


Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity

 , ,
, , {kind=link}
Abstract
:1. Introduction
2. mtDNA
2.1. The Relationship between Quantitative mtDNA Levels (and Inflammation) in Obesity and Metabolic Syndrome
2.2. mtDNA Methylation
2.3. mtDNA Mutations Accumulated during Development or in Postmitotic Tissues
2.4. mtDNA Haplogroups
2.5. mtDNA Released from Cells as Alarmins and Inflammation Inducers
2.6. Signaling Pathways Activated by mtDNA in Obesity and Metabolic Syndrome
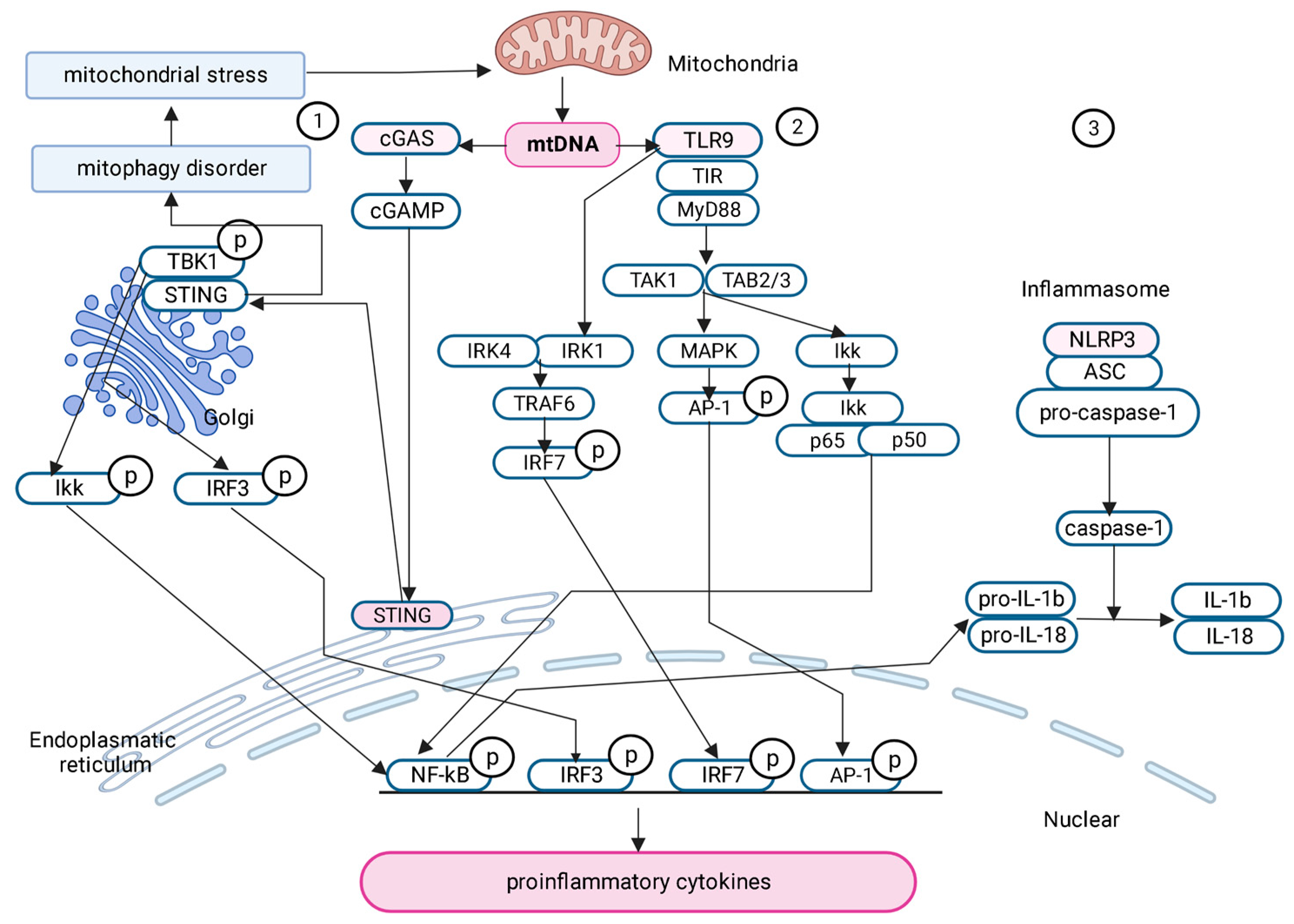
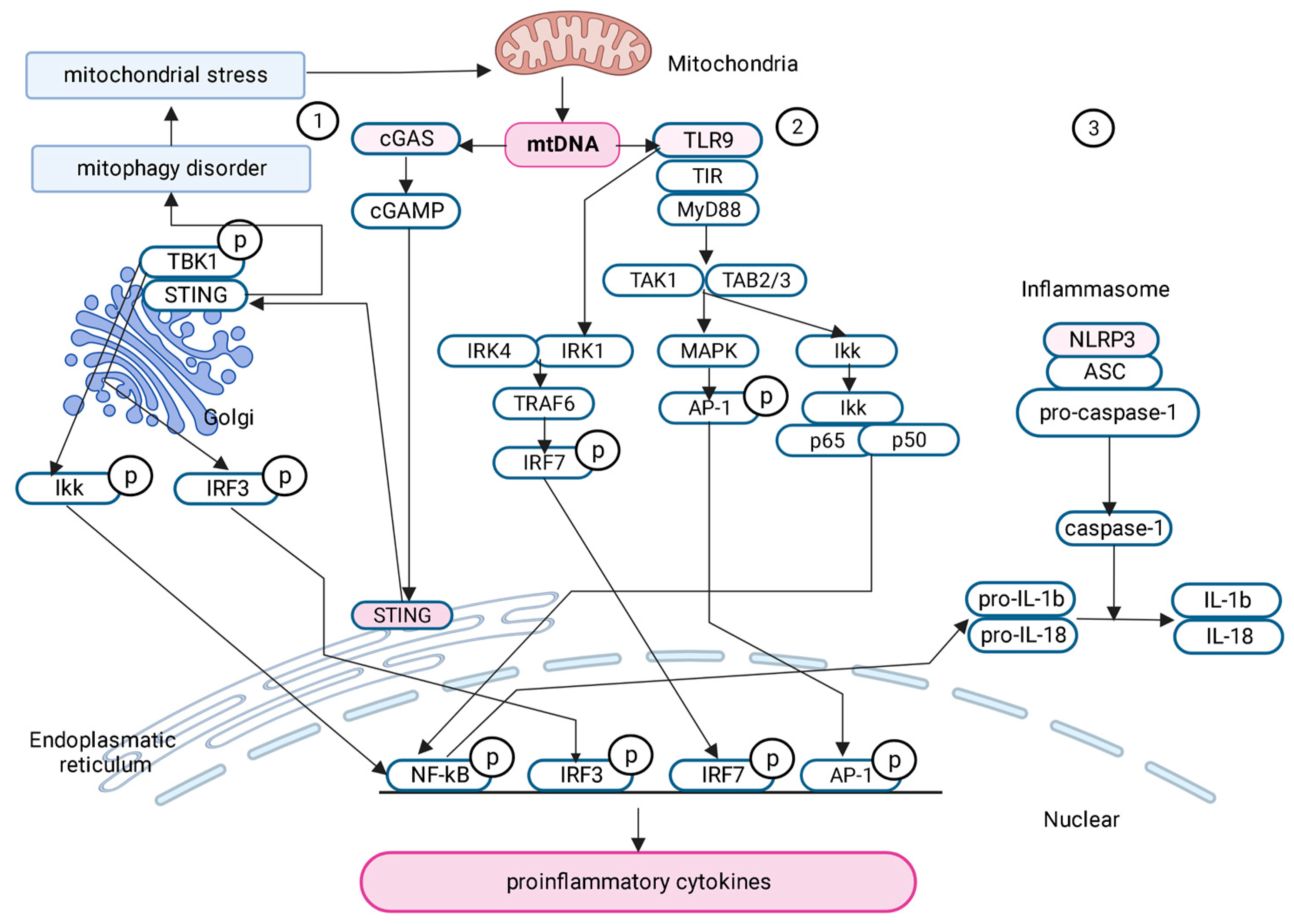
2.6.1. cGAS-STING Signaling Pathway
2.6.2. TLR9
2.6.3. Pyrin Domain of NLR Family-Containing Inflammasome 3 (NLRP3)
3. Mitochondrial Protease Lonp1
- Proteolytic digestion of oxidized proteins and turnover of specific key mitochondrial enzymes: aconitase, TFAM, StAR (steroidogenic acute regulatory protein), P450, SP -22, COX4;
- Proteinchaperone interacting with the HSP60-mtHSP70 complex, GRP78, NDUF58;
- mtDNA-binding protein involved in mtDNA replication and mitogenesis [126].
3.1. Molecular Functions of LonP1 in Obesity and Metabolic Syndrome
3.2. Nuclear Localization of Lonp1
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Révész, D.; Verhoeven, J.E.; Picard, M.; Lin, J.; Sidney, S.; Epel, E.S.; Penninx, B.W.J.H.; Puterman, E. Associations Between Cellular Aging Markers and Metabolic Syndrome: Findings From the CARDIA Study. J. Clin. Endocrinol. Metab. 2018, 103, 148–157. [Google Scholar] [CrossRef] [Green Version]
- Alkhulaifi, F.; Darkoh, C. Meal Timing, Meal Frequency and Metabolic Syndrome. Nutrients 2022, 14, 1719. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.; Lent-Schochet, D.; Ramakrishnan, N.; McLaughlin, M.; Jialal, I. Metabolic Syndrome Is an Inflammatory Disorder: A Conspiracy between Adipose Tissue and Phagocytes. Clin. Chim. Acta 2019, 496, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Ravaut, G.; Légiot, A.; Bergeron, K.-F.; Mounier, C. Monounsaturated Fatty Acids in Obesity-Related Inflammation. Int. J. Mol. Sci. 2020, 22, 330. [Google Scholar] [CrossRef]
- Mengel-From, J.; Thinggaard, M.; Dalgård, C.; Kyvik, K.O.; Christensen, K.; Christiansen, L. Mitochondrial DNA Copy Number in Peripheral Blood Cells Declines with Age and Is Associated with General Health among Elderly. Hum. Genet. 2014, 133, 1149–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Wang, Y.; Ye, K.; Picard, M.; Gu, Z. Independent Impacts of Aging on Mitochondrial DNA Quantity and Quality in Humans. BMC Genom. 2017, 18, 890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, L.; De Gaetano, A.; Mandrioli, M.; Van Tongeren, E.; Bortolotti, C.A.; Cossarizza, A.; Pinti, M. The Biology of Lonp1: More than a Mitochondrial Protease. Int. Rev. Cell Mol. Biol. 2020, 354, 1–61. [Google Scholar] [CrossRef]
- Di Rienzo, M.; Romagnoli, A.; Ciccosanti, F.; Refolo, G.; Consalvi, V.; Arena, G.; Valente, E.M.; Piacentini, M.; Fimia, G.M. AMBRA1 Regulates Mitophagy by Interacting with ATAD3A and Promoting PINK1 Stability. Autophagy 2022, 18, 1752–1762. [Google Scholar] [CrossRef]
- Zurita Rendón, O.; Shoubridge, E.A. LONP1 Is Required for Maturation of a Subset of Mitochondrial Proteins, and Its Loss Elicits an Integrated Stress Response. Mol. Cell. Biol. 2018, 38, e00412-17. [Google Scholar] [CrossRef] [Green Version]
- Hosgood, H.D.; Liu, C.-S.; Rothman, N.; Weinstein, S.J.; Bonner, M.R.; Shen, M.; Lim, U.; Virtamo, J.; Cheng, W.; Albanes, D.; et al. Mitochondrial DNA Copy Number and Lung Cancer Risk in a Prospective Cohort Study. Carcinogenesis 2010, 31, 847–849. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in Inflammation and Immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Dashti, M.; Alsaleh, H.; Eaaswarkhanth, M.; John, S.E.; Nizam, R.; Melhem, M.; Hebbar, P.; Sharma, P.; Al-Mulla, F.; Thanaraj, T.A. Delineation of Mitochondrial DNA Variants From Exome Sequencing Data and Association of Haplogroups With Obesity in Kuwait. Front. Genet. 2021, 12, 626260. [Google Scholar] [CrossRef]
- Ding, X.; Fang, T.; Pang, X.; Pan, X.; Tong, A.; Lin, Z.; Zheng, S.; Zheng, N. Mitochondrial DNA Abnormalities and Metabolic Syndrome. Front. Cell Dev. Biol. 2023, 11, 1153174. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The Metabolic Signature of Macrophage Responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef] [Green Version]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking Outside the Nucleus: Mitochondrial DNA Copy Number in Health and Disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Guha, M.; Avadhani, N.G. Mitochondrial Retrograde Signaling at the Crossroads of Tumor Bioenergetics, Genetics and Epigenetics. Mitochondrion 2013, 13, 577–591. [Google Scholar] [CrossRef] [Green Version]
- Jeng, J.-Y.; Yeh, T.-S.; Lee, J.-W.; Lin, S.-H.; Fong, T.-H.; Hsieh, R.-H. Maintenance of Mitochondrial DNA Copy Number and Expression Are Essential for Preservation of Mitochondrial Function and Cell Growth. J. Cell. Biochem. 2008, 103, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.N.; Czajka, A. Is Mitochondrial DNA Content a Potential Biomarker of Mitochondrial Dysfunction? Mitochondrion 2013, 13, 481–492. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Pulakat, L.; Whaley-Connell, A.; Sowers, J.R. Mitochondrial Biogenesis in the Metabolic Syndrome and Cardiovascular Disease. J. Mol. Med. 2010, 88, 993–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]
- Liu, J.; Lloyd, S.G. High-Fat, Low-Carbohydrate Diet Alters Myocardial Oxidative Stress and Impairs Recovery of Cardiac Function after Ischemia and Reperfusion in Obese Rats. Nutr. Res. 2013, 33, 311–321. [Google Scholar] [CrossRef] [Green Version]
- Skuratovskaia, D.; Zatolokin, P.; Vulf, M.; Mazunin, I.; Litvinova, L. Interrelation of Chemerin and TNF-α with MtDNA Copy Number in Adipose Tissues and Blood Cells in Obese Patients with and without Type 2 Diabetes. BMC Med. Genom. 2019, 12, 40. [Google Scholar] [CrossRef] [Green Version]
- Meng, S.; Wu, S.; Liang, L.; Liang, G.; Giovannucci, E.; De Vivo, I.; Nan, H. Leukocyte Mitochondrial DNA Copy Number, Anthropometric Indices, and Weight Change in US Women. Oncotarget 2016, 7, 60676–60686. [Google Scholar] [CrossRef] [Green Version]
- Skuratovskaia, D.A.; Sofronova, J.K.; Zatolokin, P.A.; Popadin, K.Y.; Vasilenko, M.A.; Litvinova, L.S.; Mazunin, I.O. Additional Evidence of the Link between MtDNA Copy Number and the Body Mass Index. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2018, 29, 1240–1244. [Google Scholar] [CrossRef]
- Liu, X.; Longchamps, R.J.; Wiggins, K.L.; Raffield, L.M.; Bielak, L.F.; Zhao, W.; Pitsillides, A.; Blackwell, T.W.; Yao, J.; Guo, X.; et al. Association of Mitochondrial DNA Copy Number with Cardiometabolic Diseases. Cell Genom. 2021, 1, 100006. [Google Scholar] [CrossRef] [PubMed]
- Koller, A.; Fazzini, F.; Lamina, C.; Rantner, B.; Kollerits, B.; Stadler, M.; Klein-Weigel, P.; Fraedrich, G.; Kronenberg, F. Mitochondrial DNA Copy Number Is Associated with All-Cause Mortality and Cardiovascular Events in Patients with Peripheral Arterial Disease. J. Intern. Med. 2020, 287, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agius, R.; Pace, N.P.; Fava, S. Reduced Leukocyte Mitochondrial Copy Number in Metabolic Syndrome and Metabolically Healthy Obesity. Front. Endocrinol. 2022, 13, 886957. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.-H.; Su, S.-L.; Hsieh, M.-C.; Cheng, W.-L.; Chang, C.-C.; Wu, H.-L.; Kuo, C.-L.; Lin, T.-T.; Liu, C.-S. Depleted Leukocyte Mitochondrial DNA Copy Number in Metabolic Syndrome. J. Atheroscler. Thromb. 2011, 18, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.-H.; Im, J.-A.; Lee, D.-C. The Relationship between Leukocyte Mitochondrial DNA Contents and Metabolic Syndrome in Postmenopausal Women. Menopause 2012, 19, 582–587. [Google Scholar] [CrossRef]
- Fazzini, F.; Lamina, C.; Raftopoulou, A.; Koller, A.; Fuchsberger, C.; Pattaro, C.; Del Greco, F.M.; Döttelmayer, P.; Fendt, L.; Fritz, J.; et al. Association of Mitochondrial DNA Copy Number with Metabolic Syndrome and Type 2 Diabetes in 14 176 Individuals. J. Intern. Med. 2021, 290, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Kim, H.K.; Ko, J.-H.; Bang, H.; Lee, D.-C. The Relationship between Leukocyte Mitochondrial DNA Copy Number and Telomere Length in Community-Dwelling Elderly Women. PLoS ONE 2013, 8, e67227. [Google Scholar] [CrossRef] [Green Version]
- Cree, L.M.; Patel, S.K.; Pyle, A.; Lynn, S.; Turnbull, D.M.; Chinnery, P.F.; Walker, M. Age-Related Decline in Mitochondrial DNA Copy Number in Isolated Human Pancreatic Islets. Diabetologia 2008, 51, 1440–1443. [Google Scholar] [CrossRef] [Green Version]
- Picard, M.; Turnbull, D.M. Linking the Metabolic State and Mitochondrial DNA in Chronic Disease, Health, and Aging. Diabetes 2013, 62, 672–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, A.A.; Sundquist, J.; Hedelius, A.; Palmér, K.; Wang, X.; Sundquist, K. Association of Mitochondrial DNA Copy Number with Prevalent and Incident Type 2 Diabetes in Women: A Population-Based Follow-up Study. Sci. Rep. 2021, 11, 4608. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liang, H.; Huang, R.; Weng, X.; Zheng, L.; Wang, Y.; Zheng, X.; Gu, Z.; Chen, F.; Shao, J.; et al. Higher Mitochondrial DNA Copy Number Is Associated with Metformin-Induced Weight Loss. Commun. Med. 2023, 3, 29. [Google Scholar] [CrossRef]
- Skuratovskaia, D.; Litvinova, L.; Vulf, M.; Zatolokin, P.; Popadin, K.; Mazunin, I. From Normal to Obesity and Back: The Associations between Mitochondrial DNA Copy Number, Gender, and Body Mass Index. Cells 2019, 8, 430. [Google Scholar] [CrossRef] [Green Version]
- Matilainen, O.; Quirós, P.M.; Auwerx, J. Mitochondria and Epigenetics—Crosstalk in Homeostasis and Stress. Trends Cell Biol. 2017, 27, 453–463. [Google Scholar] [CrossRef]
- Castegna, A.; Iacobazzi, V.; Infantino, V. The Mitochondrial Side of Epigenetics. Physiol. Genom. 2015, 47, 299–307. [Google Scholar] [CrossRef] [Green Version]
- D’Aquila, P.; Bellizzi, D.; Passarino, G. Mitochondria in Health, Aging and Diseases: The Epigenetic Perspective. Biogerontology 2015, 16, 569–585. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sumpter, J.A.; Newcomb, C.E.; Lane, J.A.; Grove, M.L.; Bressler, J.; Brody, J.A.; Floyd, J.S.; Bartz, T.M.; et al. Mitochondrial DNA Copy Number Can Influence Mortality and Cardiovascular Disease via Methylation of Nuclear DNA CpGs. Genome Med. 2020, 12, 84. [Google Scholar] [CrossRef]
- Murphy, S.K.; Huang, Z.; Hoyo, C. Differentially Methylated Regions of Imprinted Genes in Prenatal, Perinatal and Postnatal Human Tissues. PLoS ONE 2012, 7, e40924. [Google Scholar] [CrossRef] [Green Version]
- Fradin, D.; Boëlle, P.-Y.; Belot, M.-P.; Lachaux, F.; Tost, J.; Besse, C.; Deleuze, J.-F.; De Filippo, G.; Bougnères, P. Genome-Wide Methylation Analysis Identifies Specific Epigenetic Marks In Severely Obese Children. Sci. Rep. 2017, 7, 46311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, X.; Boyd-Kirkup, J.D.; McDermott, J.; Zhang, X.; Li, F.; Rong, B.; Zhang, R.; Miao, B.; Chen, P.; Cheng, H.; et al. The Stand-Biased Mitochondrial DNA Methylome And Its Regulation By DNMT3A. Genome Res. 2019, 10, 1622–1634. [Google Scholar] [CrossRef] [PubMed]
- Guantes, R.; Rastrojo, A.; Neves, R.; Lima, A.; Aguado, B.; Iborra, F.J. Global Variability in Gene Expression and Alternative Splicing Is Modulated by Mitochondrial Content. Genome Res. 2015, 25, 633–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Guo, Z.; Xu, J.; Zhang, J.; Cui, L.; Zhang, H.; Zang, S.; Ai, X. Single Nucleotide Polymorphisms In The D-Loop Region Of Mitochondrial DNA And Age-At0Onset Of Patients With Chronic Kidney Disease. Chin. Med. J. 2014, 127, 3088–3091. [Google Scholar] [PubMed]
- Stoccoro, A.; Smith, A.R.; Mosca, L.; Marocchi, A.; Gerardi, F.; Lunetta, C.; Cereda, C.; Gagliardi, S.; Lunnon, K.; Migliore, L.; et al. Reduced Mitochondrial D-Loop Methylation Levels in Sporadic Amyotrophic Lateral Sclerosis. Clin. Epigenetics 2020, 12, 137. [Google Scholar] [CrossRef]
- Mposhi, A.; Van der Wijst, M.G.; Faber, K.N.; Rots, M.G. Regulation of mitochondrial gene expression, the epigenetic enigma. Front. Biosci. (Landmark Ed) 2017, 22, 1099–1113. [Google Scholar] [CrossRef] [Green Version]
- Dostal, V.; Churchill, M.E.A. Cytosine Methylation of Mitochondrial DNA at CpG Sequences Impacts Transcription Factor A DNA Binding and Transcription. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 598–607. [Google Scholar] [CrossRef]
- Gao, J.; Wen, S.; Zhou, H.; Feng, S. De-Methylation of Displacement Loop of Mitochondrial DNA Is Associated with Increased Mitochondrial Copy Number and Nicotinamide Adenine Dinucleotide Subunit 2 Expression in Colorectal Cancer. Mol. Med. Rep. 2015, 12, 7033–7038. [Google Scholar] [CrossRef] [Green Version]
- Park, S.H.; Lee, S.Y.; Kim, S.A. Mitochondrial DNA Methylation Is Higher in Acute Coronary Syndrome Than in Stable Coronary Artery Disease. In Vivo 2021, 35, 181–189. [Google Scholar] [CrossRef]
- Low, H.C.; Chilian, W.M.; Ratnam, W.; Karupaiah, T.; Md Noh, M.F.; Mansor, F.; Ng, Z.X.; Pung, Y.F. Changes in Mitochondrial Epigenome in Type 2 Diabetes Mellitus. Br. J. Biomed. Sci. 2023, 80, 10884. [Google Scholar] [CrossRef]
- Hao, Z.; Wu, T.; Cui, X.; Zhu, P.; Tan, C.; Dou, X.; Hsu, K.W.; Lin, Y.T.; Peng, P.H.; Zang, L.S.; et al. N6-Deoxyadenosine Methylation In Mammalian Mitochondrial DNA. Mol. Cell 2020, 78, 382–395. [Google Scholar] [CrossRef]
- Bordoni, L.; Smerilli, V.; Nasuti, C.; Gabbianelli, R. Mitochondrial DNA Methylation and Copy Number Predict Body Composition in a Young Female Population. J. Transl. Med. 2019, 17, 399. [Google Scholar] [CrossRef] [Green Version]
- Bordoni, L.; Perugini, J.; Petracci, I.; Mercurio, E.D.; Lezoche, G.; Guerrieri, M.; Giordano, A.; Gabbianelli, R. Mitochondrial DNA in Visceral Adipose Tissue in Severe Obesity: From Copy Number to D-Loop Methylation. Front. Biosci. (Landmark Ed) 2022, 27, 172. [Google Scholar] [CrossRef]
- Corsi, S.; Iodice, S.; Vigna, L.; Cayir, A.; Mathers, J.C.; Bollati, V.; Byun, H.-M. Platelet Mitochondrial DNA Methylation Predicts Future Cardiovascular Outcome in Adults with Overweight and Obesity. Clin. Epigenetics 2020, 12, 29. [Google Scholar] [CrossRef] [Green Version]
- Baccarelli, A.A.; Byun, H.-M. Platelet Mitochondrial DNA Methylation: A Potential New Marker of Cardiovascular Disease. Clin. Epigenetics 2015, 7, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.D.; Linarelli, L.E.; Liu, L.; Wall, S.S.; Greenawald, M.H.; Seidel, R.W.; Estabrooks, P.A.; Almeida, F.A.; Cheng, Z. Insulin Resistance Is Associated with Epigenetic and Genetic Regulation of Mitochondrial DNA in Obese Humans. Clin. Epigenetics 2015, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khotina, V.A.; Vinokurov, A.Y.; Bagheri Ekta, M.; Sukhorukov, V.N.; Orekhov, A.N. Creation Of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomrdicines 2023, 2, 532. [Google Scholar] [CrossRef] [PubMed]
- Katada, S.; Mito, T.; Ogasawara, E.; Hayashi, J.-I.; Nakada, K. Mitochondrial DNA with a Large-Scale Deletion Causes Two Distinct Mitochondrial Disease Phenotypes in Mice. G3 (Bethesda) 2013, 3, 1545–1552. [Google Scholar] [CrossRef] [Green Version]
- Farmer, T.; Naslavsky, N.; Caplan, S. Tying Trafficking to Fusion and Fission at the Mighty Mitochondria. Traffic 2018, 19, 569–577. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. A Mitochondrial Paradigm of Metabolic and Degenerative Diseases, Aging, and Cancer: A Dawn for Evolutionary Medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, V.O.; De Rasmo, D.; Signorile, A.; Sardanelli, A.M.; Grattagliano, I.; Minerva, F.; Cardinale, G.; Portincasa, P.; Papa, S.; Palasciano, G. T16189C Mitochondrial DNA Variant Is Associated with Metabolic Syndrome in Caucasian Subjects. Nutrition 2011, 27, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Mueller, E.E.; Eder, W.; Ebner, S.; Schwaiger, E.; Santic, D.; Kreindl, T.; Stanger, O.; Paulweber, B.; Iglseder, B.; Oberkofler, H.; et al. The Mitochondrial T16189C Polymorphism Is Associated with Coronary Artery Disease in Middle European Populations. PLoS ONE 2011, 6, e16455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maassen, J.A.; ’T Hart, L.M.; Van Essen, E.; Heine, R.J.; Nijpels, G.; Jahangir Tafrechi, R.S.; Raap, A.K.; Janssen, G.M.C.; Lemkes, H.H.P.J. Mitochondrial Diabetes: Molecular Mechanisms and Clinical Presentation. Diabetes 2004, 53 (Suppl. S1), S103–S109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.; Davidson, E.; King, M.P. The Pathogenic A3243G Mutation in Human Mitochondrial TRNALeu(UUR) Decreases the Efficiency of Aminoacylation. Biochemistry 2003, 42, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Ye, W.; Chen, S.; Jin, S.; Lu, J. A Novel Heteroplasmic Mitochondrial DNA Mutation, A8890G, in a Patient with Juvenile-onset Metabolic Syndrome: A Case Report. Mol. Med. Rep. 2013, 8, 1060–1066. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C. Mitochondrial DNA Variation in Human Radiation and Disease. Cell 2015, 163, 33–38. [Google Scholar] [CrossRef] [Green Version]
- Ebner, S.; Mangge, H.; Langhof, H.; Halle, M.; Siegrist, M.; Aigner, E.; Paulmichl, K.; Paulweber, B.; Datz, C.; Sperl, W.; et al. Mitochondrial Haplogroup T Is Associated with Obesity in Austrian Juveniles and Adults. PLoS ONE 2015, 10, e0135622. [Google Scholar] [CrossRef] [Green Version]
- Nardelli, C.; Labruna, G.; Liguori, R.; Mazzaccara, C.; Ferrigno, M.; Capobianco, V.; Pezzuti, M.; Castaldo, G.; Farinaro, E.; Contaldo, F.; et al. Haplogroup T Is an Obesity Risk Factor: Mitochondrial DNA Haplotyping in a Morbid Obese Population from Southern Italy. Biomed. Res. Int. 2013, 2013, 631082. [Google Scholar] [CrossRef] [Green Version]
- Dashti, M.; Alsaleh, H.; Rodriguez-Flores, J.L.; Eaaswarkhanth, M.; Al-Mulla, F.; Thanaraj, T.A. Mitochondrial Haplogroup J Associated with Higher Risk of Obesity in the Qatari Population. Sci. Rep. 2021, 11, 1091. [Google Scholar] [CrossRef]
- Chalkia, D.; Chang, Y.-C.; Derbeneva, O.; Lvova, M.; Wang, P.; Mishmar, D.; Liu, X.; Singh, L.N.; Chuang, L.-M.; Wallace, D.C. Mitochondrial DNA Associations with East Asian Metabolic Syndrome. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Fuku, N.; Park, K.S.; Yamada, Y.; Nishigaki, Y.; Cho, Y.M.; Matsuo, H.; Segawa, T.; Watanabe, S.; Kato, K.; Yokoi, K.; et al. Mitochondrial Haplogroup N9a Confers Resistance against Type 2 Diabetes in Asians. Am. J. Hum. Genet. 2007, 80, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.-J.; Oshida, Y.; Fuku, N.; Takeyasu, T.; Fujita, Y.; Kurata, M.; Sato, Y.; Ito, M.; Tanaka, M. Mitochondrial Genome Polymorphisms Associated with Type-2 Diabetes or Obesity. Mitochondrion 2005, 5, 15–33. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Hu, N.; Zhao, Q.; Wang, B.; Zhou, H.; Fu, Q.; Shen, L.; Chen, X.; Shen, F.; Lyu, J. MtDNA Haplogroup N9a Increases the Risk of Type 2 Diabetes by Altering Mitochondrial Function and Intracellular Mitochondrial Signals. Diabetes 2018, 67, 1441–1453. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.-Q.; Pang, Y.; Yu, C.-A.; Wen, J.-Y.; Zhang, Y.-G.; Li, X.-H. Novel Mutations of Mitochondrial DNA Associated with Type 2 Diabetes in Chinese Han Population. Tohoku J. Exp. Med. 2008, 215, 377–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, Q.; Zhang, W.; Wang, H.; Guan, X.; Lu, J.; Li, W. Effects of Mitochondrial Haplogroup N9a on Type 2 Diabetes Mellitus and Its Associated Complications. Exp. Ther. Med. 2015, 10, 1918–1924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, J.-H.; Trejaut, J.A.; Yen, J.-C.; Chen, Z.-S.; Ng, W.-M.; Huang, C.-Y.; Hsu, K.-N.; Hung, K.-H.; Hsiao, Y.; Wei, Y.-H.; et al. Mitochondrial DNA Association Study of Type 2 Diabetes with or without Ischemic Stroke in Taiwan. BMC Res. Notes 2014, 7, 223. [Google Scholar] [CrossRef] [Green Version]
- Shen, F.-C.; Weng, S.-W.; Tsai, M.-H.; Su, Y.-J.; Li, S.-C.; Chang, S.-J.; Chen, J.-F.; Chang, Y.-H.; Liou, C.-W.; Lin, T.-K.; et al. Mitochondrial Haplogroups Have a Better Correlation to Insulin Requirement than Nuclear Genetic Variants for Type 2 Diabetes Mellitus in Taiwanese Individuals. J. Diabetes Investig. 2022, 13, 201–208. [Google Scholar] [CrossRef]
- Jiang, W.; Li, R.; Zhang, Y.; Wang, P.; Wu, T.; Lin, J.; Yu, J.; Gu, M. Mitochondrial DNA Mutations Associated with Type 2 Diabetes Mellitus in Chinese Uyghur Population. Sci. Rep. 2017, 7, 16989. [Google Scholar] [CrossRef] [Green Version]
- De Gaetano, A.; Solodka, K.; Zanini, G.; Selleri, V.; Mattioli, A.V.; Nasi, M.; Pinti, M. Molecular Mechanisms of MtDNA-Mediated Inflammation. Cells 2021, 10, 2898. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Hajizadeh, S.; Holme, E.; Jonsson, I.-M.; Tarkowski, A. Endogenously Oxidized Mitochondrial DNA Induces in Vivo and in Vitro Inflammatory Responses. J. Leukoc. Biol. 2004, 75, 995–1000. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Pazmandi, K.; Agod, Z.; Kumar, B.V.; Szabo, A.; Fekete, T.; Sogor, V.; Veres, A.; Boldogh, I.; Rajnavolgyi, E.; Lanyi, A.; et al. Oxidative Modification Enhances the Immunostimulatory Effects of Extracellular Mitochondrial DNA on Plasmacytoid Dendritic Cells. Free. Radic. Biol. Med. 2014, 77, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Kunkel, G.H.; Chaturvedi, P.; Tyagi, S.C. Mitochondrial Pathways to Cardiac Recovery: TFAM. Heart Fail Rev. 2016, 21, 499–517. [Google Scholar] [CrossRef] [Green Version]
- Piantadosi, C.A. Mitochondrial DNA, Oxidants, and Innate Immunity. Free. Radic. Biol. Med. 2020, 152, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, Q.; Yuan, K.; Yuan, J. MtDNA in the Pathogenesis of Cardiovascular Diseases. Dis. Markers 2021, 2021, 7157109. [Google Scholar] [CrossRef]
- Krychtiuk, K.A.; Wurm, R.; Ruhittel, S.; Lenz, M.; Huber, K.; Wojta, J.; Heinz, G.; Hülsmann, M.; Speidl, W.S. Release of Mitochondrial DNA Is Associated with Mortality in Severe Acute Heart Failure. Eur. Heart J. Acute Cardiovasc. Care 2020, 9, 419–428. [Google Scholar] [CrossRef]
- Bliksøen, M.; Mariero, L.H.; Torp, M.K.; Baysa, A.; Ytrehus, K.; Haugen, F.; Seljeflot, I.; Vaage, J.; Valen, G.; Stensløkken, K.-O. Extracellular MtDNA Activates NF-ΚB via Toll-like Receptor 9 and Induces Cell Death in Cardiomyocytes. Basic Res. Cardiol. 2016, 111, 42. [Google Scholar] [CrossRef]
- Donath, M.Y.; Meier, D.T.; Böni-Schnetzler, M. Inflammation in the Pathophysiology and Therapy of Cardiometabolic Disease. Endocr. Rev. 2019, 40, 1080–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinti, M.; Cevenini, E.; Nasi, M.; De Biasi, S.; Salvioli, S.; Monti, D.; Benatti, S.; Gibellini, L.; Cotichini, R.; Stazi, M.A.; et al. Circulating Mitochondrial DNA Increases with Age and Is a Familiar Trait: Implications for “Inflamm-Aging. ” Eur. J. Immunol. 2014, 44, 1552–1562. [Google Scholar] [CrossRef]
- Ueda, H.; Yamaguchi, O.; Taneike, M.; Akazawa, Y.; Wada-Kobayashi, H.; Sugihara, R.; Yorifuji, H.; Nakayama, H.; Omiya, S.; Murakawa, T.; et al. Administration of a TLR9 Inhibitor Attenuates the Development and Progression of Heart Failure in Mice. JACC Basic Transl. Sci. 2019, 4, 348–363. [Google Scholar] [CrossRef]
- Liu, Y.; Jesus, A.A.; Marrero, B.; Yang, D.; Ramsey, S.E.; Sanchez, G.A.M.; Tenbrock, K.; Wittkowski, H.; Jones, O.Y.; Kuehn, H.S.; et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med. 2014, 371, 507–518. [Google Scholar] [CrossRef] [Green Version]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA Stress Primes the Antiviral Innate Immune Response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP Synthase Is a Cytosolic DNA Sensor That Activates the Type I Interferon Pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.-C.; Chen, Z.J. Structural Basis of STING Binding with and Phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-Mediated, STING-Dependent Proinflammatory Gene Induction Necessitates Canonical NF-ΚB Activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Nuevo, A.; Zorzano, A. The Sensing of Mitochondrial DAMPs by Non-Immune Cells. Cell Stress 2019, 3, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of Innate Immune Adaptor Proteins MAVS, STING, and TRIF Induces IRF3 Activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [Green Version]
- Balka, K.R.; Louis, C.; Saunders, T.L.; Smith, A.M.; Calleja, D.J.; D’Silva, D.B.; Moghaddas, F.; Tailler, M.; Lawlor, K.E.; Zhan, Y.; et al. TBK1 and IKKε Act Redundantly to Mediate STING-Induced NF-ΚB Responses in Myeloid Cells. Cell Rep. 2020, 31, 107492. [Google Scholar] [CrossRef]
- Hopfner, K.-P.; Hornung, V. Molecular Mechanisms and Cellular Functions of CGAS-STING Signalling. Nat. Rev. Mol. Cell Biol. 2020, 21, 501–521. [Google Scholar] [CrossRef] [PubMed]
- Oduro, P.K.; Zheng, X.; Wei, J.; Yang, Y.; Wang, Y.; Zhang, H.; Liu, E.; Gao, X.; Du, M.; Wang, Q. The CGAS-STING Signaling in Cardiovascular and Metabolic Diseases: Future Novel Target Option for Pharmacotherapy. Acta Pharm. Sin. B 2022, 12, 50–75. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Pastukh, V.M.; Ruchko, M.V.; Simmons, J.D.; Richards, W.O.; Rachek, L.I. Plasma Mitochondrial DNA Is Elevated in Obese Type 2 Diabetes Mellitus Patients and Correlates Positively with Insulin Resistance. PLoS ONE 2019, 14, e0222278. [Google Scholar] [CrossRef] [PubMed]
- Nie, S.; Lu, J.; Wang, L.; Gao, M. Pro-Inflammatory Role of Cell-Free Mitochondrial DNA in Cardiovascular Diseases. IUBMB Life 2020, 72, 1879–1890. [Google Scholar] [CrossRef]
- Andreeva, L.; Hiller, B.; Kostrewa, D.; Lässig, C.; de Oliveira Mann, C.C.; Jan Drexler, D.; Maiser, A.; Gaidt, M.; Leonhardt, H.; Hornung, V.; et al. CGAS Senses Long and HMGB/TFAM-Bound U-Turn DNA by Forming Protein-DNA Ladders. Nature 2017, 549, 394–398. [Google Scholar] [CrossRef]
- Kumari, M.; Wang, X.; Lantier, L.; Lyubetskaya, A.; Eguchi, J.; Kang, S.; Tenen, D.; Roh, H.C.; Kong, X.; Kazak, L.; et al. IRF3 Promotes Adipose Inflammation and Insulin Resistance and Represses Browning. J. Clin. Investig. 2016, 126, 2839–2854. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Cervantes, C.; Liu, J.; He, S.; Zhou, H.; Zhang, B.; Cai, H.; Yin, D.; Hu, D.; Li, Z.; et al. DsbA-L Prevents Obesity-Induced Inflammation and Insulin Resistance by Suppressing the MtDNA Release-Activated CGAS-CGAMP-STING Pathway. Proc. Natl. Acad. Sci. USA 2017, 114, 12196–12201. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Liu, M.; Zhang, J.; Chen, H.; Dong, L.Q.; Liu, F. DsbA-L Alleviates Endoplasmic Reticulum Stress-Induced Adiponectin Downregulation. Diabetes 2010, 59, 2809–2816. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Cervantes, C.; He, S.; He, J.; Plasko, G.R.; Wen, J.; Li, Z.; Yin, D.; Zhang, C.; Liu, M.; et al. Mitochondrial Stress-Activated CGAS-STING Pathway Inhibits Thermogenic Program and Contributes to Overnutrition-Induced Obesity in Mice. Commun. Biol. 2020, 3, 257. [Google Scholar] [CrossRef]
- Zhao, P.; Wong, K.I.; Sun, X.; Reilly, S.M.; Uhm, M.; Liao, Z.; Skorobogatko, Y.; Saltiel, A.R. TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue. Cell 2018, 172, 731–743.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Tang, N.; Liu, F.-Y.; Yang, Z.; Ma, S.-Q.; An, P.; Wu, H.-M.; Fan, D.; Tang, Q.-Z. TLR9 Deficiency Alleviates Doxorubicin-Induced Cardiotoxicity via the Regulation of Autophagy. J. Cell. Mol. Med. 2020, 24, 10913–10923. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA That Escapes from Autophagy Causes Inflammation and Heart Failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; He, S.; Kong, N.; Zhu, Y.; Tang, Y.; Li, J.; Liu, Z.; Liu, J.; Gong, J. Cpg-ODN, a TLR9 Agonist, Aggravates Myocardial Ischemia/Reperfusion Injury by Activation of TLR9-P38 MAPK Signaling. Cell. Physiol. Biochem. 2018, 47, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Pelka, K.; Phulphagar, K.; Zimmermann, J.; Stahl, R.; Schmid-Burgk, J.L.; Schmidt, T.; Spille, J.-H.; Labzin, L.I.; Agrawal, S.; Kandimalla, E.R.; et al. Cutting Edge: The UNC93B1 Tyrosine-Based Motif Regulates Trafficking and TLR Responses via Separate Mechanisms. J. Immunol. 2014, 193, 3257–3261. [Google Scholar] [CrossRef] [Green Version]
- Devaraj, S.; Adams-Huet, B.; Jialal, I. Endosomal Toll-Like Receptor Status in Patients with Metabolic Syndrome. Metab. Syndr. Relat. Disord. 2015, 13, 477–480. [Google Scholar] [CrossRef]
- Nishimoto, S.; Fukuda, D.; Higashikuni, Y.; Tanaka, K.; Hirata, Y.; Murata, C.; Kim-Kaneyama, J.R.; Sato, F.; Bando, M.; Yagi, S.; et al. Obesity-induced DNA released from adipocytes stimulates chronic adipose tissue inflammation and insulin resistance. Sci. Adv. 2016, 2, e1501332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, W.; Wen, C.; Zeng, A.; Hu, X. Increased Levels of Circulating Oxidized Mitochondrial DNA Contribute to Chronic Inflammation in Metabolic Syndrome, and MitoQ-Based Antioxidant Therapy Alleviates This DNA-Induced Inflammation. Mol. Cell. Endocrinol. 2023, 560, 111812. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; He, Y.; Ming, H.; Lei, S.; Leng, Y.; Xia, Z.-Y. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J. Diabetes Res. 2019, 2019, 8151836. [Google Scholar] [CrossRef] [Green Version]
- Pahwa, R.; Singh, A.; Adams-Huet, B.; Devaraj, S.; Jialal, I. Increased Inflammasome Activity in Subcutaneous Adipose Tissue of Patients with Metabolic Syndrome. Diabetes/Metabolism Res. Rev. 2021, 37, e3383. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.K.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy Proteins Regulate Innate Immune Responses by Inhibiting the Release of Mitochondrial DNA Mediated by the NALP3 Inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ward, G.A.; McGraw, K.L.; Abbas-Aghababazadeh, F.; Meyer, B.S.; McLemore, A.F.; Vincelette, N.D.; Lam, N.B.; Aldrich, A.L.; Al Ali, N.H.; Padron, E.; et al. Oxidized Mitochondrial DNA Released after Inflammasome Activation Is a Disease Biomarker for Myelodysplastic Syndromes. Blood Adv. 2021, 5, 2216–2228. [Google Scholar] [CrossRef] [PubMed]
- Crewe, C.; Schafer, C.; Lee, I.; Kinter, M.; Szweda, L.I. Regulation of Pyruvate Dehydrogenase Kinase 4 in the Heart through Degradation by the Lon Protease in Response to Mitochondrial Substrate Availability. J. Biol. Chem. 2017, 292, 305–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sepuri, N.B.V.; Angireddy, R.; Srinivasan, S.; Guha, M.; Spear, J.; Lu, B.; Anandatheerthavarada, H.K.; Suzuki, C.K.; Avadhani, N.G. Mitochondrial LON Protease-Dependent Degradation of Cytochrome c Oxidase Subunits under Hypoxia and Myocardial Ischemia. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 519–528. [Google Scholar] [CrossRef]
- Bota, D.A.; Davies, K.J.A. Mitochondrial Lon Protease in Human Disease and Aging: Including an Etiologic Classification of Lon-Related Diseases and Disorders. Free. Radic. Biol. Med. 2016, 100, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Quirós, P.M.; Español, Y.; Acín-Pérez, R.; Rodríguez, F.; Bárcena, C.; Watanabe, K.; Calvo, E.; Loureiro, M.; Fernández-García, M.S.; Fueyo, A.; et al. ATP-Dependent Lon Protease Controls Tumor Bioenergetics by Reprogramming Mitochondrial Activity. Cell Rep. 2014, 8, 542–556. [Google Scholar] [CrossRef] [Green Version]
- Kuo, C.-Y.; Chiu, Y.-C.; Lee, A.Y.-L.; Hwang, T.-L. Mitochondrial Lon Protease Controls ROS-Dependent Apoptosis in Cardiomyocyte under Hypoxia. Mitochondrion 2015, 23, 7–16. [Google Scholar] [CrossRef]
- Nie, X.; Li, M.; Lu, B.; Zhang, Y.; Lan, L.; Chen, L.; Lu, J. Down-Regulating Overexpressed Human Lon in Cervical Cancer Suppresses Cell Proliferation and Bioenergetics. PLoS ONE 2013, 8, e81084. [Google Scholar] [CrossRef]
- Strauss, K.A.; Jinks, R.N.; Puffenberger, E.G.; Venkatesh, S.; Singh, K.; Cheng, I.; Mikita, N.; Thilagavathi, J.; Lee, J.; Sarafianos, S.; et al. CODAS Syndrome Is Associated with Mutations of LONP1, Encoding Mitochondrial AAA+ Lon Protease. Am. J. Hum. Genet. 2015, 96, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Dikoglu, E.; Alfaiz, A.; Gorna, M.; Bertola, D.; Chae, J.H.; Cho, T.-J.; Derbent, M.; Alanay, Y.; Guran, T.; Kim, O.-H.; et al. Mutations in LONP1, a Mitochondrial Matrix Protease, Cause CODAS Syndrome. Am. J. Med. Genet. A 2015, 167, 1501–1509. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Li, M.; Saito, T.; Tong, M.; Rashed, E.; Mareedu, S.; Zhai, P.; Bárcena, C.; López-Otín, C.; Yehia, G.; et al. Mitochondrial LonP1 Protects Cardiomyocytes from Ischemia/Reperfusion Injury in Vivo. J. Mol. Cell. Cardiol. 2019, 128, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottis, A.; Jovaisaite, V.; Auwerx, J. The Mitochondrial Unfolded Protein Response in Mammalian Physiology. Mamm. Genome 2014, 25, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welle, S.; Glueck, S.B. In for the Long Run: Focus on “Lifelong Voluntary Exercise in the Mouse Prevents Age-Related Alterations in Gene Expression in the Heart. ” Physiol. Genom. 2003, 12, 71–72. [Google Scholar] [CrossRef]
- Jovaisaite, V.; Mouchiroud, L.; Auwerx, J. The Mitochondrial Unfolded Protein Response, a Conserved Stress Response Pathway with Implications in Health and Disease. J. Exp. Biol. 2014, 217, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Held, N.M.; Houtkooper, R.H. Mitochondrial Quality Control Pathways as Determinants of Metabolic Health. Bioessays 2015, 37, 867–876. [Google Scholar] [CrossRef]
- Lee, J.H.; Jung, S.-B.; Lee, S.E.; Kim, J.E.; Kim, J.T.; Kang, Y.E.; Kang, S.G.; Yi, H.-S.; Ko, Y.B.; Lee, K.H.; et al. Expression of LONP1 Is High in Visceral Adipose Tissue in Obesity, and Is Associated with Glucose and Lipid Metabolism. Endocrinol. Metab. 2021, 36, 661–671. [Google Scholar] [CrossRef]
- Deshwal, S.; Fiedler, K.U.; Langer, T. Mitochondrial Proteases: Multifaceted Regulators of Mitochondrial Plasticity. Annu. Rev. Biochem. 2020, 89, 501–528. [Google Scholar] [CrossRef] [Green Version]
- Yi, H.-S.; Chang, J.Y.; Shong, M. The Mitochondrial Unfolded Protein Response and Mitohormesis: A Perspective on Metabolic Diseases. J. Mol. Endocrinol. 2018, 61, R91–R105. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.J.; Jung, S.-B.; Lee, S.E.; Kang, S.G.; Lee, J.H.; Ryu, M.J.; Chung, H.K.; Chang, J.Y.; Kim, Y.K.; Hong, H.J.; et al. An Adipocyte-Specific Defect in Oxidative Phosphorylation Increases Systemic Energy Expenditure and Protects against Diet-Induced Obesity in Mouse Models. Diabetologia 2020, 63, 837–852. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.K.; Ryu, D.; Kim, K.S.; Chang, J.Y.; Kim, Y.K.; Yi, H.-S.; Kang, S.G.; Choi, M.J.; Lee, S.E.; Jung, S.-B.; et al. Growth Differentiation Factor 15 Is a Myomitokine Governing Systemic Energy Homeostasis. J. Cell Biol. 2017, 216, 149–165. [Google Scholar] [CrossRef]
- Rigotto, G.; Basso, E. Mitochondrial Dysfunctions: A Thread Sewing Together Alzheimer’s Disease, Diabetes, and Obesity. Oxid. Med. Cell. Longev. 2019, 2019, 7210892. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Chung, K.; Lee, H.; Lee, K.; Lim, J.H.; Song, J. Downregulation of Mitochondrial Lon Protease Impairs Mitochondrial Function and Causes Hepatic Insulin Resistance in Human Liver SK-HEP-1 Cells. Diabetologia 2011, 54, 1437–1446. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, S.; Buzkova, J.; Muniandy, M.; Kaksonen, R.; Ollikainen, M.; Ismail, K.; Hakkarainen, A.; Lundbom, J.; Lundbom, N.; Vuolteenaho, K.; et al. Impaired Mitochondrial Biogenesis in Adipose Tissue in Acquired Obesity. Diabetes 2015, 64, 3135–3145. [Google Scholar] [CrossRef] [Green Version]
- Dahlman, I.; Forsgren, M.; Sjögren, A.; Nordström, E.A.; Kaaman, M.; Näslund, E.; Attersand, A.; Arner, P. Downregulation of Electron Transport Chain Genes in Visceral Adipose Tissue in Type 2 Diabetes Independent of Obesity and Possibly Involving Tumor Necrosis Factor-Alpha. Diabetes 2006, 55, 1792–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Lanza, I.R.; Swain, J.M.; Sarr, M.G.; Nair, K.S.; Jensen, M.D. Adipocyte Mitochondrial Function Is Reduced in Human Obesity Independent of Fat Cell Size. J. Clin. Endocrinol. Metab. 2014, 99, E209–E216. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Fu, T.; Guo, Q.; Zhou, D.; Sun, W.; Zhou, Z.; Chen, X.; Zhang, J.; Liu, L.; Xiao, L.; et al. Disuse-Associated Loss of the Protease LONP1 in Muscle Impairs Mitochondrial Function and Causes Reduced Skeletal Muscle Mass and Strength. Nat. Commun. 2022, 13, 894. [Google Scholar] [CrossRef]
- Fukuda, R.; Zhang, H.; Kim, J.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, C.-S.; Meng, S.; Garbis, S.D.; Moradian, A.; Taylor, R.W.; Sweredoski, M.J.; Lomenick, B.; Chan, D.C. LONP1 and MtHSP70 Cooperate to Promote Mitochondrial Protein Folding. Nat. Commun. 2021, 12, 265. [Google Scholar] [CrossRef]
- Grzybek, M.; Palladini, A.; Alexaki, V.I.; Surma, M.A.; Simons, K.; Chavakis, T.; Klose, C.; Coskun, Ü. Comprehensive and Quantitative Analysis of White and Brown Adipose Tissue by Shotgun Lipidomics. Mol. Metab. 2019, 22, 12–20. [Google Scholar] [CrossRef]
- Donohoe, F.; Wilkinson, M.; Baxter, E.; Brennan, D.J. Mitogen-Activated Protein Kinase (MAPK) and Obesity-Related Cancer. Int. J. Mol. Sci. 2020, 21, 1241. [Google Scholar] [CrossRef] [Green Version]
- Och, A.; Och, M.; Nowak, R.; Podgórska, D.; Podgórski, R. Berberine, a Herbal Metabolite in the Metabolic Syndrome: The Risk Factors, Course, and Consequences of the Disease. Molecules 2022, 27, 1351. [Google Scholar] [CrossRef]
- Matsushima, Y.; Goto, Y.-I.; Kaguni, L.S. Mitochondrial Lon Protease Regulates Mitochondrial DNA Copy Number and Transcription by Selective Degradation of Mitochondrial Transcription Factor A (TFAM). Proc. Natl. Acad. Sci. USA 2010, 107, 18410–18415. [Google Scholar] [CrossRef]
- Fu, G.K.; Markovitz, D.M. The Human LON Protease Binds to Mitochondrial Promoters in a Single-Stranded, Site-Specific, Strand-Specific Manner. Biochemistry 1998, 37, 1905–1909. [Google Scholar] [CrossRef]
- Lu, B.; Liu, T.; Crosby, J.A.; Thomas-Wohlever, J.; Lee, I.; Suzuki, C.K. The ATP-Dependent Lon Protease of Mus Musculus Is a DNA-Binding Protein That Is Functionally Conserved between Yeast and Mammals. Gene 2003, 306, 45–55. [Google Scholar] [CrossRef]
- Chen, S.-H.; Suzuki, C.K.; Wu, S.-H. Thermodynamic Characterization of Specific Interactions between the Human Lon Protease and G-Quartet DNA. Nucleic Acids Res. 2008, 36, 1273–1287. [Google Scholar] [CrossRef] [Green Version]
- Bota, D.A.; Ngo, J.K.; Davies, K.J.A. Downregulation of the Human Lon Protease Impairs Mitochondrial Structure and Function and Causes Cell Death. Free. Radic. Biol. Med. 2005, 38, 665–677. [Google Scholar] [CrossRef] [Green Version]
- Lu, B.; Yadav, S.; Shah, P.G.; Liu, T.; Tian, B.; Pukszta, S.; Villaluna, N.; Kutejová, E.; Newlon, C.S.; Santos, J.H.; et al. Roles for the Human ATP-Dependent Lon Protease in Mitochondrial DNA Maintenance. J. Biol. Chem. 2007, 282, 17363–17374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Gaetano, A.; Gibellini, L.; Bianchini, E.; Borella, R.; De Biasi, S.; Nasi, M.; Boraldi, F.; Cossarizza, A.; Pinti, M. Impaired Mitochondrial Morphology and Functionality in Lonp1wt/- Mice. J. Clin. Med. 2020, 9, 1783. [Google Scholar] [CrossRef] [PubMed]
- Polo, M.; Alegre, F.; Moragrega, A.B.; Gibellini, L.; Marti-Rodrigo, A.; Blas-Garcia, A.; Esplugues, J.V.; Apostolova, N. Lon Protease: A Novel Mitochondrial Matrix Protein in the Interconnection between Drug-Induced Mitochondrial Dysfunction and Endoplasmic Reticulum Stress. Br. J. Pharmacol. 2017, 174, 4409–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horner, S.M.; Wilkins, C.; Badil, S.; Iskarpatyoti, J.; Gale, M. Proteomic Analysis of Mitochondrial-Associated ER Membranes (MAM) during RNA Virus Infection Reveals Dynamic Changes in Protein and Organelle Trafficking. PLoS ONE 2015, 10, e0117963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-Depth Proteomic Analysis of Mammalian Mitochondria-Associated Membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Pomatto, L.C.D.; Raynes, R.; Davies, K.J.A. The Peroxisomal Lon Protease LonP2 in Aging and Disease: Functions and Comparisons with Mitochondrial Lon Protease LonP1. Biol. Rev. 2017, 92, 739–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibellini, L.; Borella, R.; De Gaetano, A.; Zanini, G.; Tartaro, D.L.; Carnevale, G.; Beretti, F.; Losi, L.; De Biasi, S.; Nasi, M.; et al. Evidence for Mitochondrial Lonp1 Expression in the Nucleus. Sci. Rep. 2022, 12, 10877. [Google Scholar] [CrossRef]
- Pinti, M.; Gibellini, L.; Liu, Y.; Xu, S.; Lu, B.; Cossarizza, A. Mitochondrial Lon Protease at the Crossroads of Oxidative Stress, Ageing and Cancer. Cell. Mol. Life Sci. 2015, 72, 4807–4824. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Dasgupta, R.; Bagchi, A. Elucidation of the Molecular Mechanism of Heat Shock Proteins and Its Correlation with K722Q Mutations in Lon Protease. Biosystems 2017, 159, 12–22. [Google Scholar] [CrossRef]
- Puri, N.; Karzai, A.W. HspQ Functions as a Unique Specificity-Enhancing Factor for the AAA+ Lon Protease. Mol. Cell 2017, 66, 672–683.e4. [Google Scholar] [CrossRef] [Green Version]
- Ngo, J.K.; Davies, K.J.A. Mitochondrial Lon Protease Is a Human Stress Protein. Free. Radic. Biol. Med. 2009, 46, 1042–1048. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.S.; Houry, W.A. Recent Advances in Targeting Human Mitochondrial AAA+ Proteases to Develop Novel Cancer Therapeutics. Adv. Exp. Med. Biol. 2019, 1158, 119–142. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todosenko, N.; Khaziakhmatova, O.; Malashchenko, V.; Yurova, K.; Bograya, M.; Beletskaya, M.; Vulf, M.; Gazatova, N.; Litvinova, L. Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity. Int. J. Mol. Sci. 2023, 24, 12012. https://doi.org/10.3390/ijms241512012
Todosenko N, Khaziakhmatova O, Malashchenko V, Yurova K, Bograya M, Beletskaya M, Vulf M, Gazatova N, Litvinova L. Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity. International Journal of Molecular Sciences. 2023; 24(15):12012. https://doi.org/10.3390/ijms241512012
Chicago/Turabian StyleTodosenko, Natalia, Olga Khaziakhmatova, Vladimir Malashchenko, Kristina Yurova, Maria Bograya, Maria Beletskaya, Maria Vulf, Natalia Gazatova, and Larisa Litvinova. 2023. "Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity" International Journal of Molecular Sciences 24, no. 15: 12012. https://doi.org/10.3390/ijms241512012
APA StyleTodosenko, N., Khaziakhmatova, O., Malashchenko, V., Yurova, K., Bograya, M., Beletskaya, M., Vulf, M., Gazatova, N., & Litvinova, L. (2023). Mitochondrial Dysfunction Associated with mtDNA in Metabolic Syndrome and Obesity. International Journal of Molecular Sciences, 24(15), 12012. https://doi.org/10.3390/ijms241512012