


Epithelial–Mesenchymal Transition Mechanisms in Chronic Airway Diseases: A Common Process to Target?

 ,
,
Abstract
1. Introduction
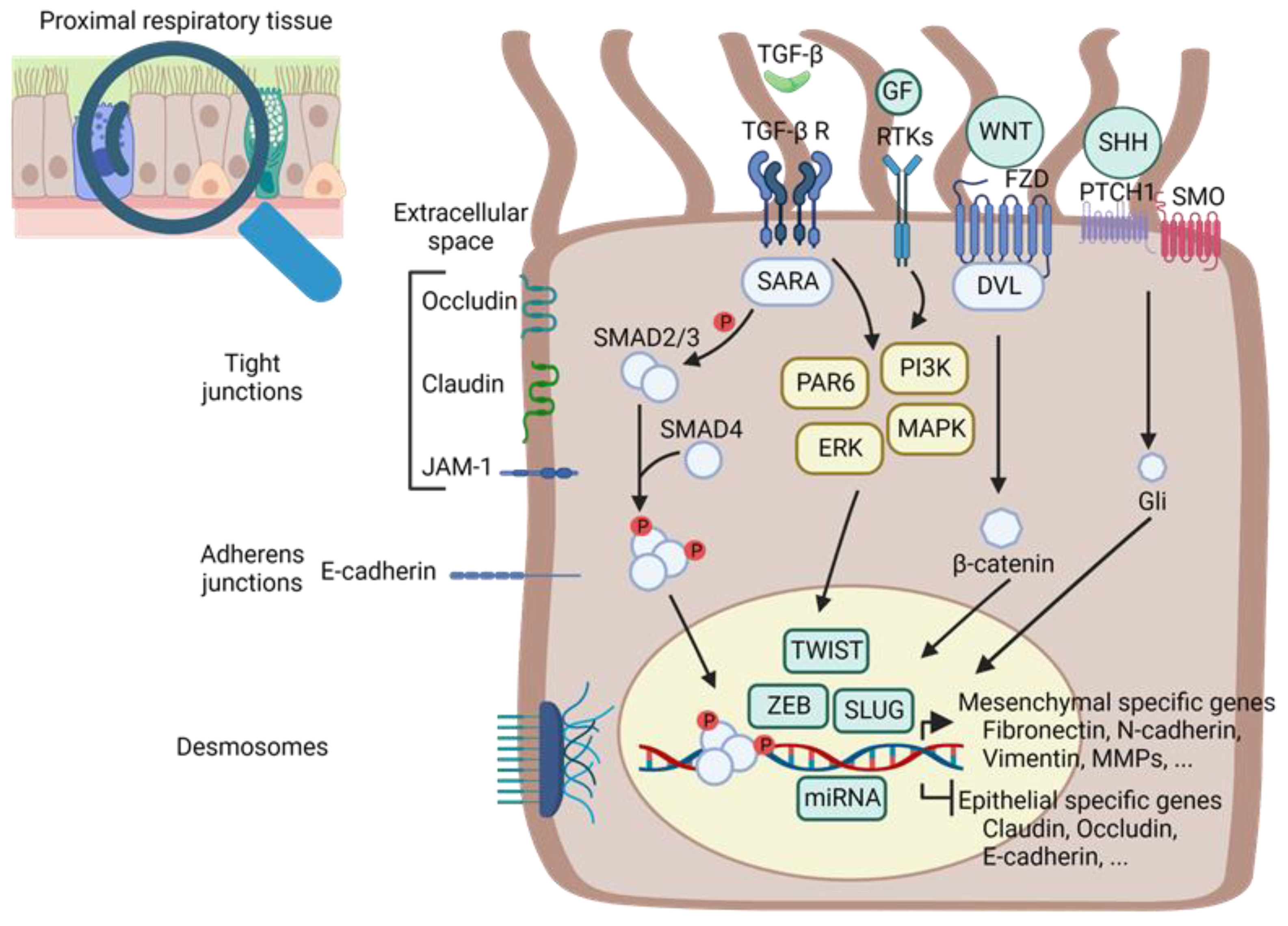
2. Common Genetic and Biochemical Mechanisms of EMT
3. Asthma
3.1. Chronic Inflammation, EMT and Airway Remodelling

{kind=link}
| Models | Techniques | EMT Program | AR | EMT Signal | EMT TFs | EMT Mark | Refs. | Year |
|---|---|---|---|---|---|---|---|---|
| HBECs of asthma and control subjects monolayer and ALI culture | RT-qPCR WB SDS-PAGE | +TGF-β1 10–50 ng/mL 72 h: spindle-shaped appearance  E-cadherin and ZO-1 (protein) E-cadherin and ZO-1 (protein) | ||||||
| IHC and IF |  fibronectin, vimentin and α-SMA (mRNA and protein) fibronectin, vimentin and α-SMA (mRNA and protein) | x | x | x | [87] | 2009 | ||
| collagen-1α1, fibrinogen, connective tissue growth factor and TGF-β1 (mRNA) Smad3-dependent process | ||||||||
| 16HBE 14o- cell line | RT-qPCR Cell proliferation WB ELISA | TGF-β1, IL-4 and IL-17A stimulation (72 h): proportion with a spindle-shape, fibroblast-like morphology with reduced cell–cell contact E-cadherin (mRNA and protein) α-SMA (mRNA and protein) pERK1/2 | x | x | x | [90] | 2013 | |
| 16HBE 14o- cell line | IF WB SDS-PAGE | TGF-β1 5 ng/mL: E-cadherin (protein) vimentin and fibronectin (protein) glycogen synthase kinase-3β (protein) | x | x | x | [93] | 2010 | |
| ALI normal HBECs | WB | +TGF-β1 + HDM 50 μg/mL: vimentin (protein) cytokeratin fibroblast-specific protein-1 (protein) delocalisation of E-cadherin | x | x | x | [93] | 2010 | |
| Male and female transgenic mice stably expressing LacZ in lung epithelial cells (SPC-Cre; R26stop-LacZ) 8–12 weeks | IF | +25 µg/day HDM intranasally (10–15 weeks): inflammation epithelial damage and thickening of the sub-epithelial contractile smooth muscle layer tissue occludin and E-cadherin (protein) vimentin, α-SMA and pro-collagen I TGF-β1 in BALFActivation of Smad-dependent TGF-β signalling pathways (p-Smad3 and SNAIL1 proteins in the nuclei) | x | x | [95] | 2011 | ||
| Male C57/BL6 mice (9 weeks) | RT-qPCR WB ELISA | +bone-marrow-derived eosinophils (intratracheal instillation): inflammation in BALF TGF-β1 in BALFtype I collagen deposition E-cadherin α-SMA and vimentin+TGF-β1 siRNA less airway remodelling | x | x | x | [98] | 2013 | |
| BEAS-2B cell line | RT-qPCR WB ELISA | +primary human eosinophils or TGF-β1 or EoL-1 cells: fibroblast-like morphology and filamentous actin forming long stress fibres E-cadherin (mRNA and protein) vimentin (mRNA and protein)Need for cell-to-cell contact for induction of EMT +PI3K and JNK inhibitors: EMT blocked | x | x | x | [98] | 2013 | |
| BEAS-2B cell line | RT-qPCR WB IF Wound-healing assay | rhTGF-β1 5 ng/mL: spindle-fibroblast-like morphology with reduced cell–cell contact α-SMA (mRNA and protein) E-cadherin (mRNA and protein) collagen type I, fibronectin-EDA and tenascin C (mRNA) motility MMP-2/9 (protein)+IL-1β: E-cadherin (mRNA tenascin C expression (mRNA)Corticosteroid pretreatment does not abrogate TGFβ1-induced EMT | x | x | x | [99] | 2009 | |
| Primary normal HBEC monolayer | RT-qPCR WB | rhTGF-β1 2 ng/mL: α-SMA (mRNA and protein) vimentin (mRNA) E-cadherin (mRNA and protein) collagen type I, fibronectin-EDA and tenascin C (mRNA) MMP-2/9 release (protein) | x | x | [99] | 2009 | ||
| BEAS-2B cell line | RT-qPCR IF WB Wound-healing cell migration assays | +IL-24 (dose–response): migratory capability Acquisition of a larger and more spindle-shaped morphology vimentin and α-SMA (mRNA and protein) E-cadherin (mRNA and protein)Activation of STAT3 and ERK1/2 phosphorylation +IL-24 + inhibitors of JAK or ERK1/2: WT phenotype restored | x | x | x | x | [102] | 2022 |
| Female wild-type SPF BALB/c mice 6–8 weeks | IHC BALF Assessment of airway hyper-responsiveness | +25 μg/day HDM-induced asthma group 5 weeks:Airway resistance and inflammation IL-24 protein Collagen deposition TGF-β1 (protein in BALF) E-cadherin (protein) vimentin and α-SMA (protein) p-STAT3 and p-ERK1/2 (protein)+HDM + si-IL-24 or rhIL-37: HDM-induced dysregulations | x | x | x | x | [102] | 2022 |
| Female C57BL/J mice 6–8 weeks CD146-KO C57BL/J mice IL-33 KO C57BL/J mice | WB IHC and IF ELISA | +25 μg intranasally administered HDM 5 weeks: inflammation in both KO mice collagen I (protein) in both KO mice | x | [136] | 2020 | |||
| MLE-12 (mouse pulmonary epithelial cell line) and A549 | WB IF RT-qPCR | +HDM 10–100 µg/mL: CD146 (mRNA and protein) MyD88, phosphorylation of NF-κB p65 and p38 (protein) TGF-β and pSMAD3 (protein) | ||||||
Primary alveolar epithelial cells from mice | +IL-33 0.1–100 ng/mL: CD146 (protein in cells and secreted)+siRNA CD146: E-cadherin; expression inversely correlated with CD146 | x | [136] | 2020 | ||||
| Normal primary HBEC monolayer | RT-qPCR IF ELISA | +TGF-β1 10 ng/mL or neutrophils of asthma patients 48 h: E-cadherin (mRNA) N-cadherin, α-SMA and vimentin (mRNA) morphological changes | x | x | [137] | 2019 | ||
| BALB/c mice 6–8 weeks | ELISA IHC RT-qPCR WB | +25 μg HDM intranasal instillation 4 weeks: TF (mRNA and protein) +HDM + shRNA TF: inflammation Improved hyperplasia collagen biomarkers of EMT reversed | x | x | x | [138] | 2021 | |
| 16HBE14o- cells | RT-qPCR Wound-healing and invasion assay | +TGF-β1 and HDM: fibronectin 1, TF and TGF-β1 (mRNA and protein) shTF reverse the changes | x | x | [138] | 2021 | ||
| 16HBE14o- cells | RT-qPCR WB | +TGF-β1: miR-448-5p (mRNA) Six1 (mRNA and protein) +TGF-β1 + miR-448-5p: TGF-β1, pSMAD3 E-cadherin (mRNA) vimentin (mRNA)+Six1 silencing: similar phenotype to miR-448-5p overexpression | x | x | [139] | 2019 | ||
| BEAS-2B cell line | WB | LPS stimulation: | ||||||
| IHC | TGF-β1, TGF-β RI and TGF-β RII (protein) +10 ng/mL TGF-β: change of morphology PAR-1 induction α-SMA (protein) | x | x | x | [140] | 2014 | ||
| Male BALB/c mice 6 weeks | IHC | +ovalbumin challenge: TGF-β1, TGF-β RI Epithelial thickening, collagen IV deposition E-cadherin (protein) α-SMA (protein)PAR-1 induction | x | x | x | [140] | 2014 |
3.2. Potential Effect of Pharmacological Treatment on EMT and Airway Remodelling
4. COPD
4.1. Chronic Inflammation in COPD
4.2. EMT and Fibrosis in COPD
| Models | Techniques | EMT Program | AR | EMT Signal | EMT TFs | EMT Mark | Refs. | Year |
|---|---|---|---|---|---|---|---|---|
| ALI HBECs (2–5 weeks) from non-smokers, smoker controls, mild, moderate and severe to very severe COPD | IHC and IF RT-qPCR WB | vimentin (mRNA and protein) for severe COPD and fibronectin (protein) E-cadherin and ZO-1 (protein) | ||||||
| ELISA | ||||||||
| +TGF-β1 vimentin and fibronectin (protein) +blocking TGF-β1 Restoring a cobblestone shape compared with the spindle shape vimentin expression | x | x | x | [171] | 2015 | |||
| ALI HBECs from non-smokers, smokers and patients with COPD | IHC and IF RT-qPCR WB ELISA | α-SMA, vimentin and collagen type I (mRNA and protein) E-cadherin and ZO-1 (mRNA and protein) KRT5 and KRT18 (mRNA) | x | [174] | 2013 | |||
| ALI HBECs (14–21 days) from non-smokers, smoker controls, mild, moderate and severe to very severe COPD | RNA-seq WB ELISA IHC and IF | Activation of WNT/β-catenin pathway with nuclear expression of β-catenin in the COPD airway epithelium vimentin (mRNA and protein) fibronectin release following WNT activation | x | x | x | [193] | 2020 | |
| HBEC monolayer | RT-qPCR WB IHC and IF ELISA | +nicotine: Wnt3a (protein and mRNA) total β-catenin (protein) E-cadherin (protein) α-SMA, MMP-9 and collagen type I (protein) | x | x | [194] | 2013 | ||
| ALI HBECs from controls lung tissue | IF RT-qPCR WB ROS | +2.5% CSE for up to 7 days: E-cadherin, ZO-1 (mRNA and protein) vimentin, collagen type I and α-SMA (mRNA and protein) GTP-Rac1 and pAKT (protein) | x | x | x | [196] | 2015 | |
| Male BALB/c mice 6 weeks | IHC WB | +CSE: collagen deposition CRAMP and vimentin E-cadherin TACE, TGF-α and EGFR (protein) | x | x | x | [197] | 2021 | |
| NCI-H292 cell line | IF | +CSE 5%: E-cadherin vimentin | x | [197] | 2021 | |||
| male C57BL/6 mice 8 weeks | IHC and IF RT-qPCR WB | 20 CS/day 12 or 24 weeks: inflammation ECM, smooth muscle thickening, goblet cell hyperplasia and mucus secretion IL-17A and C-EBPβ E-cadherin (mRNA and protein) vimentin (mRNA and protein) | x | x | [200] | 2021 | ||
| Murine bronchial epithelial cells | IF RT-qPCR WB | 20% CSE 72 h: IL-17R E-cadherin (mRNA and protein) vimentin (mRNA and protein) | x | [200] | 2021 | |||
| HSAEpiCs | RT-qPCR WB | 5% CSE 24–96 h: α-SMA, N-cadherin and uPAR (protein) E-cadherin and α-catenin (protein) | x | [201] | 2013 | |||
| BEAS-2B cell line | WB RT-qPCR | +CSE 1% 24 h: ZO-1 (protein and mRNA) vimentin (mRNA) TGF-β1 (mRNA) +TGF-β1 230 pg/mL ZO-1 (mRNA) | x | [205] | 2020 | |||
| HSAEpiC | WB RT-qPCR | +CSE 1% 24 h: vimentin (mRNA) ZO-1 (mRNA) TGF-β1 (mRNA) and p-Smad2/3 (protein) +TGF-β1 230 pg/mL, 2 h ZO-1 (protein and mRNA) | x | x | [205] | 2020 | ||
| Male and female Sprague Dawley rats (8 weeks) | ELISA IHC WB | +48 CS/day inhalation 12 weeks: IL-8, IL-6, TNF-α, sICAM-1 and ROS in BALF Airway fibrosis, airway epithelial thickness and ASM thickness α-SMA (protein) lung function TGF-β1 and p-Smad2/3 (protein) PPAR-γ (protein) | x | x | x | [206] | 2021 | |
| Male C57BL/6 mice 26–28 weeks | ELISA RT-qPCR WB | +CSE intraperitoneally injected: airway epithelium thickening, enlargement of alveolus and inflammatory cell infiltration IL-6 and TNF-α in BALF TGF-β1, Smad2 and Smad3 (protein and mRNA) | x | x | [207] | 2019 |
| Models | Techniques | EMT Program | AR | EMT Signal | EMT TFs | EMT Mark | Refs. | Year |
|---|---|---|---|---|---|---|---|---|
| Bronchial biopsy from non-smokers, smoker controls and COPD subjects | IHC | Correlation between β-catenin and SNAIL1 expression with both S100A4 and also airflow obstruction | x | x | x | [169] | 2017 | |
| Lung tissue of α1-antitrypsin-deficiency-related COPD and non- α -1 antitrypsin deficiency COPD subjects | RT-PCR | SNAIL homolog 1 in α1-antitrypsin-deficiency-related COPD group | [170] | 2012 | ||||
| Lung tissue of non-smokers, smoker controls, mild, moderate and severe-to-very-severe COPD | IHC | E-cadherin expression vimentin in large and small airways Negative correlation between vimentin and airway obstruction | x | [171] | 2015 | |||
| Lung tissue of COPD subjects | WB IHC | E-cadherin α-SMA, N-cadherin and vimentin Fragmentation and clefts in RBM | x | x | [172] | 2021 | ||
| Bronchial biopsy from COPD patients | IHC | Cytokeratin-(s) and S100A4 double staining | x | [177] | 2011 | |||
| Brushing from non-smokers, smoker controls, COPD subjects | RT-PCR IHC | TGF-β1 (protein and mRNA) Positive correlation between TGF-β1 mRNA levels and the extent of smoking history | x | [180] | 2001 | |||
| Bronchial biopsy | IHC | RBM fragmentation S100A4 and MMP-9 in RBM and/or basal epithelium +inhaled corticosteroids treatment: EGFR %RBM fragmentation S100A4 and MMP-9 in RBM and/or basal epithelium | x | x | [185] | 2014 | ||
| Lung tissue of non-smokers, smokers and patients with COPD (smoker or ex-smoker) | IHC | lamina propria and adventitia thickness in small airways of COPD subjects | ||||||
| α-SMA-positive cells (myofibroblasts) in SA collagen-1 and fibronectin deposition Negative correlation between increased SA wall thickening and decrease in airflow in the COPD groups Correlation between collagen-1 deposition in the SA lamina propria and lung function in the COPD-smokers group | x | x | [186] | 2021 | ||||
| Lung tissue of non-smokers, smoker controls, mild, moderate and severe-to-very-severe COPD | IHC and IF RT-PCR | β-catenin expression in the COPD airway epithelium β-catenin upregulation in COPD airway epithelium correlates with altered differentiation | x | x | [193] | 2020 | ||
| Lung tissue of non-smokers, smoker controls and COPD subjects | IHC | vimentin uPAR Negative correlation between FEV1% and uPAR expression Positive correlation between uPAR and the number of vimentin-positive cells | x | [201] | 2013 | |||
| Lung tissue of controls or subjects with chronic airflow limitation | IHC | vimentin and S100A4 in SA of COPD S100A4 expression associated with airflow obstruction in small airway | x | [202] | 2015 | |||
| Bronchial biopsy from non-smokers, smoker controls and COPD subjects | IHC | TGF-β1 in large airway Correlations between pSmad 2/3 and pSmad 7 expression and both S100A4 and airflow obstruction | x | x | [208] | 2017 | ||
| Lung tissue of non-COPD and patients with COPD | IHC | TGF-β1 (protein and mRNA) | x | [209] | 1998 |
4.3. Potential Effect of Treatment on EMT and Small Airway Fibrosis in COPD
5. Cystic Fibrosis and NCFB
5.1. CF Inflammation
5.2. Future Directions on the Role of EMT in CF
5.3. Potential Effect of Novel Pharmacological Treatments on EMT and CF Airway Remodelling
5.4. Inflammation and EMT in NCFB
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352, Erratum in Nat. Rev. Mol. Cell Biol. 2021, 22, 834. [Google Scholar] [CrossRef]
- Hay, E.D. An Overview of Epithelio-Mesenchymal Transformation. Acta Anat. 1995, 154, 8–20. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P. EMT: An Update. Methods Mol. Biol. 2021, 2179, 35–39. [Google Scholar] [PubMed]
- Wu, B.; Sodji, Q.H.; Oyelere, A.K. Inflammation, Fibrosis and Cancer: Mechanisms, Therapeutic Options and Challenges. Cancers 2022, 14, 552. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.Q.; Ward, C.; Muller, H.K.; Sohal, S.S.; Walters, E.H. Epithelial mesenchymal transition (EMT) and non-small cell lung cancer (NSCLC): A mutual association with airway disease. Med. Oncol. 2017, 34, 45. [Google Scholar] [CrossRef] [PubMed]
- Menju, T.; Date, H. Lung cancer and epithelial-mesenchymal transition. Gen. Thorac. Cardiovasc. Surg. 2021, 69, 781–789. [Google Scholar] [CrossRef]
- Nam, M.-W.; Kim, C.-W.; Choi, K.-C. Epithelial-Mesenchymal Transition-Inducing Factors Involved in the Progression of Lung Cancers. Biomol. Ther. 2022, 30, 213–220. [Google Scholar] [CrossRef]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef]
- Hackett, T.-L. Epithelial–mesenchymal transition in the pathophysiology of airway remodelling in asthma. Curr. Opin. Allergy Clin. Immunol. 2012, 12, 53–59. [Google Scholar] [CrossRef]
- Su, X.; Wu, W.; Zhu, Z.; Lin, X.; Zeng, Y. The effects of epithelial–mesenchymal transitions in COPD induced by cigarette smoke: An update. Respir. Res. 2022, 23, 225. [Google Scholar] [CrossRef]
- Rout-Pitt, N.; Farrow, N.; Parsons, D.; Donnelley, M. Epithelial mesenchymal transition (EMT): A universal process in lung diseases with implications for cystic fibrosis pathophysiology. Respir. Res. 2018, 19, 136. [Google Scholar] [CrossRef]
- James, A.L.; Wenzel, S. Clinical relevance of airway remodelling in airway diseases. Eur. Respir. J. 2007, 30, 134–155. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.J.; Collard, H.R.; Pardo, A.; Raghu, G.; Richeldi, L.; Selman, M.; Swigris, J.J.; Taniguchi, H.; Wells, A.U. Idiopathic pulmonary fibrosis. Nat. Rev. Dis. Primers 2017, 3, 17074. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Acloque, H.; Adams, M.S.; Fishwick, K.; Bronner-Fraser, M.; Nieto, M.A. Epithelial-mesenchymal transitions: The importance of changing cell state in development and disease. J. Clin. Investig. 2009, 119, 1438–1449. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef]
- Moulin, V.; Castilloux, G.; Auger, F.A.; Garrel, D.; O’Connor-McCourt, M.D.; Germain, L. Modulated response to cytokines of human wound healing myofibroblasts compared to dermal fibroblasts. Exp. Cell Res. 1998, 238, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Zapater, E.; Signes-Costa, J.; Montero, P.; Roger, I. Lung Fibrosis and Fibrosis in the Lungs: Is It All about Myofibroblasts? Biomedicines 2022, 10, 1423. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial–mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, 570. [Google Scholar] [CrossRef]
- Lee, J.H.; Massague, J. TGF-beta in developmental and fibrogenic EMTs. Semin. Cancer Biol. 2022, 86, 136–145. [Google Scholar] [CrossRef]
- Newfeld, S.J.; O’Connor, M.B. New aspects of TGF-beta superfamily signaling in development and disease (2022 FASEB meeting review). Fac. Rev. 2022, 11, 36. [Google Scholar] [CrossRef]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef]
- Saito, A.; Horie, M.; Nagase, T. TGF-beta Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. [Google Scholar] [CrossRef]
- Sime, P.J.; Xing, Z.; Graham, F.L.; Csaky, K.G.; Gauldie, J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J. Clin. Investig. 1997, 100, 768–776. [Google Scholar] [CrossRef]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-beta1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Hu, Y. TGF-beta1: Gentlemanly orchestrator in idiopathic pulmonary fibrosis (Review). Int. J. Mol. Med. 2021, 48. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Ribatti, D.; Lisi, S. Organ Fibrosis and Autoimmunity: The Role of Inflammation in TGFbeta-Dependent EMT. Biomolecules 2021, 11, 310. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.H.; Tham, C.L.; Harith, H.H.; Firdaus, N.; Israf, D.A. TGF-beta-induced fibrosis: A review on the underlying mechanism and potential therapeutic strategies. Eur. J. Pharmacol. 2021, 911, 174510. [Google Scholar] [CrossRef]
- Lodyga, M.; Hinz, B. TGF-beta1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef]
- Leask, A.; Abraham, D.J. TGF-beta signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef]
- Brown, R.L.; Ormsby, I.; Doetschman, T.C.; Greenhalgh, D.G. Wound healing in the transforming growth factor-beta1-deficient mouse. Wound Repair Regen. 1995, 3, 25–36. [Google Scholar] [CrossRef]
- Camara, J.; Jarai, G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair 2010, 3, 2. [Google Scholar] [CrossRef]
- Kasai, H.; Allen, J.T.; Mason, R.M.; Kamimura, T.; Zhang, Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir. Res. 2005, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki, K.; Moustakas, A. TGF-beta Signaling. Biomolecules 2020, 10, 487. [Google Scholar] [CrossRef]
- Burgy, O.; Königshoff, M. The WNT signaling pathways in wound healing and fibrosis. Matrix Biol. 2018, 68–69, 67–80. [Google Scholar] [CrossRef]
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/beta-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci. Rep. 2018, 8, 13644. [Google Scholar] [CrossRef] [PubMed]
- Effendi, W.I.; Nagano, T. The Hedgehog Signaling Pathway in Idiopathic Pulmonary Fibrosis: Resurrection Time. Int. J. Mol. Sci. 2021, 23, 171. [Google Scholar] [CrossRef] [PubMed]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Liu, J.; Wu, Z.; Liu, T.; Ullenbruch, M.R.; Ding, L.; Henke, C.A.; Bitterman, P.B.; Phan, S.H. Reemergence of Hedgehog Mediates Epithelial–Mesenchymal Crosstalk in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2015, 52, 418–428. [Google Scholar] [CrossRef]
- Cigna, N.; Farrokhi Moshai, E.; Brayer, S.; Marchal-Somme, J.; Wemeau-Stervinou, L.; Fabre, A.; Mal, H.; Leseche, G.; Dehoux, M.; Soler, P.; et al. The hedgehog system machinery controls transforming growth factor-beta-dependent myofibroblastic differentiation in humans: Involvement in idiopathic pulmonary fibrosis. Am. J. Pathol. 2012, 181, 2126–2137. [Google Scholar] [CrossRef]
- Heldin, C.-H.; Lennartsson, J.; Westermark, B. Involvement of platelet-derived growth factor ligands and receptors in tumorigenesis. J. Intern. Med. 2018, 283, 16–44. [Google Scholar] [CrossRef]
- Qin, W.; Pan, Y.; Zheng, X.; Li, D.; Bu, J.; Xu, C.; Tang, J.; Cui, R.; Lin, P.; Yu, X. MicroRNA-124 regulates TGF-alpha-induced epithelial-mesenchymal transition in human prostate cancer cells. Int. J. Oncol. 2014, 45, 1225–1231. [Google Scholar] [CrossRef]
- Li, Y.; Li, H.; Duan, Y.; Cai, X.; You, D.; Zhou, F.; Yang, C.; Tuo, X.; Liu, Z. Blockage of TGF-alpha Induced by Spherical Silica Nanoparticles Inhibits Epithelial-Mesenchymal Transition and Proliferation of Human Lung Epithelial Cells. BioMed Res. Int. 2019, 2019, 8231267. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Xiao, L.; Cui, R.; Li, D.; Zheng, X.; Zhu, L.; Sun, H.; Pan, Y.; Du, Y.; Yu, X. CX3CL1 increases invasiveness and metastasis by promoting epithelial-to-mesenchymal transition through the TACE/TGF-alpha/EGFR pathway in hypoxic androgen-independent prostate cancer cells. Oncol. Rep. 2016, 35, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Epithelial-Mesenchymal Transition by Synergy between Transforming Growth Factor-beta and Growth Factors in Cancer Progression. Diagnostics 2022, 12, 2127. [Google Scholar] [CrossRef]
- Shu, D.Y.; Lovicu, F.J. Enhanced EGF receptor-signaling potentiates TGFbeta-induced lens epithelial-mesenchymal transition. Exp. Eye Res. 2019, 185, 107693. [Google Scholar] [CrossRef]
- Schelch, K.; Wagner, C.; Hager, S.; Pirker, C.; Siess, K.; Lang, E.; Lin, R.; Kirschner, M.B.; Mohr, T.; Brcic, L.; et al. FGF2 and EGF induce epithelial–mesenchymal transition in malignant pleural mesothelioma cells via a MAPKinase/MMP1 signal. Carcinog. 2018, 39, 534–545. [Google Scholar] [CrossRef]
- Chen, R.; Jin, G.; Li, W.; McIntyre, T.M. Epidermal Growth Factor (EGF) Autocrine Activation of Human Platelets Promotes EGF Receptor-Dependent Oral Squamous Cell Carcinoma Invasion, Migration, and Epithelial Mesenchymal Transition. J. Immunol. 2018, 201, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Wang, G.; Wang, T.; Fu, B.; Zhang, Y.; Huang, L.; Deng, Y.; Chen, G.; Wu, X.; Chen, J.; et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int. J. Biol. Sci. 2020, 16, 2542–2558. [Google Scholar] [CrossRef]
- Park, M.K.; You, H.J.; Lee, H.J.; Kang, J.H.; Oh, S.H.; Kim, S.Y.; Lee, C.H. Transglutaminase-2 induces N-cadherin expression in TGF-beta1-induced epithelial mesenchymal transition via c-Jun-N-terminal kinase activation by protein phosphatase 2A down-regulation. Eur. J. Cancer 2013, 49, 1692–1705. [Google Scholar] [CrossRef]
- Shafiq, A.; Suwakulsiri, W.; Rai, A.; Chen, M.; Greening, D.W.; Zhu, H.J.; Xu, R.; Simpson, R.J. Transglutaminase-2, RNA-binding proteins and mitochondrial proteins selectively traffic to MDCK cell-derived microvesicles following H-Ras-induced epithelial-mesenchymal transition. Proteomics 2021, 21, e2000221. [Google Scholar] [CrossRef]
- Milton, A.V.; Konrad, D.B. Epithelial-mesenchymal transition and H(2)O(2) signaling—A driver of disease progression and a vulnerability in cancers. Biol. Chem. 2022, 403, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Shi, F.; Zhai, R.; Wang, H.; Li, K.; Xu, C.; Yao, W.; Zhou, F. TGF-beta promote epithelial-mesenchymal transition via NF-kappaB/NOX4/ROS signal pathway in lung cancer cells. Mol. Biol. Rep. 2021, 48, 2365–2375. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Porsbjerg, C.; Melen, E.; Lehtimaki, L.; Shaw, D. Asthma. Lancet 2023, 401, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, A.I.; Jackson, D.J.; Edwards, M.R.; Johnston, S.L. Airway Epithelial Orchestration of Innate Immune Function in Response to Virus Infection. A Focus on Asthma. Ann. Am. Thorac. Soc. 2016, 13, S55–S63. [Google Scholar] [CrossRef]
- Noble, P.B.; Pascoe, C.D.; Lan, B.; Ito, S.; Kistemaker, L.E.; Tatler, A.L.; Pera, T.; Brook, B.S.; Gosens, R.; West, A.R. Airway smooth muscle in asthma: Linking contraction and mechanotransduction to disease pathogenesis and remodelling. Pulm. Pharmacol. Ther. 2014, 29, 96–107. [Google Scholar] [CrossRef] [PubMed]
- King, G.G.; Noble, P.B. Airway remodelling in asthma: It’s not going away. Respirology 2016, 21, 203–204. [Google Scholar] [CrossRef]
- Girodet, P.O.; Allard, B.; Thumerel, M.; Begueret, H.; Dupin, I.; Ousova, O.; Lassalle, R.; Maurat, E.; Ozier, A.; Trian, T.; et al. Bronchial Smooth Muscle Remodeling in Nonsevere Asthma. Am. J. Respir. Crit. Care Med. 2016, 193, 627–633. [Google Scholar] [CrossRef]
- Prakash, Y.S. Emerging concepts in smooth muscle contributions to airway structure and function: Implications for health and disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L1113–L1140. [Google Scholar] [CrossRef]
- Holgate, S.T. Pathogenesis of asthma. Clin. Exp. Allergy 2008, 38, 872–897. [Google Scholar] [CrossRef] [PubMed]
- Mostaço-Guidolin, L.B.; Osei, E.T.; Ullah, J.; Hajimohammadi, S.; Fouadi, M.; Li, X.; Li, V.; Shaheen, F.; Yang, C.X.; Chu, F.; et al. Defective Fibrillar Collagen Organization by Fibroblasts Contributes to Airway Remodeling in Asthma. Am. J. Respir. Crit. Care Med. 2019, 200, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Osei, E.T.; Mostaço-Guidolin, L.B.; Hsieh, A.; Warner, S.M.; Al-Fouadi, M.; Wang, M.; Cole, D.J.; Maksym, G.N.; Hallstrand, T.S.; Timens, W.; et al. Epithelial-interleukin-1 inhibits collagen formation by airway fibroblasts: Implications for asthma. Sci. Rep. 2020, 10, 8721. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar]
- Le Gros, G.; Ben-Sasson, S.Z.; Seder, R.; Finkelman, F.D.; Paul, W.E. Generation of interleukin 4 (IL-4)-producing cells in vivo and in vitro: IL-2 and IL-4 are required for in vitro generation of IL-4-producing cells. J. Exp. Med. 1990, 172, 921–929. [Google Scholar] [CrossRef]
- Metcalfe, D.D.; Baram, D.; Mekori, Y.A. Mast cells. Physiol. Rev. 1997, 77, 1033–1079. [Google Scholar] [CrossRef]
- Tsartsali, L.; Hislop, A.A.; McKay, K.; James, A.L.; Elliot, J.; Zhu, J.; Rosenthal, M.; Payne, D.N.; Jeffery, P.K.; Bush, A.; et al. Development of the bronchial epithelial reticular basement membrane: Relationship to epithelial height and age. Thorax 2011, 66, 280–285. [Google Scholar] [CrossRef]
- Dunnill, M.S. The pathology of asthma, with special reference to changes in the bronchial mucosa. J. Clin. Pathol. 1960, 13, 27–33. [Google Scholar] [CrossRef]
- Roche, W.R.; Beasley, R.; Williams, J.H.; Holgate, S.T. Subepithelial fibrosis in the bronchi of asthmatics. Lancet 1989, 333, 520–524. [Google Scholar] [CrossRef]
- Broekema, M.; Timens, W.; Vonk, J.M.; Volbeda, F.; Lodewijk, M.E.; Hylkema, M.N.; Ten Hacken, N.H.; Postma, D.S. Persisting Remodeling and Less Airway Wall Eosinophil Activation in Complete Remission of Asthma. Am. J. Respir. Crit. Care Med. 2011, 183, 310–316. [Google Scholar] [CrossRef]
- Pain, M.; Bermudez, O.; Lacoste, P.; Royer, P.-J.; Botturi, K.; Tissot, A.; Brouard, S.; Eickelberg, O.; Magnan, A. Tissue remodelling in chronic bronchial diseases: From the epithelial to mesenchymal phenotype. Eur. Respir. Rev. 2014, 23, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 2002, 110, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Royce, S.G.; Cheng, V.; Samuel, C.S.; Tang, M.L. The regulation of fibrosis in airway remodeling in asthma. Mol. Cell. Endocrinol. 2012, 351, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Loffredo, L.F.; Abdala-Valencia, H.; Anekalla, K.R.; Cuervo-Pardo, L.; Gottardi, C.J.; Berdnikovs, S. Beyond epithelial-to-mesenchymal transition: Common suppression of differentiation programs underlies epithelial barrier dysfunction in mild, moderate, and severe asthma. Allergy 2017, 72, 1988–2004. [Google Scholar] [CrossRef]
- Nawijn, M.C.; Hackett, T.L.; Postma, D.S.; van Oosterhout, A.J.; Heijink, I.H. E-cadherin: Gatekeeper of airway mucosa and allergic sensitization. Trends Immunol. 2011, 32, 248–255. [Google Scholar] [CrossRef]
- Magnan, A.; Frachon, I.; Rain, B.; Peuchmaur, M.; Monti, G.; Lenot, B.; Fattal, M.; Simonneau, G.; Galanaud, P.; Emilie, D. Transforming growth factor beta in normal human lung: Preferential location in bronchial epithelial cells. Thorax 1994, 49, 789–792. [Google Scholar] [CrossRef]
- Hackett, T.L.; Warner, S.M.; Stefanowicz, D.; Shaheen, F.; Pechkovsky, D.V.; Murray, L.A.; Argentieri, R.; Kicic, A.; Stick, S.M.; Bai, T.R.; et al. Induction of epithelial-mesenchymal transition in primary airway epithelial cells from patients with asthma by transforming growth factor-beta1. Am. J. Respir. Crit. Care Med. 2009, 180, 122–133. [Google Scholar] [CrossRef]
- Halwani, R.; Al-Muhsen, S.; Al-Jahdali, H.; Hamid, Q. Role of transforming growth factor-beta in airway remodeling in asthma. Am. J. Respir. Cell Mol. Biol. 2011, 44, 127–133. [Google Scholar] [CrossRef]
- Redington, A.E.; Madden, J.; Frew, A.J.; Djukanovic, R.; Roche, W.R.; Holgate, S.T.; Howarth, P.H. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am. J. Respir. Crit. Care Med. 1997, 156, 642–647. [Google Scholar] [CrossRef]
- Ji, X.; Li, J.; Xu, L.; Wang, W.; Luo, M.; Luo, S.; Ma, L.; Li, K.; Gong, S.; He, L.; et al. IL4 and IL-17A provide a Th2/Th17-polarized inflammatory milieu in favor of TGF-beta1 to induce bronchial epithelial-mesenchymal transition (EMT). Int. J. Clin. Exp. Pathol. 2013, 6, 1481–1492. [Google Scholar]
- Evasovic, J.M.; Singer, C.A. Regulation of IL-17A and implications for TGF-beta1 comodulation of airway smooth muscle remodeling in severe asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L843–L868. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Lee, S.A.; Shin, J.M.; Park, I.H.; Lee, H.M. Glucocorticoids ameliorate TGF-beta1-mediated epithelial-to-mesenchymal transition of airway epithelium through MAPK and Snail/Slug signaling pathways. Sci. Rep. 2017, 7, 3486. [Google Scholar] [CrossRef] [PubMed]
- Heijink, I.H.; Postma, D.S.; Noordhoek, J.A.; Broekema, M.; Kapus, A. House Dust Mite–Promoted Epithelial-to-Mesenchymal Transition in Human Bronchial Epithelium. Am. J. Respir. Cell Mol. Biol. 2010, 42, 69–79. [Google Scholar] [CrossRef]
- Hackett, T.-L.; de Bruin, H.G.; Shaheen, F.; van den Berge, M.; van Oosterhout, A.J.; Postma, D.S.; Heijink, I.H. Caveolin-1 Controls Airway Epithelial Barrier Function. Implications for Asthma. Am. J. Respir. Cell Mol. Biol. 2013, 49, 662–671. [Google Scholar] [CrossRef]
- Johnson, J.R.; Roos, A.; Berg, T.; Nord, M.; Fuxe, J. Chronic Respiratory Aeroallergen Exposure in Mice Induces Epithelial-Mesenchymal Transition in the Large Airways. PLoS ONE 2011, 6, e16175. [Google Scholar] [CrossRef]
- Minshall, E.M.; Leung, D.Y.; Martin, R.J.; Song, Y.L.; Cameron, L.; Ernst, P.; Hamid, Q. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am. J. Respir. Cell Mol. Biol. 1997, 17, 326–333. [Google Scholar] [CrossRef]
- Vignola, A.M.; Chanez, P.; Chiappara, G.; Merendino, A.; Pace, E.; Rizzo, A.; la Rocca, A.M.; Bellia, V.; Bonsignore, G.; Bousquet, J. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1997, 156, 591–599. [Google Scholar] [CrossRef]
- Yasukawa, A.; Hosoki, K.; Toda, M.; Miyake, Y.; Matsushima, Y.; Matsumoto, T.; Boveda-Ruiz, D.; Gil-Bernabe, P.; Nagao, M.; Sugimoto, M.; et al. Eosinophils promote epithelial to mesenchymal transition of bronchial epithelial cells. PLoS ONE 2013, 8, e64281. [Google Scholar] [CrossRef]
- Doerner, A.M.; Zuraw, B.L. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir. Res. 2009, 10, 100. [Google Scholar] [CrossRef]
- Commins, S.; Steinke, J.W.; Borish, L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J. Allergy Clin. Immunol. 2008, 121, 1108–1111. [Google Scholar] [CrossRef]
- Zissler, U.M.; Ulrich, M.; Jakwerth, C.A.; Rothkirch, S.; Guerth, F.; Weckmann, M.; Schiemann, M.; Haller, B.; Schmidt-Weber, C.B.; Chaker, A.M. Biomatrix for upper and lower airway biomarkers in patients with allergic asthma. J. Allergy Clin. Immunol. 2018, 142, 1980–1983. [Google Scholar] [CrossRef] [PubMed]
- Feng, K.-N.; Meng, P.; Zou, X.-L.; Zhang, M.; Li, H.-K.; Yang, H.-L.; Li, H.-T.; Zhang, T.-T. IL-37 protects against airway remodeling by reversing bronchial epithelial–mesenchymal transition via IL-24 signaling pathway in chronic asthma. Respir. Res. 2022, 23, 244. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.R. Is asthma a fibrotic disease? Chest 1995, 107, 111S–117S. [Google Scholar] [CrossRef]
- Laitinen, A.; Altraja, A.; Kämpe, M.; Linden, M.; Virtanen, I.; Laitinen, L.A. Tenascin Is Increased in Airway Basement Membrane of Asthmatics and Decreased by an Inhaled Steroid. Am. J. Respir. Crit. Care Med. 1997, 156, 951–958. [Google Scholar] [CrossRef]
- Elias, J.A.; Zhu, Z.; Chupp, G.; Homer, R.J. Airway remodeling in asthma. J. Clin. Investig. 1999, 104, 1001–1006. [Google Scholar] [CrossRef]
- Zeiger, R.S.; Dawson, C.; Weiss, S. Relationships between duration of asthma and asthma severity among children in the Childhood Asthma Management Program (CAMP). J. Allergy Clin. Immunol. 1999, 103, 376–386. [Google Scholar] [CrossRef]
- Boulet, L.-P.; Turcotte, H.; Laviolette, M.; Naud, F.; Bernier, M.-C.; Martel, S.; Chakir, J. Airway hyperresponsiveness, inflammation, and subepithelial collagen deposition in recently diagnosed versus long-standing mild asthma. Influence of inhaled corticosteroids. Am. J. Respir. Crit. Care Med. 2000, 162, 1308–1313. [Google Scholar] [CrossRef]
- James, A.L.; Maxwell, P.S.; Pearce-Pinto, G.; Elliot, J.G.; Carroll, N.G. The Relationship of Reticular Basement Membrane Thickness to Airway Wall Remodeling in Asthma. Am. J. Respir. Crit. Care Med. 2002, 166, 1590–1595. [Google Scholar] [CrossRef]
- Burgess, J.K.; Mauad, T.; Tjin, G.; Karlsson, J.C.; Westergren-Thorsson, G. The extracellular matrix - the under-recognized element in lung disease? J. Pathol. 2016, 240, 397–409. [Google Scholar] [CrossRef]
- Carroll, N.G.; Perry, S.; Karkhanis, A.; Harji, S.; Butt, J.; James, A.L.; Green, F.H. The Airway Longitudinal Elastic Fiber Network and Mucosal Folding in Patients with Asthma. Am. J. Respir. Crit. Care Med. 2000, 161, 244–248. [Google Scholar] [CrossRef]
- Boser, S.R.; Mauad, T.; Araújo-Paulino, B.B.; Mitchell, I.; Shrestha, G.; Chiu, A.; Butt, J.; Kelly, M.M.; Caldini, E.; James, A.; et al. Myofibroblasts are increased in the lung parenchyma in asthma. PLoS ONE 2017, 12, e0182378. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.N.; Rogers, A.V.; Ädelroth, E.; Bandi, V.; Guntupalli, K.K.; Bush, A.; Jeffery, P.K. Early Thickening of the Reticular Basement Membrane in Children with Difficult Asthma. Am. J. Respir. Crit. Care Med. 2003, 167, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Cokugras, H.; Akcakaya, N.; Seckin; Camcioglu, Y.; Sarimurat, N.; Aksoy, F. Ultrastructural examination of bronchial biopsy specimens from children with moderate asthma. Thorax 2001, 56, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Nihlberg, K.; Larsen, K.; Hultgardh-Nilsson, A.; Malmstrom, A.; Bjermer, L.; Westergren-Thorsson, G. Tissue fibrocytes in patients with mild asthma: A possible link to thickness of reticular basement membrane? Respir. Res. 2006, 7, 50. [Google Scholar] [CrossRef]
- Snelgrove, R.J.; Patel, D.F. Zooming into the Matrix: Using Nonlinear Optical Microscopy to Visualize Collagen Remodeling in Asthmatic Airways. Am. J. Respir. Crit. Care Med. 2019, 200, 403–405. [Google Scholar] [CrossRef]
- Ward, C.; Pais, M.; Bish, R.; Reid, D.; Feltis, B.; Johns, D.; Walters, E.H. Airway inflammation, basement membrane thickening and bronchial hyperresponsiveness in asthma. Thorax 2002, 57, 309–316. [Google Scholar] [CrossRef]
- Tsurikisawa, N.; Oshikata, C.; Tsuburai, T.; Saito, H.; Sekiya, K.; Tanimoto, H.; Takeichi, S.; Mitomi, H.; Akiyama, K. Bronchial hyperresponsiveness to histamine correlates with airway remodelling in adults with asthma. Respir. Med. 2010, 104, 1271–1277. [Google Scholar] [CrossRef]
- Mostaco-Guidolin, L.; Hajimohammadi, S.; Vasilescu, D.M.; Hackett, T.-L. Application of Euclidean distance mapping for assessment of basement membrane thickness distribution in asthma. J. Appl. Physiol. (1985) 2017, 123, 473–481. [Google Scholar] [CrossRef]
- Kuo, C.; Lim, S.; King, N.J.; Johnston, S.L.; Burgess, J.K.; Black, J.L.; Oliver, B.G. Rhinovirus infection induces extracellular matrix protein deposition in asthmatic and nonasthmatic airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 300, L951–L957. [Google Scholar] [CrossRef]
- Carroll, N.; Elliot, J.; Morton, A.; James, A. The Structure of Large and Small Airways in Nonfatal and Fatal Asthma. Am. Rev. Respir. Dis. 1993, 147, 405–410. [Google Scholar] [CrossRef]
- Hirst, S.J.; Barnes, P.J.; Twort, C.H. PDGF isoform-induced proliferation and receptor expression in human cultured airway smooth muscle cells. Am. J. Physiol. 1996, 270, L415–L428. [Google Scholar] [CrossRef] [PubMed]
- Panettieri, R.A., Jr.; Goldie, R.G.; Rigby, P.J.; Eszterhas, A.J.; Hay, D.W. Endothelin-1-induced potentiation of human airway smooth muscle proliferation: An ETA receptor-mediated phenomenon. Br. J. Pharmacol. 1996, 118, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Rajah, R.; Rosenbloom, J.; Herrick, D.J. IGFBP-3 mediates TGF-beta1-induced cell growth in human airway smooth muscle cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L545–L551. [Google Scholar] [CrossRef]
- Chen, G.; Khalil, N. TGF-beta1 increases proliferation of airway smooth muscle cells by phosphorylation of map kinases. Respir. Res. 2006, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Hakonarson, H.; Maskeri, N.; Carter, C.; Chuang, S.; Grunstein, M.M. Autocrine interaction between IL-5 and IL-1beta mediates altered responsiveness of atopic asthmatic sensitized airway smooth muscle. J. Clin. Investig. 1999, 104, 657–667. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coutts, A.; Chen, G.; Stephens, N.; Hirst, S.; Douglas, D.; Eichholtz, T.; Khalil, N. Release of biologically active TGF-beta from airway smooth muscle cells induces autocrine synthesis of collagen. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L999–L1008. [Google Scholar] [CrossRef]
- Moynihan, B.J.; Tolloczko, B.; El Bassam, S.; Ferraro, P.; Michoud, M.-C.; Martin, J.G.; Laberge, S. IFN-gamma, IL-4 and IL-13 modulate responsiveness of human airway smooth muscle cells to IL-13. Respir. Res. 2008, 9, 84. [Google Scholar] [CrossRef] [PubMed]
- Grainge, C.L.; Lau, L.C.; Ward, J.A.; Dulay, V.; Lahiff, G.; Wilson, S.; Holgate, S.; Davies, D.E.; Howarth, P.H. Effect of bronchoconstriction on airway remodeling in asthma. N. Engl. J. Med. 2011, 364, 2006–2015. [Google Scholar] [CrossRef]
- Camoretti-Mercado, B.; Lockey, R.F. Airway smooth muscle pathophysiology in asthma. J. Allergy Clin. Immunol. 2021, 147, 1983–1995. [Google Scholar] [CrossRef]
- Kuyper, L.M.; Paré, P.D.; Hogg, J.C.; Lambert, R.K.; Ionescu, D.; Woods, R.; Bai, T.R. Characterization of airway plugging in fatal asthma. Am. J. Med. 2003, 115, 6–11. [Google Scholar] [CrossRef]
- Wenzel, S.E.; Vitari, C.A.; Shende, M.; Strollo, D.C.; Larkin, A.; Yousem, S.A. Asthmatic granulomatosis: A novel disease with asthmatic and granulomatous features. Am. J. Respir. Crit. Care Med. 2012, 186, 501–507. [Google Scholar] [CrossRef]
- Malmström, K.; Lohi, J.; Sajantila, A.; Jahnsen, F.L.; Kajosaari, M.; Sarna, S.; Mäkelä, M.J. Immunohistology and remodeling in fatal pediatric and adolescent asthma. Respir. Res. 2017, 18, 94. [Google Scholar] [CrossRef]
- Treho Bittar, H.E.; Doberer, D.; Mehrad, M.; Strollo, D.C.; Leader, J.K.; Wenzel, S.; Yousem, S.A. Histologic Findings of Severe/Therapy-Resistant Asthma from Video-assisted Thoracoscopic Surgery Biopsies. Am. J. Surg. Pathol. 2017, 41, 182–188. [Google Scholar] [CrossRef]
- Elliot, J.G.; Noble, P.B.; Mauad, T.; Bai, T.R.; Abramson, M.J.; McKay, K.O.; Green, F.H.Y.; James, A.L. Inflammation-dependent and independent airway remodelling in asthma. Respirology 2018, 23, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.R.; Cohn, L. Advances in mucous cell metaplasia: A plug for mucus as a therapeutic focus in chronic airway disease. Am. J. Respir. Cell Mol. Biol. 2010, 42, 268–275. [Google Scholar] [CrossRef]
- Sun, Z.; Ji, N.; Ma, Q.; Zhu, R.; Chen, Z.; Wang, Z.; Qian, Y.; Wu, C.; Hu, F.; Huang, M.; et al. Epithelial-Mesenchymal Transition in Asthma Airway Remodeling Is Regulated by the IL-33/CD146 Axis. Front. Immunol. 2020, 11, 1598. [Google Scholar] [CrossRef] [PubMed]
- Haddad, A.; Gaudet, M.; Plesa, M.; Allakhverdi, Z.; Mogas, A.K.; Audusseau, S.; Baglole, C.J.; Eidelman, D.H.; Olivenstein, R.; Ludwig, M.S.; et al. Neutrophils from severe asthmatic patients induce epithelial to mesenchymal transition in healthy bronchial epithelial cells. Respir. Res. 2019, 20, 234. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Jiang, T.; Li, P.; Dai, L.; Shi, G.; Jing, X.; Gao, S.; Jia, L.; Wu, S.; Wang, Y.; et al. Tissue factor promotes airway pathological features through epithelial-mesenchymal transition of bronchial epithelial cells in mice with house dust mite-induced asthma. Int. Immunopharmacol. 2021, 97, 107690. [Google Scholar] [CrossRef]
- Yang, Z.C.; Qu, Z.H.; Yi, M.J.; Shan, Y.C.; Ran, N.; Xu, L.; Liu, X.J. MiR-448-5p inhibits TGF-beta1-induced epithelial-mesenchymal transition and pulmonary fibrosis by targeting Six1 in asthma. J. Cell. Physiol. 2019, 234, 8804–8814. [Google Scholar] [CrossRef]
- Gong, J.-H.; Cho, I.-H.; Shin, D.; Han, S.-Y.; Park, S.-H.; Kang, Y.-H. Inhibition of airway epithelial-to-mesenchymal transition and fibrosis by kaempferol in endotoxin-induced epithelial cells and ovalbumin-sensitized mice. Lab. Investig. 2014, 94, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Mattos, W.; Lim, S.; Russell, R.; Jatakanon, A.; Chung, K.F.; Barnes, P.J. Matrix metalloproteinase-9 expression in asthma: Effect of asthma severity, allergen challenge, and inhaled corticosteroids. Chest 2002, 122, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, P.K.; Godfrey, R.W.; Ädelroth, E.; Nelson, F.; Rogers, A.; Johansson, S.-A. Effects of Treatment on Airway Inflammation and Thickening of Basement Membrane Reticular Collagen in Asthma: A Quantitative Light and Electron Microscopic Study. Am. Rev. Respir. Dis. 1992, 145, 890–899. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, C.; Hauber, H.P.; Gotfried, M.; Newman, K.; Dhanda, R.; Servi, R.J.; Ludwig, M.S.; Hamid, Q. Evidence of remodeling in peripheral airways of patients with mild to moderate asthma: Effect of hydrofluoroalkane-flunisolide. J. Allergy Clin. Immunol. 2005, 116, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, R.; Soderberg, M.; Horstedt, P.; Stenling, R. Morphological studies of bronchial mucosal biopsies from asthmatics before and after ten years of treatment with inhaled steroids. Eur. Respir. J. 1988, 1, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Phelan, P.D.; Robertson, C.F.; Olinsky, A. The Melbourne Asthma Study: 1964–1999. J. Allergy Clin. Immunol. 2002, 109, 189–194. [Google Scholar] [CrossRef]
- Sears, M.R.; Greene, J.M.; Willan, A.R.; Wiecek, E.M.; Taylor, D.R.; Flannery, E.M.; Cowan, J.O.; Herbison, G.P.; Silva, P.A.; Poulton, R. A Longitudinal, Population-Based, Cohort Study of Childhood Asthma Followed to Adulthood. N. Engl. J. Med. 2003, 349, 1414–1422. [Google Scholar] [CrossRef]
- Chen, W.-J.; Liaw, S.-F.; Lin, C.-C.; Lin, M.-W.; Chang, F.-T. Effects of Zileuton on Airway Smooth Muscle Remodeling after Repeated Allergen Challenge in Brown Norway Rats. Respiration 2013, 86, 421–429. [Google Scholar] [CrossRef]
- Hur, J.; Kang, J.Y.; Rhee, C.K.; Kim, Y.K.; Lee, S.Y. The leukotriene receptor antagonist pranlukast attenuates airway remodeling by suppressing TGF-beta signaling. Pulm. Pharmacol. Ther. 2018, 48, 5–14. [Google Scholar] [CrossRef]
- Janssen, E.M.; Dy, S.M.; Meara, A.S.; Kneuertz, P.J.; Presley, C.J.; Bridges, J.F.P. Analysis of Patient Preferences in Lung Cancer – Estimating Acceptable Tradeoffs Between Treatment Benefit and Side Effects. Patient Prefer. Adherence 2020, 14, 927–937. [Google Scholar] [CrossRef]
- Donovan, G.M.; Wang, K.C.W.; Shamsuddin, D.; Mann, T.S.; Henry, P.J.; Larcombe, A.N.; Noble, P.B. Pharmacological ablation of the airway smooth muscle layer—Mathematical predictions of functional improvement in asthma. Physiol. Rep. 2020, 8, e14451. [Google Scholar] [CrossRef]
- Clifford, R.L.; Knox, A.J. Vitamin D—A new treatment for airway remodelling in asthma? Br. J. Pharmacol. 2009, 158, 1426–1428. [Google Scholar] [CrossRef] [PubMed]
- Britt, R.D., Jr.; Thompson, M.A.; Freeman, M.R.; Stewart, A.L.; Pabelick, C.M.; Prakash, Y.S. Vitamin D Reduces Inflammation-induced Contractility and Remodeling of Asthmatic Human Airway Smooth Muscle. Ann. Am. Thorac. Soc. 2016, 13, S97–S98. [Google Scholar] [CrossRef] [PubMed]
- Britt, R.D., Jr.; Faksh, A.; Vogel, E.R.; Thompson, M.A.; Chu, V.; Pandya, H.C.; Amrani, Y.; Martin, R.J.; Pabelick, C.M.; Prakash, Y.S. Vitamin D Attenuates Cytokine-Induced Remodeling in Human Fetal Airway Smooth Muscle Cells. J. Cell. Physiol. 2015, 230, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Roth, M.; Zhao, F.; Zhong, J.; Lardinois, D.; Tamm, M. Serum IgE Induced Airway Smooth Muscle Cell Remodeling Is Independent of Allergens and Is Prevented by Omalizumab. PLoS ONE 2015, 10, e0136549. [Google Scholar] [CrossRef]
- Hoshino, M.; Ohtawa, J. Effects of Adding Omalizumab, an Anti-Immunoglobulin E Antibody, on Airway Wall Thickening in Asthma. Respiration 2012, 83, 520–528. [Google Scholar] [CrossRef]
- Tajiri, T.; Niimi, A.; Matsumoto, H.; Ito, I.; Oguma, T.; Otsuka, K.; Takeda, T.; Nakaji, H.; Inoue, H.; Iwata, T.; et al. Comprehensive efficacy of omalizumab for severe refractory asthma: A time-series observational study. Ann. Allergy Asthma Immunol. 2014, 113, 470–475.e2. [Google Scholar] [CrossRef]
- Przybyszowski, M.; Paciorek, K.; Zastrzeżyńska, W.; Gawlewicz-Mroczka, A.; Trojan-Królikowska, A.; Orłowska, A.; Soja, J.; Pawlik, W.; Sładek, K. Influence of Omalizumab Therapy on Airway Remodeling Assessed with High-Resolution Computed Tomography (HRCT) in Severe Allergic Asthma Patients. Adv. Respir. Med. 2018, 86, 282–290. [Google Scholar] [CrossRef]
- Haldar, P.; Brightling, C.E.; Hargadon, B.; Gupta, S.; Monteiro, W.; Sousa, A.; Marshall, R.P.; Bradding, P.; Green, R.H.; Wardlaw, A.J.; et al. Mepolizumab and Exacerbations of Refractory Eosinophilic Asthma. N. Engl. J. Med. 2009, 360, 973–984. [Google Scholar] [CrossRef]
- Flood-Page, P.; Menzies-Gow, A.; Phipps, S.; Ying, S.; Wangoo, A.; Ludwig, M.S.; Barnes, N.; Robinson, D.; Kay, A.B. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J. Clin. Investig. 2003, 112, 1029–1036. [Google Scholar] [CrossRef]
- Chachi, L.; Diver, S.; Kaul, H.; Rebelatto, M.C.; Boutrin, A.; Nisa, P.; Newbold, P.; Brightling, C. Computational modelling prediction and clinical validation of impact of benralizumab on airway smooth muscle mass in asthma. Eur. Respir. J. 2019, 54, 1900930. [Google Scholar] [CrossRef]
- Stolz, D.; Mkorombindo, T.; Schumann, D.M.; Agusti, A.; Ash, S.Y.; Bafadhel, M.; Bai, C.; Chalmers, J.D.; Criner, G.J.; Dharmage, S.C.; et al. Towards the elimination of chronic obstructive pulmonary disease: A Lancet Commission. Lancet 2022, 400, 921–972. [Google Scholar] [CrossRef] [PubMed]
- Agustí, A.; Hogg, J.C. Update on the Pathogenesis of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Ward, C.; Danial, W.; Wood-Baker, R.; Walters, E.H. Recent advances in understanding inflammation and remodeling in the airways in chronic obstructive pulmonary disease. Expert Rev. Respir. Med. 2013, 7, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The Nature of Small-Airway Obstruction in Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Nowrin, K.; Sohal, S.S.; Peterson, G.; Patel, R.; Walters, E.H. Epithelial-mesenchymal transition as a fundamental underlying pathogenic process in COPD airways: Fibrosis, remodeling and cancer. Expert Rev. Respir. Med. 2014, 8, 547–559. [Google Scholar] [CrossRef]
- Kheradmand, F.; Shan, M.; Xu, C.; Corry, D.B. Autoimmunity in chronic obstructive pulmonary disease: Clinical and experimental evidence. Expert Rev. Clin. Immunol. 2012, 8, 285–292. [Google Scholar] [CrossRef]
- Norrby, K. Mast cells and angiogenesis. Apmis 2002, 110, 355–371. [Google Scholar] [CrossRef]
- Soltani, A.; Ewe, Y.P.; Lim, Z.S.; Sohal, S.S.; Reid, D.; Weston, S.; Wood-Baker, R.; Walters, E.H. Mast cells in COPD airways: Relationship to bronchodilator responsiveness and angiogenesis. Eur. Respir. J. 2012, 39, 1361–1367. [Google Scholar] [CrossRef]
- Mahmood, M.Q.; Walters, E.H.; Shukla, S.D.; Weston, S.; Muller, H.K.; Ward, C.; Sohal, S.S. Beta-catenin, Twist and Snail: Transcriptional regulation of EMT in smokers and COPD, and relation to airflow obstruction. Sci. Rep. 2017, 7, 10832. [Google Scholar] [CrossRef]
- Koczulla, A.-R.; Jonigk, D.; Wolf, T.; Herr, C.; Noeske, S.; Klepetko, W.; Vogelmeier, C.; von Neuhoff, N.; Rische, J.; Wrenger, S.; et al. Krüppel-like zinc finger proteins in end-stage COPD lungs with and without severe alpha1-antitrypsin deficiency. Orphanet J. Rare Dis. 2012, 7, 29. [Google Scholar] [CrossRef]
- Gohy, S.T.; Hupin, C.; Fregimilicka, C.; Detry, B.R.; Bouzin, C.; Gaide Chevronay, H.; Lecocq, M.; Weynand, B.; Ladjemi, M.Z.; Pierreux, C.E.; et al. Imprinting of the COPD airway epithelium for dedifferentiation and mesenchymal transition. Eur. Respir. J. 2015, 45, 1258–1272. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Jiang, Y.-L.; Fei, J.; Cao, P.; Zhang, C.; Xie, G.-F.; Wang, L.-X.; Cao, W.; Fu, L.; Zhao, H. Circulatory cadmium positively correlates with epithelial-mesenchymal transition in patients with chronic obstructive pulmonary disease. Ecotoxicol. Environ. Saf. 2021, 215, 112164. [Google Scholar] [CrossRef] [PubMed]
- Shirahata, T.; Nakamura, H.; Nakajima, T.; Nakamura, M.; Chubachi, S.; Yoshida, S.; Tsuduki, K.; Mashimo, S.; Takahashi, S.; Minematsu, N.; et al. Plasma sE-cadherin and the plasma sE-cadherin/sVE-cadherin ratio are potential biomarkers for chronic obstructive pulmonary disease. Biomarkers 2018, 23, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiro, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef]
- Sohal, S.S.; Reid, D.; Soltani, A.; Ward, C.; Weston, S.; Muller, H.K.; Wood-Baker, R.; Walters, E.H. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology 2010, 15, 930–938. [Google Scholar] [CrossRef]
- Soltani, A.; Reid, D.W.; Sohal, S.S.; Wood-Baker, R.; Weston, S.; Muller, H.K.; Walters, E.H. Basement membrane and vascular remodelling in smokers and chronic obstructive pulmonary disease: A cross-sectional study. Respir. Res. 2010, 11, 105. [Google Scholar] [CrossRef]
- Sohal, S.S.; Reid, D.; Soltani, A.; Ward, C.; Weston, S.; Muller, H.K.; Wood-Baker, R.; Walters, E.H. Evaluation of epithelial mesenchymal transition in patients with chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 130. [Google Scholar] [CrossRef]
- Gonzalez, D.M.; Medici, D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci. Signal. 2014, 7, re8. [Google Scholar] [CrossRef]
- Zuo, H.; Cattani-Cavalieri, I.; Valença, S.S.; Musheshe, N.; Schmidt, M. Function of cAMP scaffolds in obstructive lung disease: Focus on epithelial-to-mesenchymal transition and oxidative stress. Br. J. Pharmacol. 2019, 176, 2402–2415. [Google Scholar] [CrossRef]
- Takizawa, H.; Tanaka, M.; Takami, K.; Ohtoshi, T.; Ito, K.; Satoh, M.; Okada, Y.; Yamasawa, F.; Nakahara, K.; Umeda, A. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am. J. Respir. Crit. Care Med. 2001, 163, 1476–1483. [Google Scholar] [CrossRef]
- Schupp, J.C.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage Activation in Acute Exacerbation of Idiopathic Pulmonary Fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, X.M.; Ng, Y.Y.; Ma, F.Y.; Zhou, S.; Zhang, Y.; Yang, C.; Huang, X.R.; Xiao, J.; Wang, Y.Y.; et al. TGF-beta/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 2016, 7, 8809–8822. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Chang, Y.; Zhang, C.; Xiong, Y.; Wang, X.; Ma, X.; Wang, Z.; Li, H.; Shimosawa, T.; Pei, L.; et al. UUO induces lung fibrosis with macrophage-myofibroblast transition in rats. Int. Immunopharmacol. 2021, 93, 107396. [Google Scholar] [CrossRef]
- Hallgren, O.; Rolandsson, S.; Andersson-Sjöland, A.; Nihlberg, K.; Wieslander, E.; Kvist-Reimer, M.; Dahlbäck, M.; Eriksson, L.; Bjermer, L.; Erjefält, J.S.; et al. Enhanced ROCK1 dependent contractility in fibroblast from chronic obstructive pulmonary disease patients. J. Transl. Med. 2012, 10, 171. [Google Scholar] [CrossRef] [PubMed]
- Sohal, S.S.; Soltani, A.; Reid, D.; Ward, C.; Wills, K.E.; Muller, H.K.; Walters, E.H. A randomized controlled trial of inhaled corticosteroids (ICS) on markers of epithelial–mesenchymal transition (EMT) in large airway samples in COPD: An exploratory proof of concept study. Int. J. Chronic Obstruct. Pulmon. Dis. 2014, 9, 533–542. [Google Scholar] [CrossRef]
- Eapen, M.S.; Lu, W.; Hackett, T.L.; Singhera, G.K.; Mahmood, M.Q.; Hardikar, A.; Ward, C.; Walters, E.H.; Sohal, S.S. Increased myofibroblasts in the small airways, and relationship to remodelling and functional changes in smokers and COPD patients: Potential role of epithelial-mesenchymal transition. ERJ Open Res. 2021, 7, 00876-2020. [Google Scholar] [CrossRef]
- Togo, S.; Holz, O.; Liu, X.; Sugiura, H.; Kamio, K.; Wang, X.; Kawasaki, S.; Ahn, Y.; Fredriksson, K.; Skold, C.M.; et al. Lung Fibroblast Repair Functions in Patients with Chronic Obstructive Pulmonary Disease Are Altered by Multiple Mechanisms. Am. J. Respir. Crit. Care Med. 2008, 178, 248–260. [Google Scholar] [CrossRef]
- Barnes, P.J. Small airway fibrosis in COPD. Int. J. Biochem. Cell Biol. 2019, 116, 105598. [Google Scholar] [CrossRef]
- Lynch, M.D.; Watt, F.M. Fibroblast heterogeneity: Implications for human disease. J. Clin. Investig. 2018, 128, 26–35. [Google Scholar] [CrossRef]
- Marginean, C.; Popescu, M.S.; Vladaia, M.; Tudorascu, D.; Pirvu, D.C.; Petrescu, F. Involvement of Oxidative Stress in COPD. Curr. Health Sci. J. 2018, 44, 48–55. [Google Scholar]
- Antar, S.A.; Ashour, N.A.; Marawan, M.E.; Al-Karmalawy, A.A. Fibrosis: Types, Effects, Markers, Mechanisms for Disease Progression, and Its Relation with Oxidative Stress, Immunity, and Inflammation. Int. J. Mol. Sci. 2023, 24, 4004. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Borok, Z. TGF-beta-induced EMT: Mechanisms and implications for fibrotic lung disease. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L525–L534. [Google Scholar] [CrossRef] [PubMed]
- Carlier, F.M.; Dupasquier, S.; Ambroise, J.; Detry, B.; Lecocq, M.; Biétry–Claudet, C.; Boukala, Y.; Gala, J.-L.; Bouzin, C.; Verleden, S.E.; et al. Canonical WNT pathway is activated in the airway epithelium in chronic obstructive pulmonary disease. Ebiomedicine 2020, 61, 103034. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zou, Y.; Zhao, Z.; Li, B.; Ran, P. Nicotine-induced epithelial-mesenchymal transition via Wnt/beta-catenin signaling in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L199–L209. [Google Scholar] [CrossRef]
- Kneidinger, N.; Yildirim, A.O.; Callegari, J.; Takenaka, S.; Stein, M.M.; Dumitrascu, R.; Bohla, A.; Bracke, K.R.; Morty, R.E.; Brusselle, G.G.; et al. Activation of the WNT/beta-catenin pathway attenuates experimental emphysema. Am. J. Respir. Crit. Care Med. 2011, 183, 723–733. [Google Scholar] [CrossRef]
- Milara, J.; Peiro, T.; Serrano, A.; Artigues, E.; Aparicio, J.; Tenor, H.; Sanz, C.; Cortijo, J. Simvastatin Increases the Ability of Roflumilast N-oxide to Inhibit Cigarette Smoke-Induced Epithelial to Mesenchymal Transition in Well-differentiated Human Bronchial Epithelial Cells in vitro. COPD 2015, 12, 320–331. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, Y.; Zhu, Y.; Li, C.; Zhou, L.; Li, X.; Zhang, F.; Qiu, X.; Qu, Y. Cathelicidin induces epithelial-mesenchymal transition to promote airway remodeling in smoking-related chronic obstructive pulmonary disease. Ann. Transl. Med. 2021, 9, 223. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, Y.; Sun, C.; Wang, Q.; Yang, Z.; Pan, X.; Zhu, M.; Xiao, W. The human cathelicidin LL-37 enhances airway mucus production in chronic obstructive pulmonary disease. Biochem. Biophys. Res. Commun. 2014, 443, 103–109. [Google Scholar] [CrossRef]
- Sun, C.; Zhu, M.; Yang, Z.; Pan, X.; Zhang, Y.; Wang, Q.; Xiao, W. LL-37 secreted by epithelium promotes fibroblast collagen production: A potential mechanism of small airway remodeling in chronic obstructive pulmonary disease. Lab. Investig. 2014, 94, 991–1002. [Google Scholar] [CrossRef]
- Chu, S.; Ma, L.; Wu, Y.; Zhao, X.; Xiao, B.; Pan, Q. C-EBPbeta mediates in cigarette/IL-17A-induced bronchial epithelial-mesenchymal transition in COPD mice. BMC Pulm. Med. 2021, 21, 376. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Zhang, Y.; Zhang, Y.; Xiao, W. The role of uPAR in epithelial-mesenchymal transition in small airway epithelium of patients with chronic obstructive pulmonary disease. Respir. Res. 2013, 14, 67. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, M.Q.; Sohal, S.S.; Shukla, S.D.; Hardikar, A.; Noor, W.D.; Muller, H.K.; Knight, D.A.; Walters, E.H.; Ward, C. Epithelial mesenchymal transition in smokers: Large versus small airways and relation to airflow obstruction. Int. J. Chronic Obstruct. Pulmon. Dis. 2015, 10, 1515–1524. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xiao, W.; Jiang, Y.; Wang, H.; Xu, X.; Ma, D.; Chen, H.; Wang, X. Levels of Components of the Urokinase-Type Plasminogen Activator System are Related to Chronic Obstructive Pulmonary Disease Parenchymal Destruction and Airway Remodelling. J. Int. Med. Res. 2012, 40, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Young, R.P.; Whittington, C.F.; Hopkins, R.J.; Hay, B.A.; Epton, M.J.; Black, P.N.; Gamble, G.D. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur. Respir. J. 2010, 36, 1375–1382. [Google Scholar] [CrossRef]
- Wu, N.; Wu, Z.; Sun, J.; Yan, M.; Wang, B.; Du, X.; Liu, Y. Small airway remodeling in diabetic and smoking chronic obstructive pulmonary disease patients. Aging 2020, 12, 7927–7944. [Google Scholar] [CrossRef]
- Pan, K.; Lu, J.; Song, Y. Artesunate ameliorates cigarette smoke-induced airway remodelling via PPAR-gamma/TGF-beta1/Smad2/3 signalling pathway. Respir. Res. 2021, 22, 91. [Google Scholar] [CrossRef]
- Wang, W.; Zha, G.; Zou, J.J.; Wang, X.; Li, C.N.; Wu, X.J. Berberine Attenuates Cigarette Smoke Extract-induced Airway Inflammation in Mice: Involvement of TGF-beta1/Smads Signaling Pathway. Curr. Med. Sci. 2019, 39, 748–753. [Google Scholar] [CrossRef]
- Mahmood, M.Q.; Reid, D.; Ward, C.; Muller, H.K.; Knight, D.A.; Sohal, S.S.; Walters, E.H. Transforming growth factor (TGF) β(1) and Smad signalling pathways: A likely key to EMT-associated COPD pathogenesis. Respirology 2017, 22, 133–140. [Google Scholar] [CrossRef]
- de Boer, W.I.; van Schadewijk, A.; Sont, J.K.; Sharma, H.S.; Stolk, J.; Hiemstra, P.S.; van Krieken, J.H. Transforming growth factor beta1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 158, 1951–1957. [Google Scholar] [CrossRef]
- Ge, F.; Feng, Y.; Huo, Z.; Li, C.; Wang, R.; Wen, Y.; Gao, S.; Peng, H.; Wu, X.; Liang, H.; et al. Inhaled corticosteroids and risk of lung cancer among chronic obstructive pulmonary disease patients: A comprehensive analysis of nine prospective cohorts. Transl. Lung Cancer Res. 2021, 10, 1266–1276. [Google Scholar] [CrossRef]
- Zhu, L.; Xu, F.; Kang, X.; Zhou, J.; Yao, Q.; Lin, Y.; Zhang, W. The antioxidant N-acetylcysteine promotes immune response and inhibits epithelial-mesenchymal transition to alleviate pulmonary fibrosis in chronic obstructive pulmonary disease by suppressing the VWF/p38 MAPK axis. Mol. Med. 2021, 27, 97. [Google Scholar] [CrossRef] [PubMed]
- Martorana, P.A.; Beume, R.; Lucattelli, M.; Wollin, L.; Lungarella, G. Roflumilast fully prevents emphysema in mice chronically exposed to cigarette smoke. Am. J. Respir. Crit. Care Med. 2005, 172, 848–853. [Google Scholar] [CrossRef]
- Milara, J.; Peiró, T.; Serrano, A.; Guijarro, R.; Zaragozá, C.; Tenor, H.; Cortijo, J. Roflumilast N-oxide inhibits bronchial epithelial to mesenchymal transition induced by cigarette smoke in smokers with COPD. Pulm. Pharmacol. Ther. 2014, 28, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, Y.; Zhou, Z.; Huang, M.; Deng, W.; Wang, Y.; Zhou, X.; Chen, L.; Li, Y.; Zeng, T.; et al. Celecoxib inhibits the epithelial-to-mesenchymal transition in bladder cancer via the miRNA-145/TGFBR2/Smad3 axis. Int. J. Mol. Med. 2019, 44, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, Y.; Imanishi, Y.; Ozawa, H.; Sakamoto, K.; Fujii, R.; Shigetomi, S.; Habu, N.; Otsuka, K.; Sato, Y.; Sekimizu, M.; et al. Selective EP2 and Cox-2 inhibition suppresses cell migration by reversing epithelial-to-mesenchymal transition and Cox-2 overexpression and E-cadherin downregulation are implicated in neck metastasis of hypopharyngeal cancer. Am. J. Transl. Res. 2020, 12, 1096–1113. [Google Scholar] [PubMed]
- Paller, C.; Pu, H.; Begemann, D.E.; Wade, C.A.; Hensley, P.J.; Kyprianou, N. TGF-beta receptor I inhibitor enhances response to enzalutamide in a pre-clinical model of advanced prostate cancer. Prostate 2019, 79, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Bobal, P.; Lastovickova, M.; Bobalova, J. The Role of ATRA, Natural Ligand of Retinoic Acid Receptors, on EMT-Related Proteins in Breast Cancer: Minireview. Int. J. Mol. Sci. 2021, 22, 13345. [Google Scholar] [CrossRef]
- Blaschuk, O.W. Potential Therapeutic Applications of N-Cadherin Antagonists and Agonists. Front. Cell Dev. Biol. 2022, 10, 866200. [Google Scholar] [CrossRef]
- Lv, Q.; Wang, J.; Xu, C.; Huang, X.; Ruan, Z.; Dai, Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3beta/beta-catenin and TGF-beta1/Smad2/3 signaling pathways. Mol. Med. 2020, 26, 49. [Google Scholar] [CrossRef]
- Ihara, H.; Mitsuishi, Y.; Kato, M.; Takahashi, F.; Tajima, K.; Hayashi, T.; Hidayat, M.; Winardi, W.; Wirawan, A.; Hayakawa, D.; et al. Nintedanib inhibits epithelial-mesenchymal transition in A549 alveolar epithelial cells through regulation of the TGF-beta/Smad pathway. Respir. Investig. 2020, 58, 275–284. [Google Scholar] [CrossRef]
- Shteinberg, M.; Haq, I.J.; Polineni, D.; Davies, J.C. Cystic fibrosis. Lancet 2021, 397, 2195–2211. [Google Scholar] [CrossRef] [PubMed]
- Françoise, A.; Héry-Arnaud, G. The Microbiome in Cystic Fibrosis Pulmonary Disease. Genes 2020, 11, 536. [Google Scholar] [CrossRef] [PubMed]
- Bedrossian, C.W.; Greenberg, S.D.; Singer, D.B.; Hansen, J.J.; Rosenberg, H.S. The lung in cystic fibrosis. A quantitative study including prevalence of pathologic findings among different age groups. Hum. Pathol. 1976, 7, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Durieu, I.; Peyrol, S.; Gindre, D.; Bellon, G.; Durand, D.V.; Pacheco, Y. Subepithelial Fibrosis and Degradation of the Bronchial Extracellular Matrix in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 1998, 158, 580–588. [Google Scholar] [CrossRef]
- Burgel, P.-R.; Montani, D.; Danel, C.; Dusser, D.J.; Nadel, J.A. A morphometric study of mucins and small airway plugging in cystic fibrosis. Thorax 2007, 62, 153–161. [Google Scholar] [CrossRef]
- Hays, S.R.; Ferrando, R.E.; Carter, R.; Wong, H.H.; Woodruff, P.G. Structural changes to airway smooth muscle in cystic fibrosis. Thorax 2005, 60, 226–228. [Google Scholar] [CrossRef][Green Version]
- Hilliard, T.N.; Regamey, N.; Shute, J.K.; Nicholson, A.G.; Alton, E.W.; Bush, A.; Davies, J.C. Airway remodelling in children with cystic fibrosis. Thorax 2007, 62, 1074–1080. [Google Scholar] [CrossRef]
- Boucher, R.C. Muco-Obstructive Lung Diseases. Reply. N. Engl. J. Med. 2019, 381, e20. [Google Scholar]
- De Rose, V. Mechanisms and markers of airway inflammation in cystic fibrosis. Eur. Respir. J. 2002, 19, 333–340. [Google Scholar] [CrossRef]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum Biomarkers of Inflammation and Lung Function Decline in Children with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Sly, P.D.; Gangell, C.L.; Chen, L.; Ware, R.S.; Ranganathan, S.; Mott, L.S.; Murray, C.P.; Stick, S.M.; Investigators, A.C. Risk factors for bronchiectasis in children with cystic fibrosis. N. Engl. J. Med. 2013, 368, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.-M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Buckley, A.G.; Kicic-Starcevich, E.; Lannigan, F.J.; et al. Alpha-1 Antitrypsin Mitigates the Inhibition of Airway Epithelial Cell Repair by Neutrophil Elastase. Am. J. Respir. Cell Mol. Biol. 2016, 54, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Doumas, S.; Kolokotronis, A.; Stefanopoulos, P. Anti-Inflammatory and Antimicrobial Roles of Secretory Leukocyte Protease Inhibitor. Infect. Immun. 2005, 73, 1271–1274. [Google Scholar] [CrossRef] [PubMed]
- Twigg, M.S.; Brockbank, S.; Lowry, P.; FitzGerald, S.P.; Taggart, C.; Weldon, S. The Role of Serine Proteases and Antiproteases in the Cystic Fibrosis Lung. Mediat. Inflamm. 2015, 2015, 293053. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.; Ye, S.; Pedrina, J.; Dlugolenski, D.; Stambas, J. Extracellular Matrix Enzymes and Immune Cell Biology. Front. Mol. Biosci. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Devereux, G.; Steele, S.; Jagelman, T.; Fielding, S.; Muirhead, R.; Brady, J.; Grierson, C.; Brooker, R.; Winter, J.; Fardon, T.; et al. An observational study of matrix metalloproteinase (MMP)-9 in cystic fibrosis. J. Cyst. Fibros. 2014, 13, 557–563. [Google Scholar] [CrossRef]
- Sagel, S.D.; Kapsner, R.K.; Osberg, I. Induced sputum matrix metalloproteinase-9 correlates with lung function and airway inflammation in children with cystic fibrosis. Pediatr. Pulmonol. 2005, 39, 224–232. [Google Scholar] [CrossRef]
- Esposito, R.; Mirra, D.; Spaziano, G.; Panico, F.; Gallelli, L.; D’Agostino, B. The Role of MMPs in the Era of CFTR Modulators: An Additional Target for Cystic Fibrosis Patients? Biomolecules 2023, 13, 350. [Google Scholar] [CrossRef]
- Garratt, L.W.; Sutanto, E.N.; Ling, K.-M.; Looi, K.; Iosifidis, T.; Martinovich, K.M.; Shaw, N.C.; Kicic-Starcevich, E.; Knight, D.A.; Ranganathan, S.; et al. Matrix metalloproteinase activation by free neutrophil elastase contributes to bronchiectasis progression in early cystic fibrosis. Eur. Respir. J. 2015, 46, 384–394. [Google Scholar] [CrossRef]
- Duszyk, M.; Shu, Y.; Sawicki, G.; Radomski, A.; Man, S.F.; Radomski, M.W. Inhibition of matrix metalloproteinase MMP-2 activates chloride current in human airway epithelial cells. Can. J. Physiol. Pharmacol. 1999, 77, 529–535. [Google Scholar] [CrossRef]
- Adam, D.; Roux-Delrieu, J.; Luczka, E.; Bonnomet, A.; Lesage, J.; Mérol, J.-C.; Polette, M.; Abély, M.; Coraux, C. Cystic fibrosis airway epithelium remodelling: Involvement of inflammation. J. Pathol. 2015, 235, 408–419. [Google Scholar] [CrossRef]
- Bruscia, E.M.; Zhang, P.X.; Barone, C.; Scholte, B.J.; Homer, R.; Krause, D.S.; Egan, M.E. Increased susceptibility of Cftr−/− mice to LPS-induced lung remodeling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L711–L719. [Google Scholar] [CrossRef] [PubMed]
- Conese, M.; Di Gioia, S. Pathophysiology of Lung Disease and Wound Repair in Cystic Fibrosis. Pathophysiology 2021, 28, 155–188. [Google Scholar] [CrossRef]
- Southey, M.C.; Batten, L.; Andersen, C.R.; McCredie, M.R.; Giles, G.G.; Dite, G.; Hopper, J.L.; Venter, D.J. CFTR deltaF508 carrier status, risk of breast cancer before the age of 40 and histological grading in a population-based case-control study. Int. J. Cancer 1998, 79, 487–489. [Google Scholar] [CrossRef]
- Zhang, J.T.; Jiang, X.H.; Xie, C.; Cheng, H.; Da Dong, J.; Wang, Y.; Fok, K.L.; Zhang, X.H.; Sun, T.T.; Tsang, L.L.; et al. Downregulation of CFTR promotes epithelial-to-mesenchymal transition and is associated with poor prognosis of breast cancer. Biochim. Biophys. Acta 2013, 1833, 2961–2969. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Dong, F.; Gao, H.; Guo, Y.; Li, H.; Yang, F.; Zhao, P.; Dai, Y.; Wang, J.; Zhou, W.; et al. Promoter hypermethylation of the CFTR gene as a novel diagnostic and prognostic marker of breast cancer. Cell Biol. Int. 2020, 44, 603–609. [Google Scholar] [CrossRef]
- Amaral, M.D.; Quaresma, M.C.; Pankonien, I. What Role Does CFTR Play in Development, Differentiation, Regeneration and Cancer? Int. J. Mol. Sci. 2020, 21, 3133. [Google Scholar] [CrossRef]
- Cohen, J.C.; Larson, J.E.; Killeen, E.; Love, D.; Takemaru, K. CFTR and Wnt/beta-catenin signaling in lung development. BMC Dev. Biol. 2008, 8, 70. [Google Scholar] [CrossRef]
- Sun, H.; Wang, Y.; Zhang, J.; Chen, Y.; Liu, Y.; Lin, Z.; Liu, M.; Sheng, K.; Liao, H.; Tsang, K.S.; et al. CFTR mutation enhances Dishevelled degradation and results in impairment of Wnt-dependent hematopoiesis. Cell Death Dis. 2018, 9, 275. [Google Scholar] [CrossRef]
- Quaresma, M.C.; Pankonien, I.; Clarke, L.A.; Sousa, L.S.; Silva, I.A.L.; Railean, V.; Doušová, T.; Fuxe, J.; Amaral, M.D. Mutant CFTR Drives TWIST1 mediated epithelial–mesenchymal transition. Cell Death Dis. 2020, 11, 920. [Google Scholar] [CrossRef]
- Collin, A.M.; Lecocq, M.; Detry, B.; Carlier, F.M.; Bouzin, C.; de Sany, P.; Hoton, D.; Verleden, S.; Froidure, A.; Pilette, C.; et al. Loss of ciliated cells and altered airway epithelial integrity in cystic fibrosis. J. Cyst. Fibros. 2021, 20, e129–e139. [Google Scholar] [CrossRef] [PubMed]
- Nyabam, S.; Wang, Z.; Thibault, T.; Oluseyi, A.; Basar, R.; Marshall, L.; Griffin, M. A novel regulatory role for tissue transglutaminase in epithelial-mesenchymal transition in cystic fibrosis. Biochim. Biophys. Acta 2016, 1863, 2234–2244. [Google Scholar] [CrossRef] [PubMed]
- Clarke, L.A.; Botelho, H.M.; Sousa, L.; Falcao, A.O.; Amaral, M.D. Transcriptome meta-analysis reveals common differential and global gene expression profiles in cystic fibrosis and other respiratory disorders and identifies CFTR regulators. Genomics 2015, 106, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.-L.; et al. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef]
- Bombieri, C.; Seia, M.; Castellani, C. Genotypes and Phenotypes in Cystic Fibrosis and Cystic Fibrosis Transmembrane Regulator–Related Disorders. Semin. Respir. Crit. Care Med. 2015, 36, 180–193. [Google Scholar] [CrossRef]
- Bustamante, A.E.; Fernández, L.T.; Rivas, L.C.; Mercado-Longoria, R. Disparities in cystic fibrosis survival in Mexico: Impact of socioeconomic status. Pediatr. Pulmonol. 2021, 56, 1566–1572. [Google Scholar] [CrossRef]
- Oates, G.R.; Baker, E.; Rowe, S.M.; Gutierrez, H.H.; Schechter, M.S.; Morgan, W.; Harris, W.T. Tobacco smoke exposure and socioeconomic factors are independent predictors of pulmonary decline in pediatric cystic fibrosis. J. Cyst. Fibros. 2020, 19, 783–790. [Google Scholar] [CrossRef]
- Merlo, C.A.; Boyle, M.P. Modifier genes in cystic fibrosis lung disease. J. Lab. Clin. Med. 2003, 141, 237–241. [Google Scholar] [CrossRef]
- Drumm, M.L.; Konstan, M.W.; Schluchter, M.D.; Handler, A.; Pace, R.; Zou, F.; Zariwala, M.; Fargo, D.; Xu, A.; Dunn, J.M.; et al. Genetic Modifiers of Lung Disease in Cystic Fibrosis. N. Engl. J. Med. 2005, 353, 1443–1453. [Google Scholar] [CrossRef]
- Taylor-Cousar, J.L.; Zariwala, M.A.; Burch, L.H.; Pace, R.G.; Drumm, M.L.; Calloway, H.; Fan, H.; Weston, B.W.; Wright, F.A.; Knowles, M.R.; et al. Histo-Blood Group Gene Polymorphisms as Potential Genetic Modifiers of Infection and Cystic Fibrosis Lung Disease Severity. PLoS ONE 2009, 4, e4270. [Google Scholar] [CrossRef] [PubMed]
- Guillot, L.; Beucher, J.; Tabary, O.; Le Rouzic, P.; Clement, A.; Corvol, H. Lung disease modifier genes in cystic fibrosis. Int. J. Biochem. Cell Biol. 2014, 52, 83–93. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Panjwani, N.; Avolio, J.; Ouyang, H.; Keshavjee, S.; Rommens, J.M.; Gonska, T.; Moraes, T.J.; Strug, L.J. Expression of cystic fibrosis lung disease modifier genes in human airway models. J. Cyst. Fibros. 2022, 21, 616–622. [Google Scholar] [CrossRef]
- Mésinèle, J.; Ruffin, M.; Guillot, L.; Corvol, H. Modifier Factors of Cystic Fibrosis Phenotypes: A Focus on Modifier Genes. Int. J. Mol. Sci. 2022, 23, 14205. [Google Scholar] [CrossRef] [PubMed]
- Sepahzad, A.; Morris-Rosendahl, D.J.; Davies, J.C. Cystic Fibrosis Lung Disease Modifiers and Their Relevance in the New Era of Precision Medicine. Genes 2021, 12, 562. [Google Scholar] [CrossRef]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef]
- Harris, W.T.; Kelly, D.R.; Zhou, Y.; Wang, D.; MacEwen, M.; Hagood, J.S.; Clancy, J.P.; Ambalavanan, N.; Sorscher, E.J. Myofibroblast differentiation and enhanced TGF-B signaling in cystic fibrosis lung disease. PLoS ONE 2013, 8, e70196. [Google Scholar] [CrossRef]
- Brazova, J.; Sismova, K.; Vavrova, V.; Bartosova, J.; Macek, M., Jr.; Lauschman, H.; Sediva, A. Polymorphisms of TGF-beta1 in cystic fibrosis patients. Clin. Immunol. 2006, 121, 350–357. [Google Scholar] [CrossRef]
- Faria, E.J.; Faria, I.C.; Ribeiro, J.D.; Ribeiro, A.F.; Hessel, G.; Bertuzzo, C.S. Association of MBL2, TGF-beta1 and CD14 gene polymorphisms with lung disease severity in cystic fibrosis. J. Bras. Pneumol. 2009, 35, 334–342. [Google Scholar] [CrossRef]
- Trojan, T.; Alejandre Alcazar, M.A.; Fink, G.; Thomassen, J.C.; Maessenhausen, M.V.; Rietschel, E.; Schneider, P.M.; van Koningsbruggen-Rietschel, S. The effect of TGF-β(1) polymorphisms on pulmonary disease progression in patients with cystic fibrosis. BMC Pulm. Med. 2022, 22, 183. [Google Scholar] [CrossRef]
- Corvol, H.; Boelle, P.Y.; Brouard, J.; Knauer, N.; Chadelat, K.; Henrion-Caude, A.; Flamant, C.; Muselet-Charlier, C.; Boule, M.; Fauroux, B.; et al. Genetic variations in inflammatory mediators influence lung disease progression in cystic fibrosis. Pediatr. Pulmonol. 2008, 43, 1224–1232. [Google Scholar] [CrossRef]
- Arkwright, P.D.; Laurie, S.; Super, M.; Pravica, V.; Schwarz, M.J.; Webb, A.K.; Hutchinson, I.V. TGF-β(1) genotype and accelerated decline in lung function of patients with cystic fibrosis. Thorax 2000, 55, 459–462. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ghigo, A.; De Santi, C.; Hart, M.; Mitash, N.; Swiatecka-Urban, A. Cell signaling and regulation of CFTR expression in cystic fibrosis cells in the era of high efficiency modulator therapy. J. Cyst. Fibros. 2023, 22, S12–S16. [Google Scholar] [CrossRef]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Noah, T.L. Transforming growth factor-β(1) in bronchoalveolar lavage fluid from children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Harris, W.T.; Muhlebach, M.S.; Oster, R.A.; Knowles, M.R.; Clancy, J.P.; Noah, T.L. Plasma TGF-β(1) in pediatric cystic fibrosis: Potential biomarker of lung disease and response to therapy. Pediatr. Pulmonol. 2011, 46, 688–695. [Google Scholar] [CrossRef]
- Thomassen, J.C.; Trojan, T.; Walz, M.; Vohlen, C.; Fink, G.; Rietschel, E.; Alejandre Alcazar, M.A.; van Koningsbruggen-Rietschel, S. Reduced neutrophil elastase inhibitor elafin and elevated transforming growth factor-β(1) are linked to inflammatory response in sputum of cystic fibrosis patients with Pseudomonas aeruginosa. ERJ Open Res. 2021, 7, 00636-2020. [Google Scholar] [CrossRef]
- Sagwal, S.; Chauhan, A.; Kaur, J.; Prasad, R.; Singh, M.; Singh, M. Association of Serum TGF-beta1 Levels with Different Clinical Phenotypes of Cystic Fibrosis Exacerbation. Lung 2020, 198, 377–383. [Google Scholar] [CrossRef]
- Eickmeier, O.; Boom, L.; Schreiner, F.; Lentze, M.J.; NGampolo, D.; Schubert, R.; Zielen, S.; Schmitt-Grohé, S. Transforming Growth Factorβ1 Genotypes in Relation to TGFbeta1, Interleukin-8, and Tumor Necrosis Factor Alpha in Induced Sputum and Blood in Cystic Fibrosis. Mediat. Inflamm. 2013, 2013, 913135. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Peterson-Carmichael, S.L.; Harris, W.T.; Goel, R.; Noah, T.L.; Johnson, R.; Leigh, M.W.; Davis, S.D. Association of lower airway inflammation with physiologic findings in young children with cystic fibrosis. Pediatr. Pulmonol. 2009, 44, 503–511. [Google Scholar] [CrossRef]
- Stolzenburg, L.R.; Wachtel, S.; Dang, H.; Harris, A. miR-1343 attenuates pathways of fibrosis by targeting the TGF-beta receptors. Biochem. J. 2016, 473, 245–256. [Google Scholar] [CrossRef]
- Corvol, H.; Rousselet, N.; Thompson, K.E.; Berdah, L.; Cottin, G.; Foussigniere, T.; Longchampt, E.; Fiette, L.; Sage, E.; Prunier, C.; et al. FAM13A is a modifier gene of cystic fibrosis lung phenotype regulating rhoa activity, actin cytoskeleton dynamics and epithelial-mesenchymal transition. J. Cyst. Fibros. 2018, 17, 190–203. [Google Scholar] [CrossRef]
- Quaresma, M.C.; Botelho, H.M.; Pankonien, I.; Rodrigues, C.S.; Pinto, M.C.; Costa, P.R.; Duarte, A.; Amaral, M.D. Exploring YAP1-centered networks linking dysfunctional CFTR to epithelial–mesenchymal transition. Life Sci. Alliance 2022, 5, e202101326. [Google Scholar] [CrossRef] [PubMed]
- Sousa, L.; Pankonien, I.; Simões, F.B.; Chanson, M.; Amaral, M.D. Impact of KLF4 on Cell Proliferation and Epithelial Differentiation in the Context of Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 6717. [Google Scholar] [CrossRef] [PubMed]
- McBennett, K.A.; Davis, P.B.; Konstan, M.W. Increasing life expectancy in cystic fibrosis: Advances and challenges. Pediatr. Pulmonol. 2022, 57, S5–S12. [Google Scholar] [CrossRef] [PubMed]
- Adam, D.; Bilodeau, C.; Sognigbé, L.; Maillé, E.; Ruffin, M.; Brochiero, E. CFTR rescue with VX-809 and VX-770 favors the repair of primary airway epithelial cell cultures from patients with class II mutations in the presence of Pseudomonas aeruginosa exoproducts. J. Cyst. Fibros. 2018, 17, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Conese, M. Elexacaftor/Tezacaftor/Ivacaftor Accelerates Wound Repair in Cystic Fibrosis Airway Epithelium. J. Pers. Med. 2022, 12, 1577. [Google Scholar] [CrossRef] [PubMed]
- Bec, R.; Reynaud-Gaubert, M.; Arnaud, F.; Naud, R.; Dufeu, N.; Di Bisceglie, M.; Coiffard, B.; Gaubert, J.-Y.; Bermudez, J.; Habert, P. Chest computed tomography improvement in patients with cystic fibrosis treated with elexacaftor-tezacaftor-ivacaftor: Early report. Eur. J. Radiol. 2022, 154. [Google Scholar] [CrossRef]
- Maselli, D.J.; Amalakuhan, B.; Keyt, H.; Diaz, A.A. Suspecting non-cystic fibrosis bronchiectasis: What the busy primary care clinician needs to know. Int. J. Clin. Pract. 2017, 71, e12924. [Google Scholar] [CrossRef]
- Bergin, D.A.; Hurley, K.; Mehta, A.; Cox, S.; Ryan, D.; O’Neill, S.J.; Reeves, E.P.; McElvaney, N.G. Airway inflammatory markers in individuals with cystic fibrosis and non-cystic fibrosis bronchiectasis. J. Inflamm. Res. 2013, 6, 1–11. [Google Scholar] [CrossRef]
- Chalmers, J.D.; Polverino, E.; Crichton, M.L.; Ringshausen, F.C.; De Soyza, A.; Vendrell, M.; Burgel, P.R.; Haworth, C.S.; Loebinger, M.R.; Dimakou, K.; et al. Bronchiectasis in Europe: Data on disease characteristics from the European Bronchiectasis registry (EMBARC). Lancet Respir. Med. 2023, 11, 637–649. [Google Scholar] [CrossRef]
- Taylor, S.L.; Rogers, G.B.; Chen, A.C.; Burr, L.D.; McGuckin, M.A.; Serisier, D.J. Matrix Metalloproteinases Vary with Airway Microbiota Composition and Lung Function in Non–Cystic Fibrosis Bronchiectasis. Ann. Am. Thorac. Soc. 2015, 12, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Keir, H.R.; Shoemark, A.; Dicker, A.J.; Perea, L.; Pollock, J.; Giam, Y.H.; Suarez-Cuartin, G.; Crichton, M.L.; Lonergan, M.; Oriano, M.; et al. Neutrophil extracellular traps, disease severity, and antibiotic response in bronchiectasis: An international, observational, multicohort study. Lancet Respir. Med. 2021, 9, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.-H.; Chou, P.-C.; Chou, C.-L.; Ho, S.-C.; Joa, W.-C.; Chen, L.-F.; Sheng, T.-F.; Lin, H.-C.; Wang, T.-Y.; Chang, P.-J.; et al. Matrix Metalloproteinase-1 Polymorphism (-1607G) and Disease Severity in Non-Cystic Fibrosis Bronchiectasis in Taiwan. PLoS ONE 2013, 8, e66265. [Google Scholar] [CrossRef] [PubMed]

| Models | Techniques | EMT Program | AR | EMT Signal | EMT TFs | EMT Mark | Refs. | Year |
|---|---|---|---|---|---|---|---|---|
| Primary HBECs (from explanted CF lung or from control subjects who underwent lung surgery) | TEER IF RT-qPCR WB Wound healing | N-cadherin and vimentin (protein) TEER | x | x | [250] | 2020 | ||
| CFBE41o-wt or -F508del-CFTR HEK 293T cells | IF WB Wound healing | N-cadherin vimentin and collagen I (protein) ZO-1 and CX31, while claudin-1 and Desmoplakin I/II Multilayered organisation for CF versus monolayer for control EMT markers localisation differences Ki-67-positive cells in basal cell layers time to close the wounds TEER TWIST1 (protein)+CFTR modulator treatments N-cadherin and vimentin TWIST1+response to TGF-β1: No difference in response CK18 and N-cadherin (protein)WT more resistant to EMT induction than CF TWIST1 shRNA knockdown in HEK293T cells: vimentin inhibition | x | x | x | [250] | 2020 | |
| CF versus control lung tissues | IF RT-qPCR | occludin, tight junction protein 1/zonula occludens-1, connexin 43, connexin 26 and cytokeratin 18 (mRNA) vimentin (mRNA) Polygonal flat cells on the CF epithelial layer surface | ||||||
| Fewer cylindrical-shaped columnar cells and several cell layers in CF tissue TWIST1 and ZEB1 (mRNA) Positive staining for SNAILl + Slug and ZEB1 Partial EMT | x | x | [250] | 2020 | ||||
| CF versus control lung tissue | IHC | MUC5AC (protein) β-tubulin (protein) vimentin-positive spindle-shaped Thickening of the RBM | x | x | [251] | 2021 | ||
| ALI HBECs (CF and controls lung tissue 2 weeks) | TEER WB ELISA RT-qPCR IHC | MUC5AC (protein) MCIDAS, MYB and FOXJ1 (mRNA) TEER E-cadherin and occludin Spindle-shaped cells and thickness of epithelium +CFTR inhibition or Pseudomonas infection in control culture No EMT induction | x | x | ||||
| IB3-1 cells F508del/W1282X C38 cells as control | Cell migration assay WB IHC RT-qPCR ELISA | fibronectin, N-cadherin and the transcription repressor Slug in IB3 cells compared to the C38 cells migratory phenotype TGF-β1 (mRNA and protein) +TGF-β receptor inhibitor fibronectin (protein)+TG2 inhibitors in IB3-1 cells Inhibition of cell migration TGF-β1 (protein) | x | x | x | x | [252] | 2016 |
| ALI HBECs 14 days | WB | +TGF-β1 3 ng/mL 48 h: TG2 Fibronectin and N-cadherin E-cadherin (protein) +TG2 overexpression: fibronectin, N-cadherin, Slug (protein) E-cadherin (protein) TEER | x | [252] | 2016 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mottais, A.; Riberi, L.; Falco, A.; Soccal, S.; Gohy, S.; De Rose, V. Epithelial–Mesenchymal Transition Mechanisms in Chronic Airway Diseases: A Common Process to Target? Int. J. Mol. Sci. 2023, 24, 12412. https://doi.org/10.3390/ijms241512412
Mottais A, Riberi L, Falco A, Soccal S, Gohy S, De Rose V. Epithelial–Mesenchymal Transition Mechanisms in Chronic Airway Diseases: A Common Process to Target? International Journal of Molecular Sciences. 2023; 24(15):12412. https://doi.org/10.3390/ijms241512412
Chicago/Turabian StyleMottais, Angélique, Luca Riberi, Andrea Falco, Simone Soccal, Sophie Gohy, and Virginia De Rose. 2023. "Epithelial–Mesenchymal Transition Mechanisms in Chronic Airway Diseases: A Common Process to Target?" International Journal of Molecular Sciences 24, no. 15: 12412. https://doi.org/10.3390/ijms241512412
APA StyleMottais, A., Riberi, L., Falco, A., Soccal, S., Gohy, S., & De Rose, V. (2023). Epithelial–Mesenchymal Transition Mechanisms in Chronic Airway Diseases: A Common Process to Target? International Journal of Molecular Sciences, 24(15), 12412. https://doi.org/10.3390/ijms241512412