Google Scholar] [CrossRef] [Green Version]
- Chew, Y.P.; Ellis, M.; Wilkie, S.; Mittnacht, S. pRB phosphorylation mutants reveal role of pRB in regulating S phase completion by a mechanism independent of E2F. Oncogene 1998, 17, 2177–2186. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.J.; Park, S.M.; Hwang, E.M.; Baek, K.E.; Kim, I.K.; Nam, I.K.; Im, M.J.; Park, S.H.; Bae, S.; Park, J.Y.; et al. Gadd45b mediates Fas-induced apoptosis by enhancing the interaction between p38 and retinoblastoma tumor suppressor. J. Biol. Chem. 2010, 285, 25500–25505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaBaer, J.; Garrett, M.D.; Stevenson, L.F.; Slingerland, J.M.; Sandhu, C.; Chou, H.S.; Fattaey, A.; Harlow, E. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997, 11, 847–862. [Google Scholar] [CrossRef] [Green Version]
- Tran, T.T.; Rane, C.K.; Zito, C.R.; Weiss, S.A.; Jessel, S.; Lucca, L.; Lu, B.Y.; Oria, V.O.; Adeniran, A.; Chiang, V.L.; et al. Clinical Significance of PDCD4 in Melanoma by Subcellular Expression and in Tumor-Associated Immune Cells. Cancers 2021, 13, 1049. [Google Scholar] [CrossRef]
- Nakashima, H.; Hamamura, K.; Houjou, T.; Taguchi, R.; Yamamoto, N.; Mitsudo, K.; Tohnai, I.; Ueda, M.; Urano, T.; Furukawa, K.; et al. Overexpression of caveolin-1 in a human melanoma cell line results in dispersion of ganglioside GD3 from lipid rafts and alteration of leading edges, leading to attenuation of malignant properties. Cancer Sci. 2007, 98, 512–520. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF—The first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautron, A.; Migault, M.; Bachelot, L.; Corre, S.; Galibert, M.D.; Gilot, D. Human TYRP1: Two functions for a single gene? Pigment. Cell Melanoma Res. 2021, 34, 836–852. [Google Scholar] [CrossRef] [PubMed]
- Sinnberg, T.; Menzel, M.; Ewerth, D.; Sauer, B.; Schwarz, M.; Schaller, M.; Garbe, C.; Schittek, B. β-Catenin signaling increases during melanoma progression and promotes tumor cell survival and chemoresistance. PLoS ONE 2011, 6, e23429. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Koyano, T.; Namba, M.; Kobayashi, T.; Nakakuni, K.; Nakano, D.; Fukushima, M.; Nishiyama, A.; Matsuyama, M. The p21 dependent G2 arrest of the cell cycle in epithelial tubular cells links to the early stage of renal fibrosis. Sci. Rep. 2019, 9, 12059. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.O.; Davidson, J.M.; Duronio, R.J. Endoreplication: Polyploidy with purpose. Genes Dev. 2009, 23, 2461–2477. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Meikle, S.; Peyssonnaux, C.; Grindlay, J.; Kaiser, C.; Steen, H.; Shaw, P.E.; Mischak, H.; Eychène, A.; Kolch, W. A Raf-1 mutant that dissociates MEK/extracellular signal-regulated kinase activation from malignant transformation and differentiation but not proliferation. Mol. Cell. Biol. 2003, 23, 1983–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berens, E.B.; Holy, J.M.; Riegel, A.T.; Wellstein, A. A Cancer Cell Spheroid Assay to Assess Invasion in a 3D Setting. J. Vis. Exp. 2015, 105, e53409. [Google Scholar] [CrossRef]
- De Wever, O.; Hendrix, A.; De Boeck, A.; Westbroek, W.; Braems, G.; Emami, S.; Sabbah, M.; Gespach, C.; Bracke, M. Modeling and quantification of cancer cell invasion through collagen type I matrices. Int. J. Dev. Biol. 2010, 54, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Turriziani, B.; Garcia-Munoz, A.; Pilkington, R.; Raso, C.; Kolch, W.; von Kriegsheim, A. On-beads digestion in conjunction with data-dependent mass spectrometry: A shortcut to quantitative and dynamic interaction proteomics. Biology 2014, 3, 320–332. [Google Scholar] [CrossRef] [Green Version]
- Howard, J.; Wynne, K.; Moldenhauer, E.; Clarke, P.; Maguire, C.; Bollard, S.; Yin, X.; Brennan, L.; Mooney, L.; Fitzsimons, S.; et al. A comparative analysis of extracellular vesicles (EVs) from human and feline plasma. Sci. Rep. 2022, 12, 10851. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [Green Version]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Krstic, A.; Neve, A.; Casalou, C.; Rauch, N.; Wynne, K.; Cassidy, H.; McCann, A.; Kavanagh, E.; McCann, B.; et al. Kinase Suppressor of RAS 1 (KSR1) Maintains the Transformed Phenotype of BRAFV600E Mutant Human Melanoma Cells. Int. J. Mol. Sci. 2023, 24, 11821. https://doi.org/10.3390/ijms241411821
Liu Z, Krstic A, Neve A, Casalou C, Rauch N, Wynne K, Cassidy H, McCann A, Kavanagh E, McCann B, et al. Kinase Suppressor of RAS 1 (KSR1) Maintains the Transformed Phenotype of BRAFV600E Mutant Human Melanoma Cells. International Journal of Molecular Sciences. 2023; 24(14):11821. https://doi.org/10.3390/ijms241411821
Chicago/Turabian StyleLiu, Zhi, Aleksandar Krstic, Ashish Neve, Cristina Casalou, Nora Rauch, Kieran Wynne, Hilary Cassidy, Amanda McCann, Emma Kavanagh, Brendan McCann, and et al. 2023. "Kinase Suppressor of RAS 1 (KSR1) Maintains the Transformed Phenotype of BRAFV600E Mutant Human Melanoma Cells" International Journal of Molecular Sciences 24, no. 14: 11821. https://doi.org/10.3390/ijms241411821
APA StyleLiu, Z., Krstic, A., Neve, A., Casalou, C., Rauch, N., Wynne, K., Cassidy, H., McCann, A., Kavanagh, E., McCann, B., Blanco, A., Rauch, J., & Kolch, W. (2023). Kinase Suppressor of RAS 1 (KSR1) Maintains the Transformed Phenotype of BRAFV600E Mutant Human Melanoma Cells. International Journal of Molecular Sciences, 24(14), 11821. https://doi.org/10.3390/ijms241411821