
Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Correctors on the Expression of Full-Length WT and F508del CFTR and CFTR Single Domain mRNAs
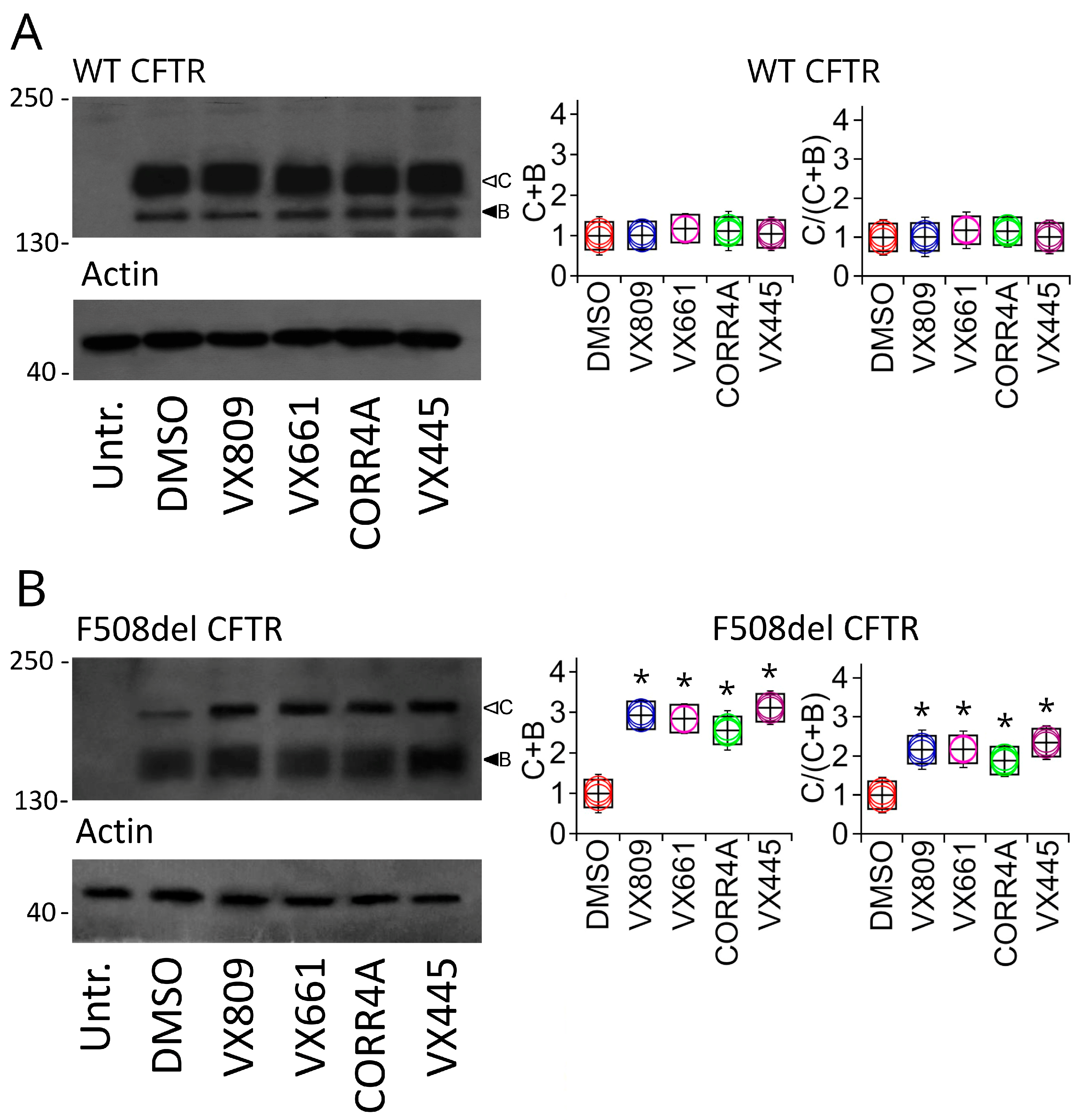
2.2. Effects of Correctors on the Functional Expression of Full-Length WT and F508del CFTR Protein
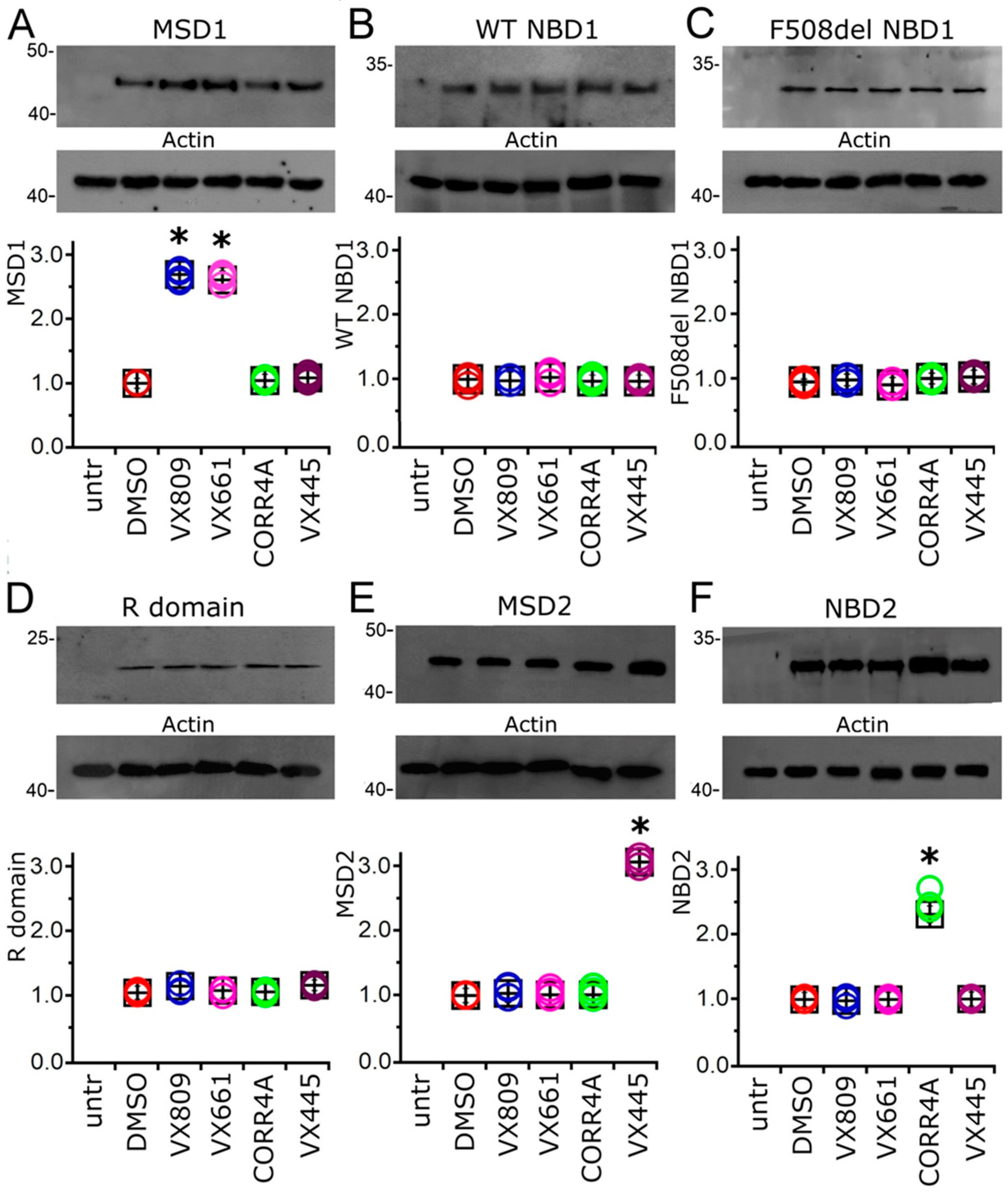
2.3. Effect of Correctors on the Expression of CFTR Single Domains
2.4. Effect of Tested Correctors on the Stability of NBD2
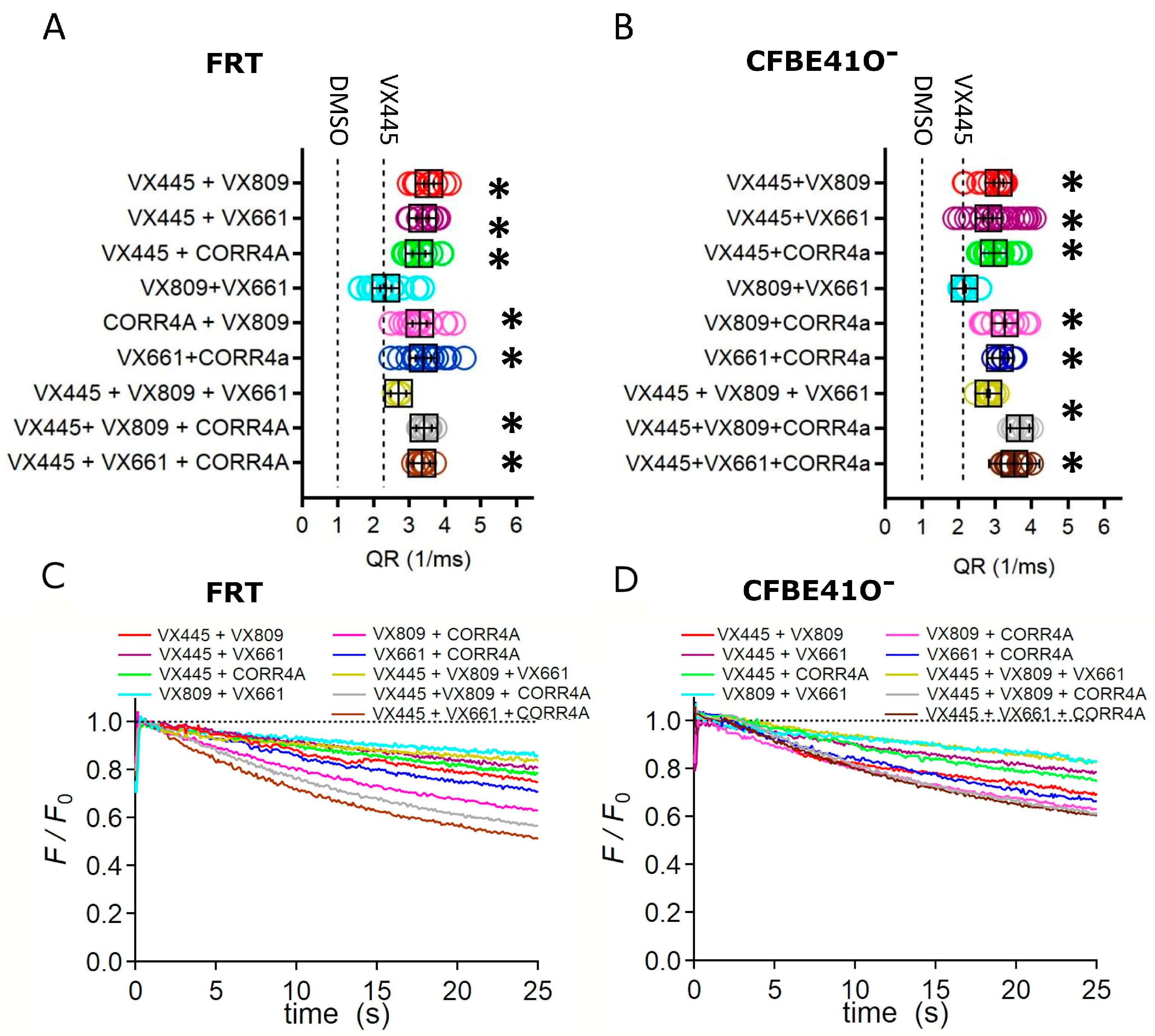
2.5. Effect of Combinations of Correctors on the Channel Activity of Full-Length F508del CFTR Protein
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Generation and Expression of CFTR Constructs
4.4. RNA Isolation, Reverse Transcription, and Quantitative Real-Time Polymerase Chain Reaction
4.5. Western Blot
4.6. Cycloheximide Chase Assay
4.7. YFP-Based Assay
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Riordan, J.; Rommens, J.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Kerem, B.; Rommens, J.M.; Buchanan, J.A.; Markiewicz, D.; Cox, T.K.; Chakravarti, A.; Buchwald, M.; Tsui, L.C. Identification of the cystic fibrosis gene: Genetic analysis. Science 1989, 245, 1073–1080. [Google Scholar] [CrossRef]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Cutting, G.R. Cystic fibrosis genetics: From molecular understanding to clinical application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, Z.; Csanády, L.; Gadsby, D.C.; Chen, J. Molecular Structure of the Human CFTR Ion Channel. Cell 2017, 169, 85–95.e8. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Skach, W.R. Mechanisms of CFTR Folding at the Endoplasmic Reticulum. Front. Pharmacol. 2012, 3, 201. [Google Scholar] [CrossRef]
- Farinha, C.M.; Canato, S. From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. [Google Scholar] [CrossRef]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar] [CrossRef]
- Ward, C.L.; Omura, S.; Kopito, R.R. Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 1995, 83, 121–127. [Google Scholar] [CrossRef]
- Younger, J.M.; Chen, L.; Ren, H.-Y.; Rosser, M.F.; Turnbull, E.L.; Fan, C.-Y.; Patterson, C.; Cyr, D.M. Sequential Quality-Control Checkpoints Triage Misfolded Cystic Fibrosis Transmembrane Conductance Regulator. Cell 2006, 126, 571–582. [Google Scholar] [CrossRef]
- Skach, W.R. Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney Int. 2000, 57, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Zolin, A.; Cuppens, H.; Olesen, H.V.; Viviani, L. The relative frequency of CFTR mutation classes in European patients with cystic fibrosis. J. Cyst. Fibros. 2014, 13, 403–409. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.H.; Gregory, R.J.; Marshall, J.; Paul, S.; Souza, D.W.; White, G.A.; O’Riordan, C.R.; Smith, A.E. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 1990, 63, 827–834. [Google Scholar] [CrossRef]
- Lukacs, G.L.; Verkman, A.S. CFTR: Folding, misfolding and correcting the ΔF508 conformational defect. Trends Mol. Med. 2012, 18, 81–91. [Google Scholar] [CrossRef]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef]
- Serohijos, A.W.; Hegedus, T.; Aleksandrov, A.A.; He, L.; Cui, L.; Dokholyan, N.V.; Riordan, J.R. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc. Natl. Acad. Sci. USA 2008, 105, 3256–3261. [Google Scholar] [CrossRef]
- Mendoza, J.L.; Schmidt, A.; Li, Q.; Nuvaga, E.; Barrett, T.; Bridges, R.J.; Feranchak, A.P.; Brautigam, C.A.; Thomas, P.J. Requirements for efficient correction of ΔF508 CFTR revealed by analyses of evolved sequences. Cell 2012, 148, 164–174. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Barrière, H.; Bagdány, M.; Rabeh, W.M.; Du, K.; Höhfeld, J.; Young, J.C.; Lukacs, G.L. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010, 329, 805–810. [Google Scholar] [CrossRef]
- Swiatecka-Urban, A.; Brown, A.; Moreau-Marquis, S.; Renuka, J.; Coutermarsh, B.; Barnaby, R.; Karlson, K.H.; Flotte, T.R.; Fukuda, M.; Langford, G.M.; et al. The short apical membrane half-life of rescued ΔF508-cystic fibrosis transmembrane conductance regulator (CFTR) results from accelerated endocytosis of ΔF508-CFTR in polarized human airway epithelial cells. J. Biol. Chem. 2005, 280, 36762–36772. [Google Scholar] [CrossRef] [PubMed]
- Fajac, I.; De Boeck, K. New horizons for cystic fibrosis treatment. Pharmacol Ther. 2017, 170, 205–211. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.J.; Simon, A.; et al. Mechanism-based corrector combination restores ∆F508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef] [PubMed]
- Odolczyk, N.; Fritsch, J.; Norez, C.; Servel, N.; da Cunha, M.F.; Bitam, S.; Kupniewska, A.; Wiszniewski, L.; Colas, J.; Tarnowski, K.; et al. Discovery of novel potent DeltaF508-CFTR correctors that target the nucleotide binding domain. EMBO Mol. Med. 2013, 5, 1484–1501. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S.; Ramsey, B.W.; Boyle, M.P.; Konstan, M.W.; Huang, X.; Marigowda, G.; Waltz, D.; Wainwright, C.E. VX-809 TRAFFIC and TRANSPORT Study Groups. Efficacy and safety of lumacaftor/ivacaftor combination therapy in patients with cystic fibrosis homozygous for Phe508del CFTR by pulmonary function subgroup: A pooled analysis. Lancet Respir. Med. 2016, 4, 617. [Google Scholar] [CrossRef]
- Davies, J.C.; Moskowitz, S.M.; Brown, C.; Horsley, A.; Mall, M.A.; McKone, E.F.; Plant, B.J.; Prais, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; et al. VXS VX-659-Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis and One or Two Phe508del Alleles. N. Engl. J. Med. 2018, 379, 1599–1611. [Google Scholar] [CrossRef]
- Almughem, F.A.; Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alharbi, W.S.; Alshahrani, M.Y.; Alshehri, A.A. Cystic fibrosis: Overview of the current development trends and innovative therapeutic strategies. Pharmaceutics 2020, 12, 616. [Google Scholar] [CrossRef]
- Keating, D.; Marigowda, G.; Burr, L.; Daines, C.; Mall, M.A.; McKone, E.F.; Ramsey, B.W.; Rowe, S.M.; Sass, L.A.; Tullis, E.; et al. VXS VX-445-Tezacaftor-Ivacaftor in patients with cystic fibrosis and one or two Phe508del alleles. N. Engl. J. Med. 2018, 379, 1612–1620. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural identification of a hotspot on CFTR for potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharmacol. 2013, 86, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef] [PubMed]
- Amico, G.; Brandas, C.; Moran, O.; Baroni, D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. Int. J. Mol. Sci. 2019, 20, 5463. [Google Scholar] [CrossRef]
- Veit, G.; Roldan, A.; Hancock, M.A.; Da Fonte, D.F.; Xu, H.; Hussein, M.; Frenkiel, S.; Matouk, E.; Velkov, T.; Lukacs, G.L. Allosteric folding correction of F508del and rare CFTR mutants by elexacaftor-tezacaftor-ivacaftor (Trikafta) combination. JCI Insight 2020, 5, e139983. [Google Scholar] [CrossRef] [PubMed]
- Capurro, V.; Tomati, V.; Sondo, E.; Renda, M.; Borrelli, A.; Pastorino, C.; Guidone, D.; Venturini, A.; Giraudo, A.; Mandrup Bertozzi, S.; et al. Partial Rescue of F508del-CFTR Stability and Trafficking Defects by Double Corrector Treatment. Int. J. Mol. Sci. 2021, 22, 5262. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Gramegna, A.; Contarini, M.; Aliberti, S.; Casciaro, R.; Blasi, F.; Castellani, C. From Ivacaftor to Triple Combination: A Systematic Review of Efficacy and Safety of CFTR Modulators in People with Cystic Fibrosis. Int. J. Mol. Sci. 2020, 21, 5882. [Google Scholar] [CrossRef]
- Griese, M.; Costa, S.; Linnemann, R.W.; Mall, M.A.; McKone, E.F.; Polineni, D.; Quon, B.S.; Ringshausen, F.C.; Taylor-Cousar, J.L.; Withers, N.J.; et al. Safety and Efficacy of Elexacaftor/Tezacaftor/Ivacaftor for 24 Weeks or Longer in People with Cystic Fibrosis and One or More F508del Alleles: Interim Results of an Open-Label Phase 3 Clinical Trial. Am. J. Respir. Crit. Care Med. 2021, 203, 381–385. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Laselva, O.; Bartlett, C.; Gunawardena, T.N.A.; Ouyang, H.; Eckford, P.D.W.; Moraes, T.J.; Bear, C.E.; Gonska, T. Rescue of multiple class II CFTR mutations by elexacaftor + tezacaftor + ivacaftor mediated in part by the dual activities of Elexacaftor as both corrector and potentiator. Eur. Respir. 2021, 57, 2002774. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular structures reveal synergistic rescue of Δ508 CFTR by Trikafta modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Bithiazole correctors rescue CFTR mutants by two different mechanisms. Biochemistry 2013, 52, 5161–5163. [Google Scholar] [CrossRef] [PubMed]
- Grove, D.E.; Rosser, M.F.N.; Ren, H.Y.; Naren, A.P.; Cyr, D.M. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol. Biol. Cell 2009, 20, 4059–4069. [Google Scholar] [CrossRef]
- Brandas, C.; Ludovico, A.; Parodi, A.; Moran, O.; Millo, E.; Cichero, E.; Baroni, D. NBD2 Is Required for the Rescue of Mutant F508del CFTR by a Thiazole-Based Molecule: A Class II Corrector for the Multi-Drug Therapy of Cystic Fibrosis. Biomolecules 2021, 11, 1417. [Google Scholar] [CrossRef] [PubMed]
- Costa, E.; Girotti, S.; Pauro, F.; Leufkens, H.G.M.; Cipolli, M. The impact of FDA and EMA regulatory decision-making process on the access to CFTR modulators for the treatment of cystic fibrosis. Orphanet J. Rare Dis. 2022, 17, 188. [Google Scholar] [CrossRef]
- Shaughnessy, C.A.; Zeitlin, P.L.; Bratcher, P.E. Elexacaftor is a CFTR potentiator and acts synergistically with ivacaftor during acute and chronic treatment. Sci. Rep. 2021, 11, 19810, Erratum in Sci. Rep. 2021, 11, 21295. [Google Scholar] [CrossRef]
- Veit, G.; Vaccarin, C.; Lukacs, G.L. Elexacaftor co-potentiates the activity of F508del and gating mutants of CFTR. J. Cyst. Fibros. Off. J. Eur. Cyst. Fibros. Soc. 2021, 20, 895–898. [Google Scholar] [CrossRef]
- Galietta, L.; Haggie, P.; Verkman, A. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001, 499, 220–224. [Google Scholar] [CrossRef]
- Galietta, L.V.; Jayaraman, S.; Verkman, A.S. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am. J. Physiol. Cell Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bongiorno, R.; Ludovico, A.; Moran, O.; Baroni, D. Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2. Int. J. Mol. Sci. 2023, 24, 12838. https://doi.org/10.3390/ijms241612838
Bongiorno R, Ludovico A, Moran O, Baroni D. Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2. International Journal of Molecular Sciences. 2023; 24(16):12838. https://doi.org/10.3390/ijms241612838
Chicago/Turabian StyleBongiorno, Roberta, Alessandra Ludovico, Oscar Moran, and Debora Baroni. 2023. "Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2" International Journal of Molecular Sciences 24, no. 16: 12838. https://doi.org/10.3390/ijms241612838
APA StyleBongiorno, R., Ludovico, A., Moran, O., & Baroni, D. (2023). Elexacaftor Mediates the Rescue of F508del CFTR Functional Expression Interacting with MSD2. International Journal of Molecular Sciences, 24(16), 12838. https://doi.org/10.3390/ijms241612838