
Decoding the Gene Regulatory Network of Muscle Stem Cells in Mouse Duchenne Muscular Dystrophy: Revelations from Single-Nuclei RNA Sequencing Analysis

Abstract
:1. Introduction
2. Results
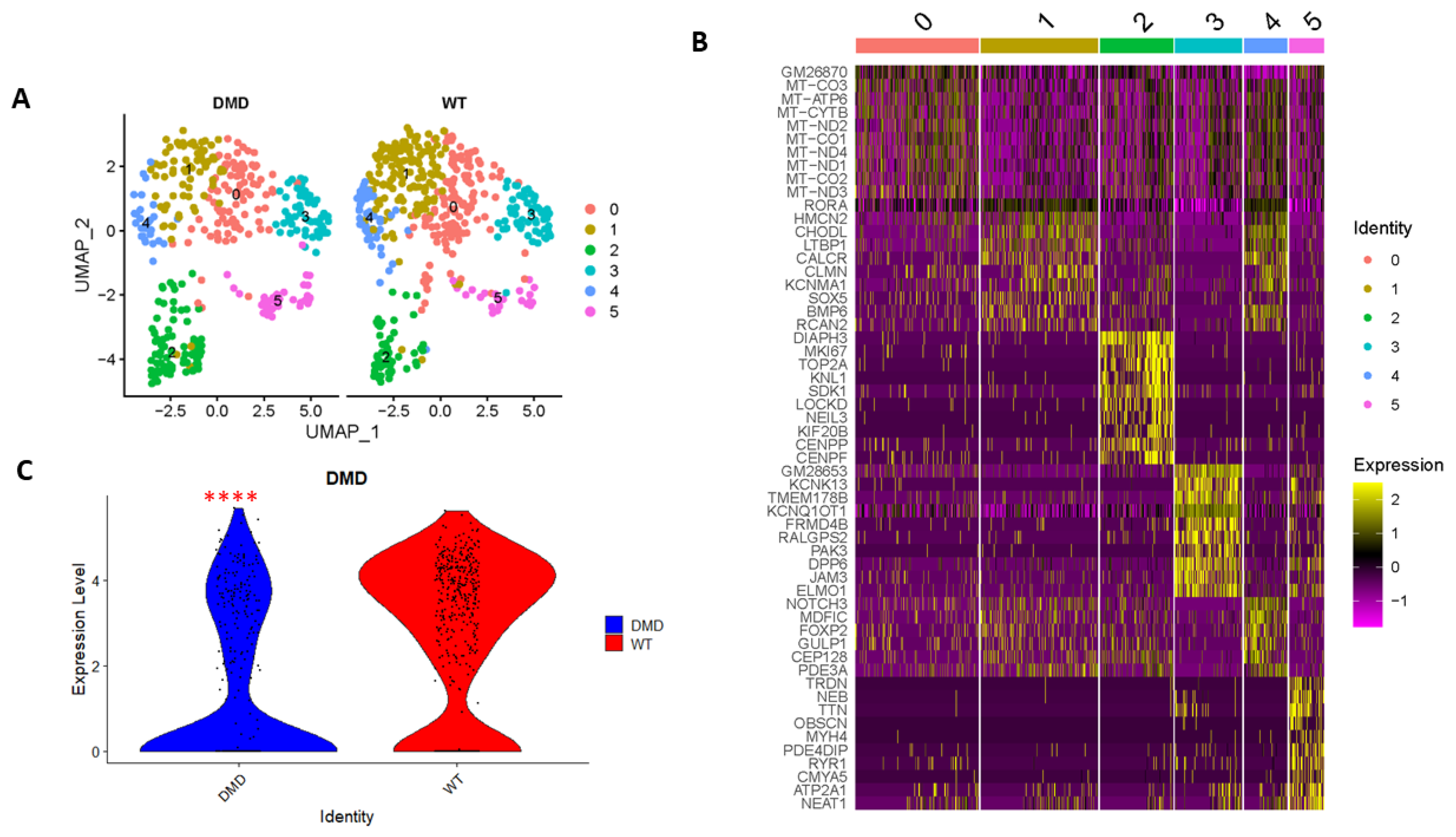
2.1. Single Cell Transcriptomics Reveals MuSC Clusters
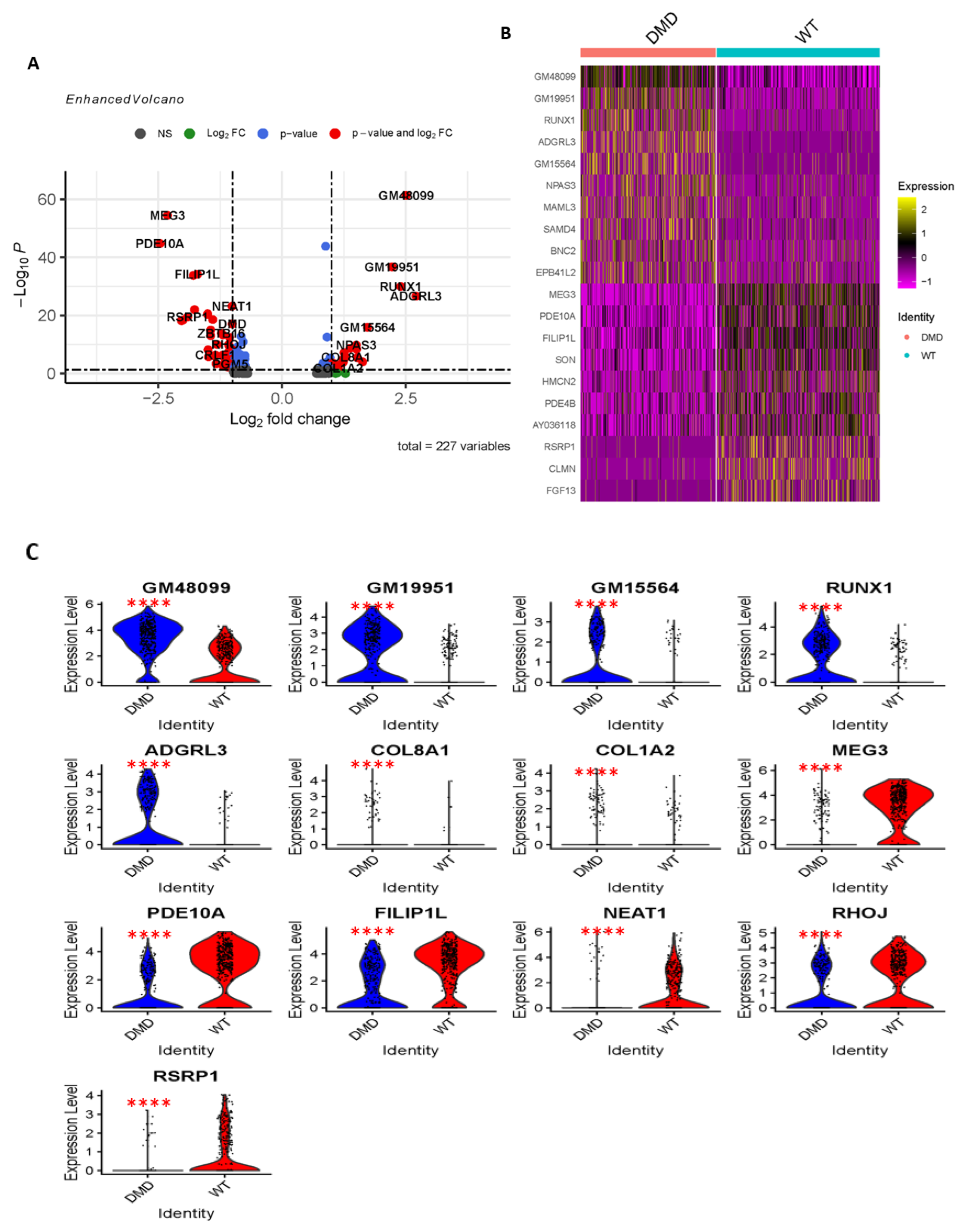
2.2. Differential Gene Expression and Functional Enrichment Analysis of MuSCs in DMDmut Versus Control Muscles
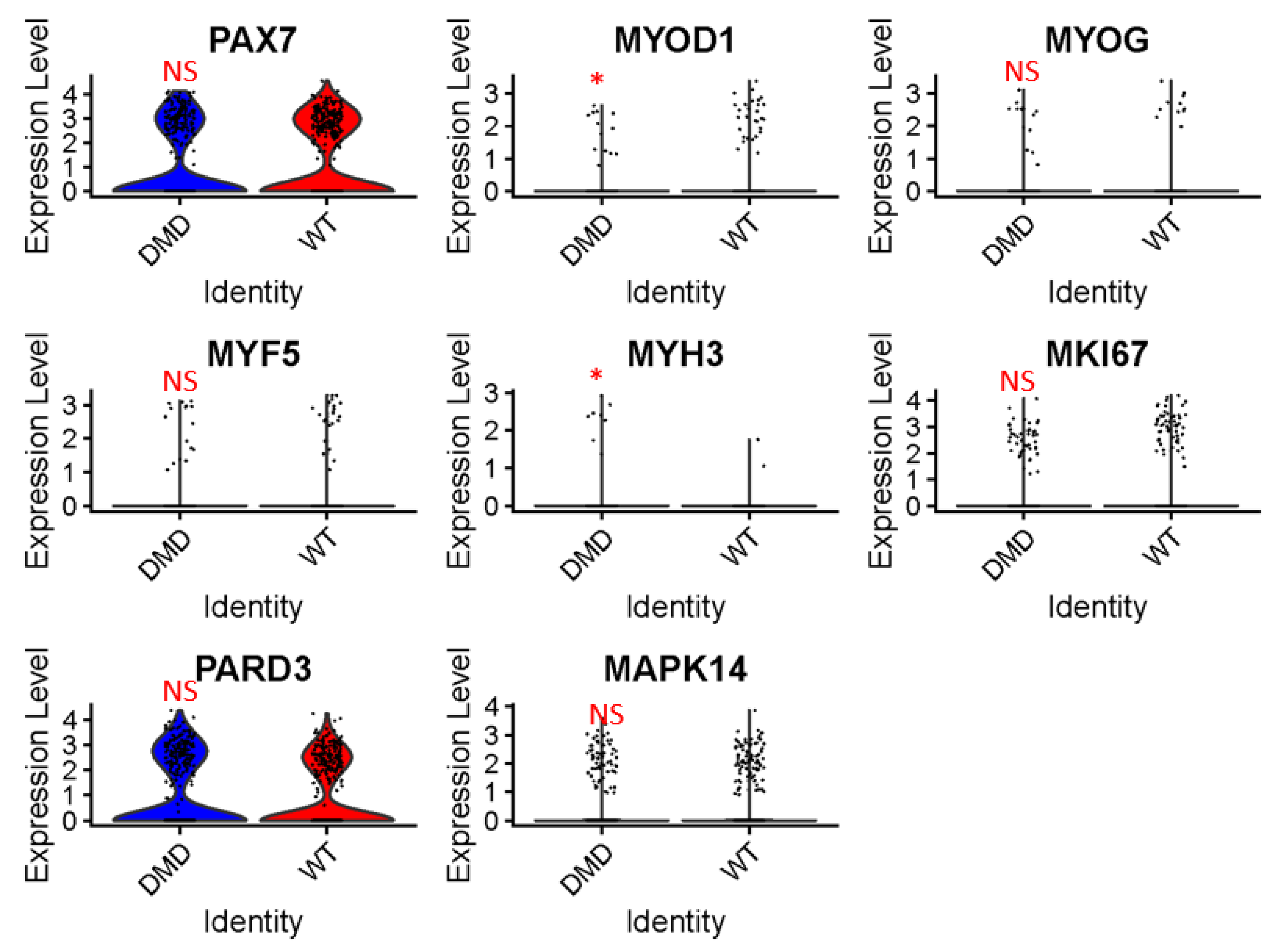
2.3. Expression Analysis of Muscle Differentiation and Proliferation Genes in DMD Mutant MuSCs
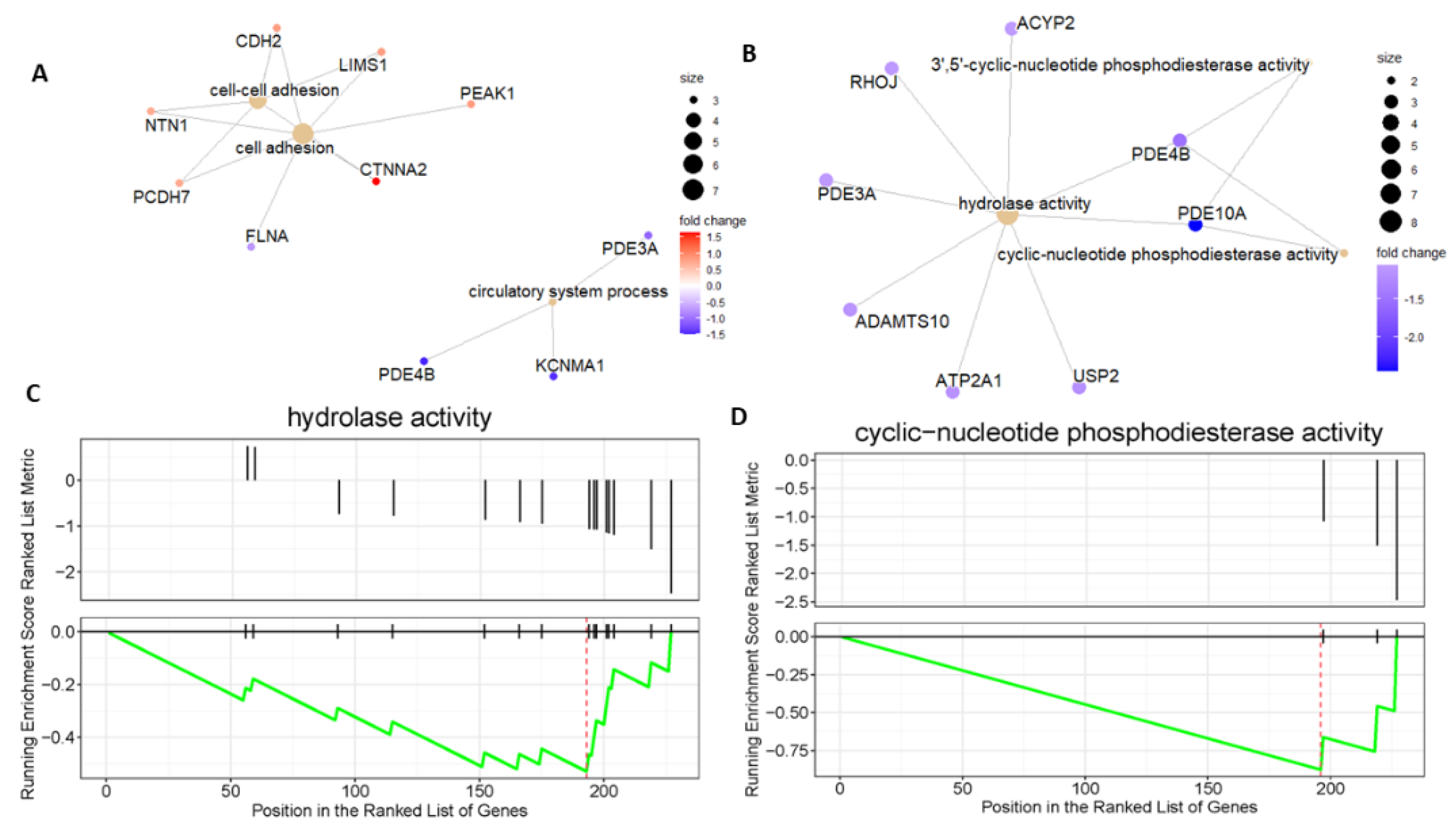
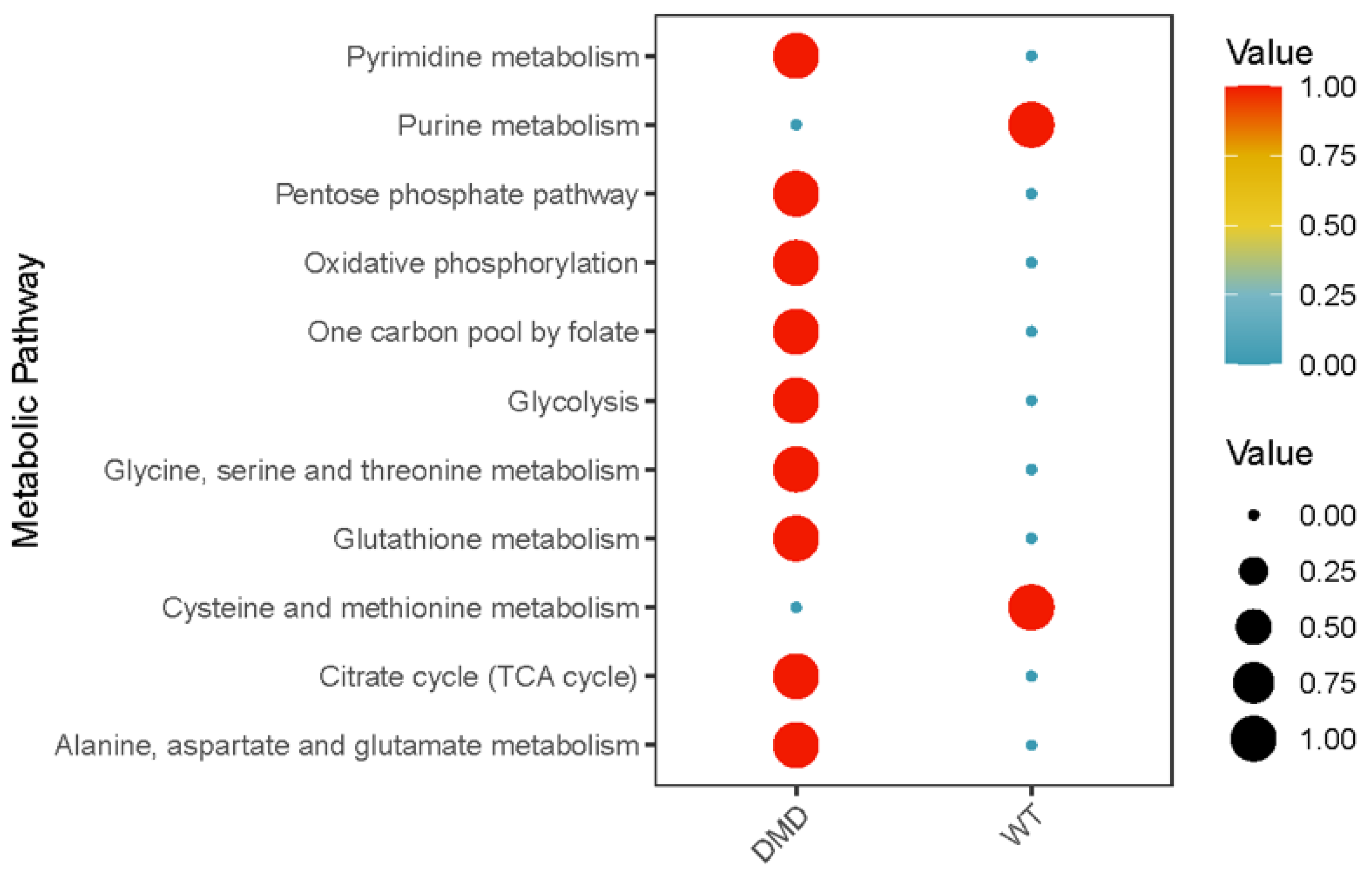
2.4. Differential Metabolic Pathways of MuSCs within DMDmut and Control Muscles
3. Discussion
4. Materials and Methods
4.1. SnRNA-Seq Datasets
4.2. SnRNA-Seq Data Analysis
4.3. Gene Ontology and Gene Set Enrichment Analysis
4.4. Single-Cell Metabolic Analysis
4.5. Statistical Analysis
5. Conclusions and Limitations
- (1)
- Our study primarily focuses on an “-omics” analysis of DMD function in MuSCs. It is indeed important to emphasize that the results of our bioinformatic analysis have not been experimentally validated, and further insights are required to fully assess their value. For example, our study reported the discovery of new LncRNAs and metabolic mechanisms associated with DMD mutation in MuSCs. However, none of these proposed mechanisms underwent validation;
- (2)
- It is essential to acknowledge that the findings should be interpreted with caution, as the observed effects might be influenced by the early stage of the disease and the ongoing compensatory mechanisms. Future studies focusing on older animals or specifically investigating senescence-related pathways will provide further insights into the progressive nature of MuSC dysfunction in DMD.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gumerson, J.D.; Michele, D.E. The Dystrophin-Glycoprotein Complex in the Prevention of Muscle Damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Shen, Y.; Su, X.; Weintraub, N.L.; Tang, Y. Effective restoration of dystrophin expression in iPSC Mdx-derived muscle progenitor cells using the CRISPR/Cas9 system and homology-directed repair technology. Comput. Struct. Biotechnol. J. 2020, 18, 765–773. [Google Scholar] [CrossRef]
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in Duchenne muscular dystrophy: Pathogenesis and therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.H.; Kuhr, C.; Tapscott, S.J.; Storb, R. Hematopoietic Cell Transplantation Provides an Immune-Tolerant Platform for Myoblast Transplantation in Dystrophic Dogs. Mol. Ther. 2008, 16, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.F.; Bonilla, E.; Martucci, G.; Moraes, C.T.; Hays, A.P.; Dimauro, S. Immunocytochemical study of dystrophin in muscle cultures from patients with Duchenne muscular dystrophy and unaffected control patients. Am. J. Pathol. 1988, 132, 410–416. [Google Scholar] [PubMed]
- Huard, J.; Labrecque, C.; Dansereau, G.; Robitaille, L.; Tremblay, J.P. Dystrophin expression in myotubes formed by the fusion of normal and dystrophic myoblasts. Muscle Nerve 1991, 14, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Wein, N.; Vulin, A.; Falzarano, M.S.; Szigyarto, C.A.-K.; Maiti, B.; Findlay, A.; Heller, K.N.; Uhlén, M.; Bakthavachalu, B.; Messina, S.; et al. Translation from a DMD exon 5 IRES results in a functional dystrophin isoform that attenuates dystrophinopathy in humans and mice. Nat. Med. 2014, 20, 992–1000. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Lin, M.; Tang, Y.; Wang, B.; Wang, J.H.-C. A novel functional assessment of the differentiation of micropatterned muscle cells. J. Biomech. 2008, 41, 3349–3353. [Google Scholar] [CrossRef] [Green Version]
- Jelinkova, S.; Fojtik, P.; Kohutova, A.; Vilotic, A.; Marková, L.; Pesl, M.; Jurakova, T.; Kruta, M.; Vrbsky, J.; Gaillyova, R.; et al. Dystrophin Deficiency Leads to Genomic Instability in Human Pluripotent Stem Cells via NO Synthase-Induced Oxidative Stress. Cells 2019, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.; Kuroda, K.; Le Grand, F.; Rudnicki, M.A. Asymmetric Self-Renewal and Commitment of Satellite Stem Cells in Muscle. Cell 2007, 129, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite Cells in Muscular Dystrophy—Lost in Polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemello, F.; Wang, Z.; Li, H.; McAnally, J.R.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 29691–29701. [Google Scholar] [CrossRef]
- Dort, J.; Orfi, Z.; Fabre, P.; Molina, T.; Conte, T.C.; Greffard, K.; Pellerito, O.; Bilodeau, J.-F.; Dumont, N.A. Resolvin-D2 targets myogenic cells and improves muscle regeneration in Duchenne muscular dystrophy. Nat. Commun. 2021, 12, 6264. [Google Scholar] [CrossRef] [PubMed]
- Taglietti, V.; Kefi, K.; Rivera, L.; Bergiers, O.; Cardone, N.; Coulpier, F.; Gioftsidi, S.; Drayton-Libotte, B.; Hou, C.; Authier, F.-J.; et al. Thyroid-stimulating hormone receptor signaling restores skeletal muscle stem cell regeneration in rats with muscular dystrophy. Sci. Transl. Med. 2023, 15, eadd5275. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.R.; Waitzberg, D.L.; Da Silva, I.D.C.G.; Jr, T.C.T.; Barros, L.; Canuto, G.A.B.; Faccio, A.T.; Yamaguchi, L.F.; Kato, M.J.; Tavares, M.F.M.; et al. Distinct pattern of one-carbon metabolism, a nutrient-sensitive pathway, in invasive breast cancer: A metabolomic study. Oncotarget 2020, 11, 1637–1652. [Google Scholar] [CrossRef]
- Tian, X.; Yu, Q.; Yao, D.; Shao, L.; Liang, Z.; Jia, F.; Li, X.; Hui, T.; Dai, R. New Insights into the Response of Metabolome of Escherichia coli O157:H7 to Ohmic Heating. Front. Microbiol. 2018, 9, 2936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalo-Gobernado, R.; Ayuso, M.I.; Sansone, L.; Bernal-Jiménez, J.J.; Ramos-Herrero, V.D.; Sánchez-García, E.; Ramos, T.L.; Abia, R.; Muriana, F.J.G.; Bermúdez, B.; et al. Neuroprotective Effects of Diets Containing Olive Oil and DHA/EPA in a Mouse Model of Cerebral Ischemia. Nutrients 2019, 11, 1109. [Google Scholar] [CrossRef] [Green Version]
- Au, K.S.; Findley, T.O.; Northrup, H. Finding the genetic mechanisms of folate deficiency and neural tube defects-Leaving no stone unturned. Am. J. Med. Genet. Part A 2017, 173, 3042–3057. [Google Scholar] [CrossRef]
- Serrano, N.C.; Quintero-Lesmes, D.C.; Becerra-Bayona, S.M.; Guio, E.; Beltran, M.; Paez, M.C.; Ortiz, R.; Saldarriaga, W.; Diaz, L.A.; Monterrosa, Á.; et al. Association of pre-eclampsia risk with maternal levels of folate, homocysteine and vitamin B12 in Colombia: A case-control study. PLoS ONE 2018, 13, e0208137. [Google Scholar] [CrossRef]
- Kamat, P.K.; Kalani, A.; Tyagi, S.C.; Tyagi, N. Hydrogen Sulfide Epigenetically Attenuates Homocysteine-Induced Mitochondrial Toxicity Mediated through NMDA Receptor in Mouse Brain Endothelial (bEnd3) Cells. J. Cell. Physiol. 2014, 230, 378–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capatina, N.; Burton, G.J.; Yung, H.W. Elevated homocysteine activates unfolded protein responses and causes aberrant trophoblast differentiation and mouse blastocyst development. Physiol. Rep. 2022, 10, e15467. [Google Scholar] [CrossRef] [PubMed]
- Almeida-Becerril, T.; Rodríguez-Cruz, M.; Villa-Morales, J.; Sánchez-Mendoza, C.R.; Galeazzi-Aguilar, J.E. Circulating Nrf2, Glutathione, and Malondialdehyde Correlate with Disease Severity in Duchenne Muscular Dystrophy. Antioxidants 2023, 12, 871. [Google Scholar] [CrossRef] [PubMed]
- Haenggi, T.; Fritschy, J.-M. Role of dystrophin and utrophin for assembly and function of the dystrophin glycoprotein complex in non-muscle tissue. Cell. Mol. Life Sci. 2006, 63, 1614–1631. [Google Scholar] [CrossRef]
- Austin, R.C.; Morris, G.E.; Howard, P.; Klamut, H.J.; Ray, P.N. Expression and synthesis of alternatively spliced variants of Dp71 in adult human brain. Neuromuscul. Disord. 2000, 10, 187–193. [Google Scholar] [CrossRef]
- Aleman, V.; Osorio, B.; Chavez, O.; Rendon, A.; Mornet, D.; Martinez, D. Subcellular localization of Dp71 dystrophin isoforms in cultured hippocampal neurons and forebrain astrocytes. Histochem. Cell Biol. 2001, 115, 243–254. [Google Scholar] [CrossRef]
- Ueda, H.; Tsukahara, S.; Kobayashi, T.; Ohno, S. Immunocytochemical Study of Dystrophin-Related Protein in the Rat Retina. Ophthalmic Res. 2009, 27, 219–226. [Google Scholar] [CrossRef]
- Haenggi, T.; Schaub, M.C.; Fritschy, J.-M. Molecular heterogeneity of the dystrophin-associated protein complex in the mouse kidney nephron: Differential alterations in the absence of utrophin and dystrophin. Cell Tissue Res. 2005, 319, 299–313. [Google Scholar] [CrossRef]
- Loh, N.Y.; Newey, E.S.; Davies, E.K.; Blake, D.J. Assembly of multiple dystrobrevin-containing complexes in the kidney. J. Cell Sci. 2000, 113 Pt 15, 2715–2724. [Google Scholar] [CrossRef]
- Shen, Y.; Kim, I.-M.; Tang, Y. Identification of Novel Gene Regulatory Networks for Dystrophin Protein in Vascular Smooth Muscle Cells by Single-Nuclear Transcriptome Analysis. Cells 2023, 12, 892. [Google Scholar] [CrossRef]
- Shen, Y.; Kim, I.-M.; Hamrick, M.; Tang, Y. Uncovering the Gene Regulatory Network of Endothelial Cells in Mouse Duchenne Muscular Dystrophy: Insights from Single-Nuclei RNA Sequencing Analysis. Biology 2023, 12, 422. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.T.; Beavo, J.A. Cyclic Nucleotide Phosphodiesterases: Molecular Regulation to Clinical Use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, M.F.; Faucz, F.R.; Bimpaki, E.; Horvath, A.; Levy, I.; de Alexandre, R.B.; Ahmad, F.; Manganiello, V.; Stratakis, C.A. Clinical and Molecular Genetics of the Phosphodiesterases (PDEs). Endocr. Rev. 2014, 35, 195–233. [Google Scholar] [CrossRef] [PubMed]
- Borneman, R.M.; Gavin, E.; Musiyenko, A.; Richter, W.; Lee, K.J.; Crossman, D.K.; Andrews, J.F.; Wilhite, A.M.; McClellan, S.; Aragon, I.; et al. Phosphodiesterase 10A (PDE10A) as a novel target to suppress β-catenin and RAS signaling in epithelial ovarian cancer. J. Ovarian Res. 2022, 15, 120. [Google Scholar] [CrossRef]
- Lambert, M.R.; Spinazzola, J.M.; Widrick, J.J.; Pakula, A.; Conner, J.R.; Chin, J.E.; Owens, J.M.; Kunkel, L.M. PDE10A Inhibition Reduces the Manifestation of Pathology in DMD Zebrafish and Represses the Genetic Modifier PITPNA. Mol. Ther. 2021, 29, 1086–1101. [Google Scholar] [CrossRef] [PubMed]
- Hitachi, K.; Honda, M.; Tsuchida, K. The Functional Role of Long Non-Coding RNA in Myogenesis and Skeletal Muscle Atrophy. Cells 2022, 11, 2291. [Google Scholar] [CrossRef] [PubMed]
- Neguembor, M.V.; Jothi, M.; Gabellini, D. Long noncoding RNAs, emerging players in muscle differentiation and disease. Skelet. Muscle 2014, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Li, L.; Shi, G.; Chen, L.; Fang, C.; Li, M.; Li, C. MEG3 Promotes Differentiation of Porcine Satellite Cells by Sponging miR-423-5p to Relieve Inhibiting Effect on SRF. Cells 2020, 9, 449. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Zuo, H.; Jin, J.; Lv, W.; Xu, Z.; Fan, Y.; Zhang, J.; Zuo, B. Long noncoding RNA Neat1 modulates myogenesis by recruiting Ezh2. Cell Death Dis. 2019, 10, 505. [Google Scholar] [CrossRef] [Green Version]
- Na, Y.; Huang, G.; Wu, J. The Role of RUNX1 in NF1-Related Tumors and Blood Disorders. Mol. Cells 2020, 43, 153–159. [Google Scholar] [CrossRef]
- Umansky, K.B.; Gruenbaum-Cohen, Y.; Tsoory, M.; Feldmesser, E.; Goldenberg, D.; Brenner, O.; Groner, Y. Runx1 Transcription Factor Is Required for Myoblasts Proliferation during Muscle Regeneration. PLOS Genet. 2015, 11, e1005457. [Google Scholar] [CrossRef] [Green Version]
- Deltcheva, E.; Nimmo, R. RUNX transcription factors at the interface of stem cells and cancer. Biochem. J. 2017, 474, 1755–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barros-Álvarez, X.; Nwokonko, R.M.; Vizurraga, A.; Matzov, D.; He, F.; Papasergi-Scott, M.M.; Robertson, M.J.; Panova, O.; Yardeni, E.H.; Seven, A.B.; et al. The tethered peptide activation mechanism of adhesion GPCRs. Nature 2022, 604, 757–762. [Google Scholar] [CrossRef]
- Vecchia, E.D.; Mortimer, N.; Palladino, V.S.; Kittel-Schneider, S.; Lesch, K.P.; Reif, A.; Schenck, A.; Norton, W.H.J. Cross-species models of attention-deficit/hyperactivity disorder and autism spectrum disorder: Lessons from CNTNAP2, ADGRL3, and PARK2. Psychiatr. Genet. 2019, 29, 1–17. [Google Scholar] [CrossRef]
- Liu, J.-W.; Li, H.; Zhang, Y. Npas3 regulates stemness maintenance of radial glial cells and neuronal migration in the developing mouse cerebral cortex. Front. Cell. Neurosci. 2022, 16, 865681. [Google Scholar] [CrossRef]
- Lieber, R.L.; Binder-Markey, B.I. Biochemical and structural basis of the passive mechanical properties of whole skeletal muscle. J. Physiol. 2021, 599, 3809–3823. [Google Scholar] [CrossRef]
- Brashear, S.E.; Wohlgemuth, R.P.; Hu, L.-Y.; Jbeily, E.H.; Christiansen, B.A.; Smith, L.R. Collagen cross-links scale with passive stiffness in dystrophic mouse muscles, but are not altered with administration of a lysyl oxidase inhibitor. PLoS ONE 2022, 17, e0271776. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Liang, Z.; Xu, J.; Li, W.; Zhao, D.; Zhao, Y.; Yan, C. Activation of the wnt/β-Catenin Signaling Pathway in Polymyositis, Dermatomyositis and Duchenne Muscular Dystrophy. J. Clin. Neurol. 2016, 12, 351–360. [Google Scholar] [CrossRef]
- Kwon, M.; Rubio, G.; Wang, H.; Riedlinger, G.; Adem, A.; Zhong, H.; Slegowski, D.; Post-Zwicker, L.; Chidananda, A.; Schrump, D.S.; et al. Smoking-associated Downregulation of FILIP1L Enhances Lung Adenocarcinoma Progression Through Mucin Production, Inflammation, and Fibrosis. Cancer Res. Commun. 2022, 2, 1197–1213. [Google Scholar] [CrossRef] [PubMed]
- Chapman, K.M.; Medrano, G.A.; Jaichander, P.; Chaudhary, J.; Waits, A.E.; Nobrega, M.A.; Hotaling, J.M.; Ober, C.; Hamra, F.K. Targeted Germline Modifications in Rats Using CRISPR/Cas9 and Spermatogonial Stem Cells. Cell Rep. 2015, 10, 1828–1835. [Google Scholar] [CrossRef] [Green Version]
- Debaugnies, M.; Rodríguez-Acebes, S.; Blondeau, J.; Parent, M.-A.; Zocco, M.; Song, Y.; de Maertelaer, V.; Moers, V.; Latil, M.; Dubois, C.; et al. RHOJ controls EMT-associated resistance to chemotherapy. Nature 2023, 616, 168–175. [Google Scholar]
- Rahman, F.A.; Angus, S.A.; Stokes, K.; Karpowicz, P.; Krause, M.P. Impaired ECM Remodeling and Macrophage Activity Define Necrosis and Regeneration Following Damage in Aged Skeletal Muscle. Int. J. Mol. Sci. 2020, 21, 4575. [Google Scholar] [CrossRef]
- Juhas, M.; Bursac, N. Roles of adherent myogenic cells and dynamic culture in engineered muscle function and maintenance of satellite cells. Biomaterials 2014, 35, 9438–9446. [Google Scholar] [CrossRef] [Green Version]
- Ryall, J.G.; Dell’Orso, S.; Derfoul, A.; Juan, A.; Zare, H.; Feng, X.; Clermont, D.; Koulnis, M.; Gutierrez-Cruz, G.; Sartorelli, V.; et al. The NAD+-Dependent SIRT1 Deacetylase Translates a Metabolic Switch into Regulatory Epigenetics in Skeletal Muscle Stem Cells. Cell Stem Cell 2015, 16, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacharya, D.; Scimè, A. Mitochondrial Function in Muscle Stem Cell Fates. Front. Cell Dev. Biol. 2020, 8, 480. [Google Scholar] [PubMed]
- Guess, M.G.; Barthel, K.K.B.; Harrison, B.C.; Leinwand, L.A. miR-30 Family microRNAs Regulate Myogenic Differentiation and Provide Negative Feedback on the microRNA Pathway. PLoS ONE 2015, 10, e0118229. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster | Gene |
|---|---|
| 0 | GM26870 MT-CO3 MT-ATP6 MT-CYTB MT-ND2 MT-CO1 MT-ND4 MT-ND1 MT-CO2 MT-ND3 |
| 1 | RORA HMCN2 CHODL LTBP1 CALCR CLMN KCNMA1 SOX5 BMP6 RCAN2 |
| 2 | DIAPH3 MKI67 TOP2A KNL1 SDK1 LOCKD NEIL3 KIF20B CENPP CENPF |
| 3 | GM28653 KCNK13 TMEM178B KCNQ1OT1 FRMD4B RALGPS2 PAK3 DPP6 JAM3 |
| 4 | CHODL NOTCH3 MDFIC CALCR BMP6 FOXP2 GULP1 CEP128 PDE3A KCNMA1 |
| 5 | TRDN NEB TTN OBSCN MYH4 PDE4DIP RYR1 CMYA5 ATP2A1 NEAT1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Kim, I.-M.; Tang, Y. Decoding the Gene Regulatory Network of Muscle Stem Cells in Mouse Duchenne Muscular Dystrophy: Revelations from Single-Nuclei RNA Sequencing Analysis. Int. J. Mol. Sci. 2023, 24, 12463. https://doi.org/10.3390/ijms241512463
Shen Y, Kim I-M, Tang Y. Decoding the Gene Regulatory Network of Muscle Stem Cells in Mouse Duchenne Muscular Dystrophy: Revelations from Single-Nuclei RNA Sequencing Analysis. International Journal of Molecular Sciences. 2023; 24(15):12463. https://doi.org/10.3390/ijms241512463
Chicago/Turabian StyleShen, Yan, Il-Man Kim, and Yaoliang Tang. 2023. "Decoding the Gene Regulatory Network of Muscle Stem Cells in Mouse Duchenne Muscular Dystrophy: Revelations from Single-Nuclei RNA Sequencing Analysis" International Journal of Molecular Sciences 24, no. 15: 12463. https://doi.org/10.3390/ijms241512463