
β2-Adrenergic Receptor Mediated Inhibition of T Cell Function and Its Implications for CAR-T Cell Therapy


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
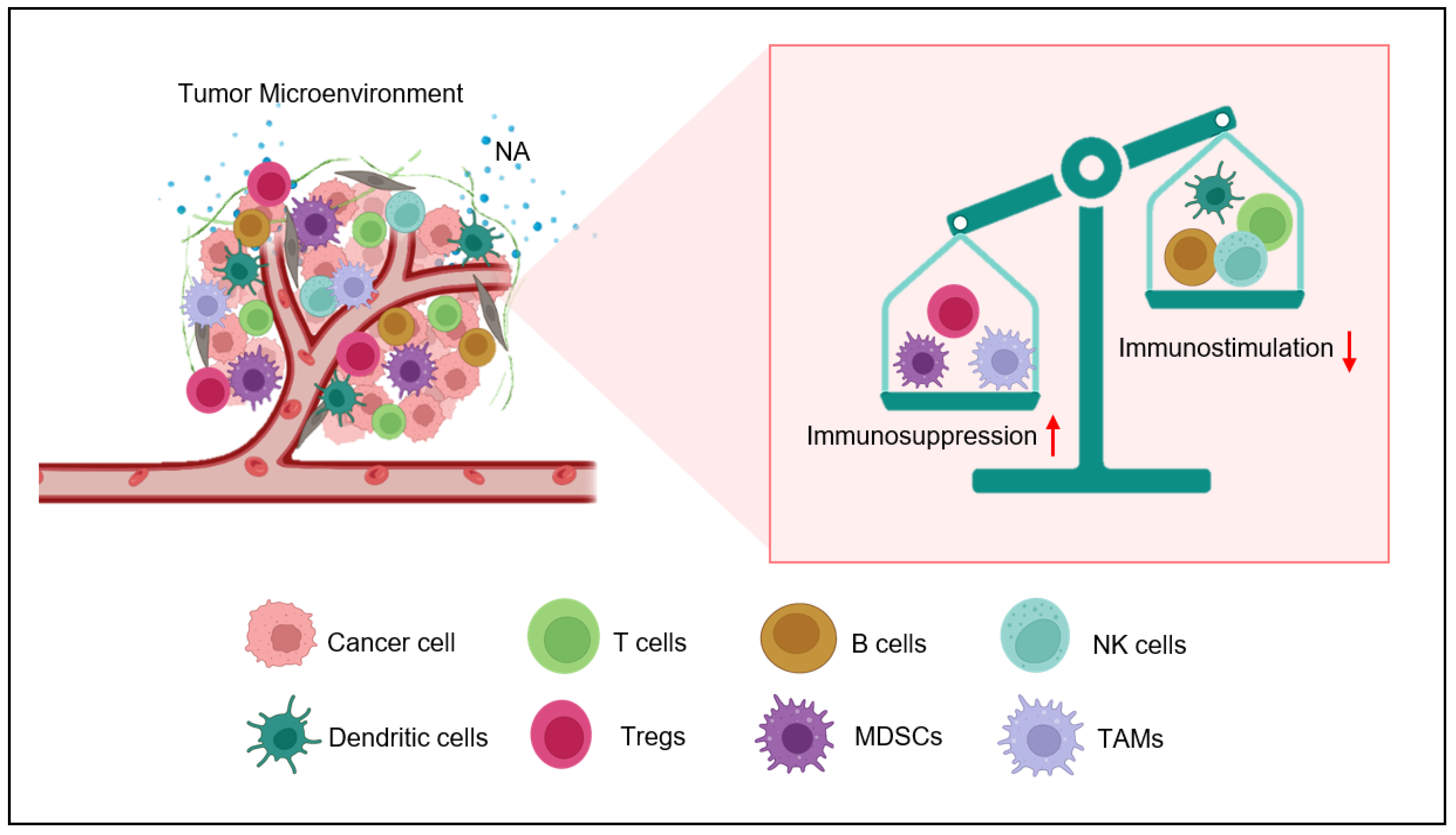
2. Nor-Adrenaline in Tumor Microenvironment
3. Adrenergic Stress Endorses Immunosuppressive Tumor Milieu Development
4. Eminent Role of β2-AR in T Cells
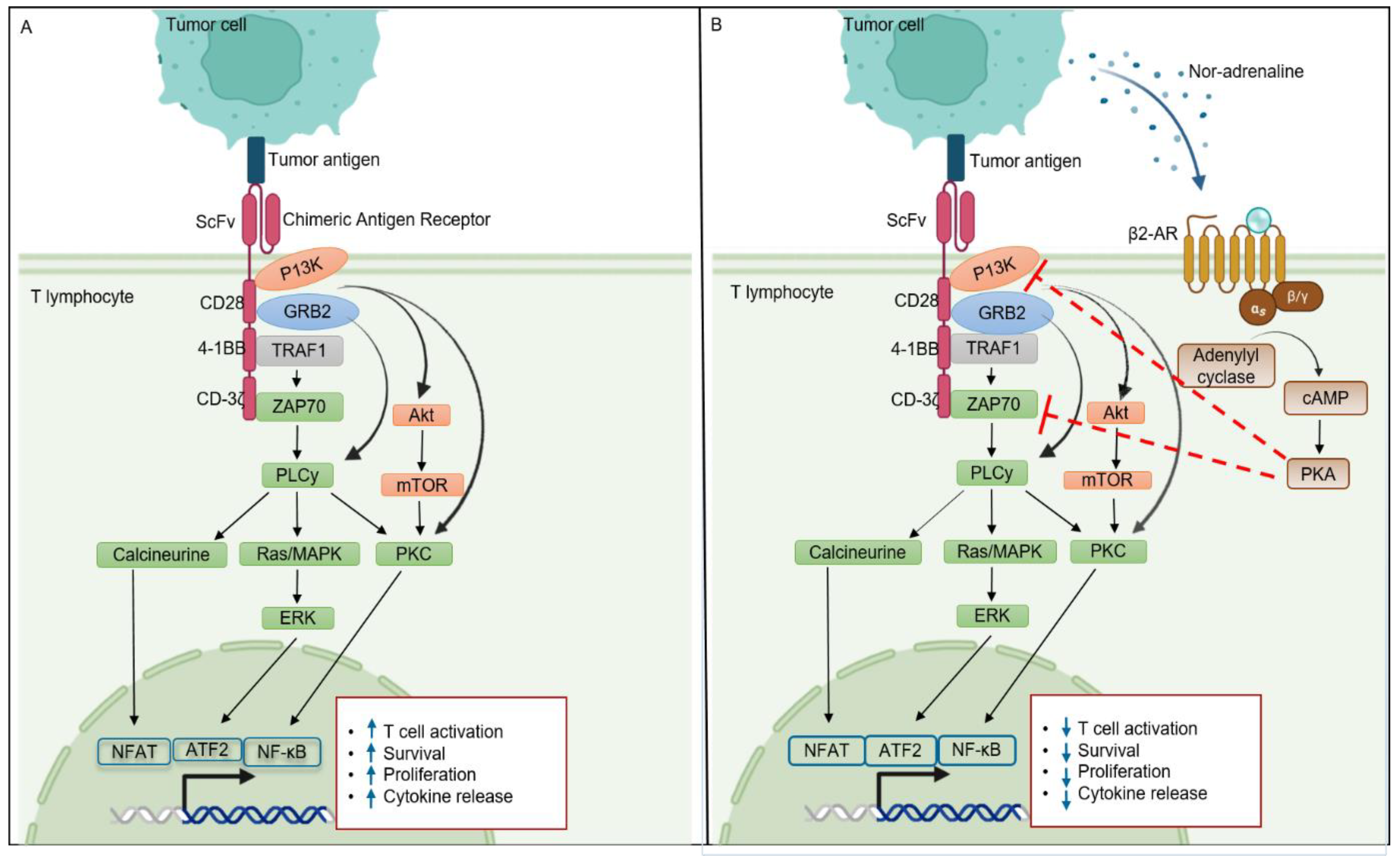
5. β2-AR and T Cell Signaling
6. β2-AR as a Potential Target for Enhancing Cancer Immunotherapy Efficacy
6.1. β2-AR and T Cell Co-Stimulation
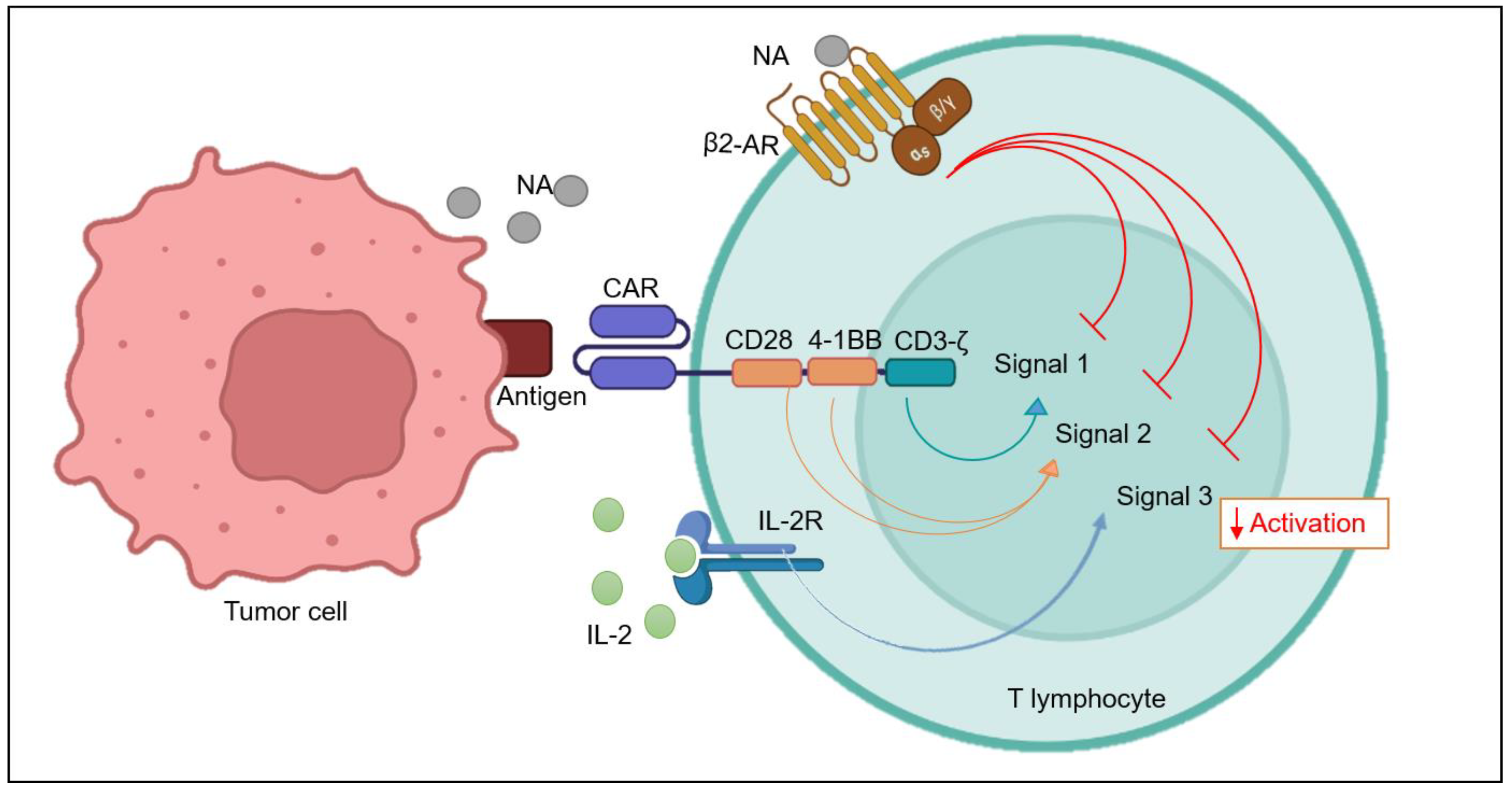
6.2. β2-AR and T Cell Activation Signals
6.3. β2-AR Self-Regulates T Cell Activation
6.4. β2-AR and IL-2 Signaling
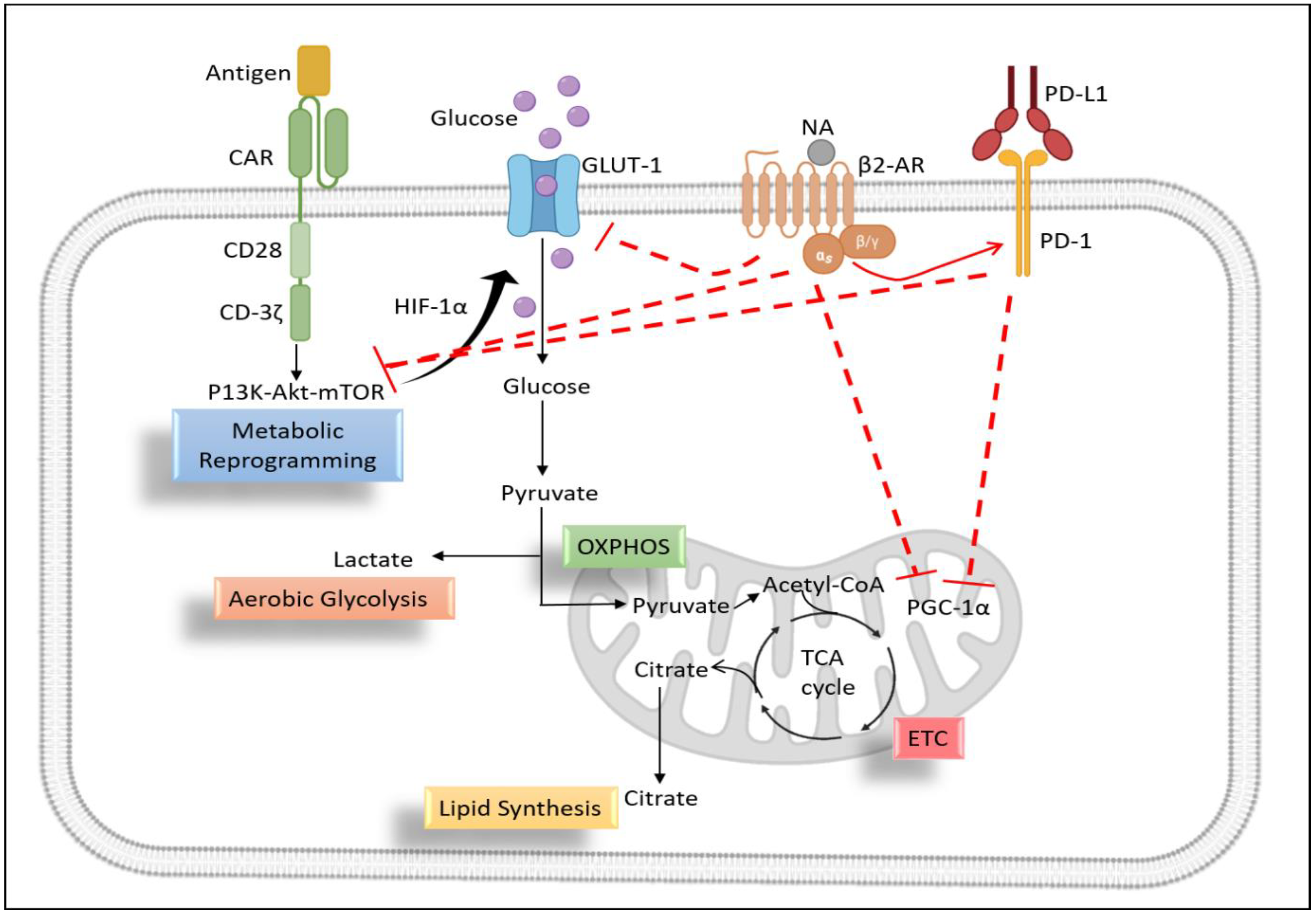
6.5. Metabolic Profiling of T Lymphocytes
6.5.1. Role of β2-AR in T Cell Glycolysis
6.5.2. Role of β2-AR in Mitochondrial Metabolism of T Cells
6.6. β2-AR Mediated Redox Signaling in T Cells
7. Perspective for CAR-T Cell Therapy
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lymperopoulos, A. Physiology and pharmacology of the cardiovascular adrenergic system. Front. Physiol. 2013, 4, 240. [Google Scholar] [CrossRef]
- Behl, T.; Kaur, I.; Sehgal, A.; Singh, S.; Makeen, H.A.; Albratty, M.; Alhazmi, H.A.; Bhatia, S.; Bungau, S. The Locus Coeruleus—Noradrenaline system: Looking into Alzheimer’s therapeutics with rose coloured glasses. Biomed. Pharmacother. 2022, 151, 113179. [Google Scholar] [CrossRef]
- Molina, P.E. Neurobiology of the stress response: Contribution of the sympathetic nervous system to the neuroimmune axis in traumatic injury. Shock 2005, 24, 3–10. [Google Scholar] [CrossRef]
- Chrousos, G.P. Stress and disorders of the stress system. Nat. Rev. Endocrinol. 2009, 5, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Zhu, Z.; Yang, C.; Wang, L.; Ding, G.; Jiang, F. Epinephrine increases malignancy of breast cancer through p38 MAPK signaling pathway in depressive disorders. Int. J. Clin. Exp. Pathol. 2019, 12, 1932–1946. [Google Scholar]
- Irwin, M.R.; Miller, A.H. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav. Immun. 2007, 21, 374–383. [Google Scholar] [CrossRef]
- Graff, R.M.; Kunz, H.E.; Agha, N.H.; Baker, F.L.; Laughlin, M.; Bigley, A.B.; Markofski, M.M.; LaVoy, E.C.; Katsanis, E.; Bond, R.A.; et al. β2-Adrenergic receptor signaling mediates the preferential mobilization of differentiated subsets of CD8+ T-cells, NK-cells and non-classical monocytes in response to acute exercise in humans. Brain Behav. Immun. 2018, 74, 143–153. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Araujo, L.P.; Maricato, J.T.; Nascimento, V.M.; Guereschi, M.G.; Rezende, R.M.; Quintana, F.J.; Basso, A.S. Norepinephrine Controls Effector T Cell Differentiation through β2-Adrenergic Receptor–Mediated Inhibition of NF-κB and AP-1 in Dendritic Cells. J. Immunol. 2016, 196, 637–644. [Google Scholar] [CrossRef]
- Xiao, L.; Li, X.; Fang, C.; Yu, J.; Chen, T. Neurotransmitters: Promising immune modulators in the tumor microenvironment. Front. Immunol. 2023, 14, 1118637. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.; Quintas, C.; Gonçalves, J.; Fresco, P. Contribution of adrenergic mechanisms for the stress-induced breast cancer carcinogenesis. J. Cell. Physiol. 2022, 237, 2107–2127. [Google Scholar] [CrossRef] [PubMed]
- Croasdell, A.; Duffney, P.F.; Kim, N.; Lacy, S.H.; Sime, P.J.; Phipps, R.P. PPARγ and the innate immune system mediate the resolution of inflammation. PPAR Res. 2015, 2015, 549691. [Google Scholar] [CrossRef] [PubMed]
- Apavaloaei, A.; Hardy, M.P.; Thibault, P.; Perreault, C. The origin and immune recognition of tumor-specific antigens. Cancers 2020, 12, 2607. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef]
- Ellis, G.I.; Sheppard, N.C.; Riley, J.L. Genetic engineering of T cells for immunotherapy. Nat. Rev. Genet. 2021, 22, 427–447. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; He, J.; Liu, L.; Wang, J.; Wang, S.; Liu, L.; Gao, L.; Gao, L.; Liu, Y.; Kong, P.; et al. CD19-directed fast CART therapy for relapsed/refractory acute lymphoblastic leukemia: From bench to bedside. Blood 2019, 134, 1340. [Google Scholar] [CrossRef]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Behl, T.; Islam, M.R.; Alam, M.N.; Islam, M.M.; Albarrati, A.; Albratty, M.; Meraya, A.M.; Bungau, S.G. Emerging Management Approach for the Adverse Events of Immunotherapy of Cancer. Molecules 2022, 27, 3798. [Google Scholar] [CrossRef]
- Dogan, A.; Siegel, D.; Tran, N.; Fu, A.; Fowler, J.; Belani, R.; Landgren, O. B-cell maturation antigen expression across hematologic cancers: A systematic literature review. Blood Cancer J. 2020, 10, 73. [Google Scholar] [CrossRef]
- Yan, T.; Zhu, L.; Chen, J. Current advances and challenges in CAR T-Cell therapy for solid tumors: Tumor-associated antigens and the tumor microenvironment. Exp. Hematol. Oncol. 2023, 12, 14. [Google Scholar] [CrossRef]
- El-Sayes, N.; Vito, A.; Mossman, K. Tumor heterogeneity: A great barrier in the age of cancer immunotherapy. Cancers 2021, 13, 806. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Song, X.; Jin, Y.; Li, F.; Yu, H.; Cao, C.; Jiang, Q. Carts for solid tumors: Feasible or infeasible? Oncol. Res. Treat. 2017, 40, 540–546. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Zhou, Y.; Li, Z.; Farooq, M.A.; Ajmal, I.; Zhang, H.; Zhang, L.; Tao, L.; Yao, J.; Du, B.; et al. Co-expression of IL-7 improves NKG2D-based CAR T cell therapy on prostate cancer by enhancing the expansion and inhibiting the apoptosis and exhaustion. Cancers 2020, 12, 1969. [Google Scholar] [CrossRef]
- Gao, Y.; Lin, H.; Guo, D.; Cheng, S.; Zhou, Y.; Zhang, L.; Yao, J.; Farooq, M.A.; Ajmal, I.; Duan, Y.; et al. Suppression of 4.1 R enhances the potency of NKG2D-CAR T cells against pancreatic carcinoma via activating ERK signaling pathway. Oncogenesis 2021, 10, 62. [Google Scholar] [CrossRef]
- Sanders, V.M.; Straub, R.H. Norepinephrine, the β-adrenergic receptor, and immunity. Brain Behav. Immun. 2002, 16, 290–332. [Google Scholar] [CrossRef]
- Sanders, V.M. The beta2-adrenergic receptor on T and B lymphocytes: Do we understand it yet? Brain Behav. Immun. 2012, 26, 195–200. [Google Scholar] [CrossRef]
- Wang, W.; Cao, X. Beta-adrenergic signaling in tumor immunology and immunotherapy. Crit. Rev. Immunol. 2019, 39, 93–103. [Google Scholar] [CrossRef]
- Wrobel, L.J.; Gayet-Ageron, A.; Le Gal, F.A. Effects of beta-blockers on melanoma microenvironment and disease survival in human. Cancers 2020, 12, 1094. [Google Scholar] [CrossRef]
- Lorton, D.; Bellinger, D.L. Molecular mechanisms underlying β-adrenergic receptor-mediated cross-talk between sympathetic neurons and immune cells. Int. J. Mol. Sci. 2015, 16, 5635–5665. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Ammons, D.T.; MacDonald, C.R.; Chow, L.; Repasky, E.A.; Dow, S. Chronic Adrenergic Stress and Generation of Myeloid-Derived Suppressor Cells: Implications for Cancer Immunotherapy in Dogs. Vet. Comp. Oncol. 2023, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Koinis, F.; Xagara, A.; Chantzara, E.; Leontopoulou, V.; Aidarinis, C.; Kotsakis, A. Myeloid-derived suppressor cells in prostate cancer: Present knowledge and future perspectives. Cells 2022, 11, 20. [Google Scholar] [CrossRef] [PubMed]
- Ağaç, D.; Estrada, L.D.; Maples, R.; Hooper, L.V.; Farrar, J.D. The β2-adrenergic receptor controls inflammation by driving rapid IL-10 secretion. Brain Behav. Immun. 2018, 74, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Zahalka, A.H.; Arnal-Estapé, A.; Maryanovich, M.; Nakahara, F.; Cruz, C.D.; Finley, L.W.; Frenette, P.S. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science 2017, 358, 321–326. [Google Scholar] [CrossRef]
- Gardner, K.P.; Cristofanilli, M.; Chumsri, S.; Lapidus, R.; Tang, C.M.; Raghavakaimal, A.; Adams, D.L. Beta 2-Adrenergic Receptor in Circulating Cancer-Associated Cells Predicts for Increases in Stromal Macrophages in Circulation and Patient Survival in Metastatic Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7299. [Google Scholar] [CrossRef]
- Lamkin, D.M.; Ho, H.Y.; Ong, T.H.; Kawanishi, C.K.; Stoffers, V.L.; Ahlawat, N.; Ma, J.C.; Arevalo, J.M.; Cole, S.W.; Sloan, E.K. β-Adrenergic-stimulated macrophages: Comprehensive localization in the M1-M2 spectrum. Brain Behav. Immun. 2016, 57, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Thapa, S.; Cao, X. Nervous regulation: Beta-2-adrenergic signaling in immune homeostasis, cancer immunotherapy, and autoimmune diseases. Cancer Immunol. Immunother. 2023, 72, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Qiao, G. Understanding the Mechanisms by Which β-Adrenergic Stress Signaling Impairs T-Cell Immune Responses. Ph.D. Dissertation, State University of New York at Buffalo, Buffalo, NY, USA, 2021. [Google Scholar]
- Kavelaars, A. Regulated expression of α-1 adrenergic receptors in the immune system. Brain Behav. Immun. 2002, 16, 799–807. [Google Scholar] [CrossRef]
- Kalinichenko, V.V.; Mokyr, M.B.; Graf, L.H.; Cohen, R.L.; Chambers, D.A. Norepinephrine-mediated inhibition of antitumor cytotoxic T lymphocyte generation involves a β-adrenergic receptor mechanism and decreased TNF-α gene expression. J. Immunol. 1999, 163, 2492–2499. [Google Scholar] [CrossRef]
- Casale, T.B.; Kaliner, M. Demonstration that circulating human blood cells have no detectable alpha1-adrenergic receptors by radioligand binding analysis. J. Allergy Clin. Immunol. 1984, 74, 812–818. [Google Scholar] [CrossRef]
- Van Der Voort, C.R.; Kavelaars, A.; Van De Pol, M.; Heijnen, C.J. Noradrenaline induces phosphorylation of ERK-2 in human peripheral blood mononuclear cells after induction of α1-adrenergic receptors. J. Neuroimmunol. 2000, 108, 82–91. [Google Scholar] [CrossRef]
- Grisanti, L.A.; Perez, D.M.; Porter, J.E. Modulation of immune cell function by α1-adrenergic receptor activation. Curr. Top. Membr. 2011, 67, 113–138. [Google Scholar] [CrossRef] [PubMed]
- Nissen, M.D.; Sloan, E.K.; Mattarollo, S.R. β-Adrenergic Signaling Impairs Antitumor CD8+ T-cell Responses to B-cell Lymphoma ImmunotherapyβAR Signaling Impairs T cell–Mediated Immunotherapy. Cancer Immunol. Res. 2018, 6, 98–109. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; He, Y.; Chen, F.; Zhang, F.; Shao, D.; Wang, Z. Leveraging β-Adrenergic Receptor Signaling Blockade for Improved Cancer Immunotherapy Through Biomimetic Nanovaccine. Small 2023, 19, e2207029. [Google Scholar] [CrossRef] [PubMed]
- Estrada, L.D.; Ağaç, D.; Farrar, J.D. Sympathetic neural signaling via the β2-adrenergic receptor suppresses T-cell receptor-mediated human and mouse CD8+ T-cell effector function. Eur. J. Immunol. 2016, 46, 1948–1958. [Google Scholar] [CrossRef]
- Slota, C.; Shi, A.; Chen, G.; Bevans, M.; Weng, N.P. Norepinephrine preferentially modulates memory CD8 T cell function inducing inflammatory cytokine production and reducing proliferation in response to activation. Brain Behav. Immun. 2015, 46, 168–179. [Google Scholar] [CrossRef]
- Grebe, K.M.; Hickman, H.D.; Irvine, K.R.; Takeda, K.; Bennink, J.R.; Yewdell, J.W. Sympathetic nervous system control of anti-influenza CD8+ T cell responses. Proc. Natl. Acad. Sci. USA 2009, 106, 5300–5305. [Google Scholar] [CrossRef]
- Chen, M.; Qiao, G.; Hylander, B.L.; Mohammadpour, H.; Wang, X.Y.; Subjeck, J.R.; Singh, A.K.; Repasky, E.A. Adrenergic stress constrains the development of anti-tumor immunity and abscopal responses following local radiation. Nat. Commun. 2020, 11, 1821. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; Liu, H.; Kokolus, K.M.; Eng, J.W.L.; Messmer, M.N.; et al. β-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8+ T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef]
- Qiao, G.; Bucsek, M.J.; Winder, N.M.; Chen, M.; Giridharan, T.; Olejniczak, S.H.; Hylander, B.L.; Repasky, E.A. β-Adrenergic signaling blocks murine CD8+ T-cell metabolic reprogramming during activation: A mechanism for immunosuppression by adrenergic stress. Cancer Immunol. Immunother. 2019, 68, 11–22. [Google Scholar] [CrossRef]
- Wehbi, V.L.; Taskén, K. Molecular mechanisms for cAMP-mediated immunoregulation in T cells–role of anchored protein kinase a signaling units. Front. Immunol. 2016, 7, 222. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Jones, L.L.; Geiger, T.L. Modulation of PI3K signaling to improve CAR T cell function. Oncotarget 2018, 9, 35807. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Ma, Y.C.; Benjamin, J.; Littman, D.; Chao, M.V.; Huang, X.Y. Apoptotic signaling through the β-adrenergic receptor: A new Gs effector pathway. J. Biol. Chem. 2000, 275, 20726–20733. [Google Scholar] [CrossRef] [PubMed]
- Weinkove, R.; George, P.; Dasyam, N.; McLellan, A.D. Selecting costimulatory domains for chimeric antigen receptors: Functional and clinical considerations. Clin. Transl. Immunol. 2019, 8, e1049. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, G.A.; Gabrilovich, D.; Sotomayor, E.M. Immunosuppressive strategies that are mediated by tumor cells. Annu. Rev. Immunol. 2007, 25, 267–296. [Google Scholar] [CrossRef] [PubMed]
- Bartik, M.M.; Brooks, W.H.; Roszman, T.L. Modulation of T cell proliferation by stimulation of the β-adrenergic receptor: Lack of correlation between inhibition of T cell proliferation and cAMP accumulation. Cell. Immunol. 1993, 148, 408–421. [Google Scholar] [CrossRef]
- Harrison, A.J.; Du, X.; von Scheidt, B.; Kershaw, M.H.; Slaney, C.Y. Enhancing co-stimulation of CAR T cells to improve treatment outcomes in solid cancers. Immunother. Adv. 2021, 1, ltab016. [Google Scholar] [CrossRef]
- Giardino Torchia, M.L.; Conze, D.B.; Jankovic, D.; Ashwell, J.D. Balance between NF-κB p100 and p52 regulates T cell costimulation dependence. J. Immunol. 2013, 190, 549–555. [Google Scholar] [CrossRef]
- Boucher, J.C.; Li, G.; Kotani, H.; Cabral, M.L.; Morrissey, D.; Lee, S.B.; Spitler, K.; Beatty, N.J.; Cervantes, E.V.; Shrestha, B.; et al. CD28 Costimulatory Domain–Targeted Mutations Enhance Chimeric Antigen Receptor T-cell Function. Cancer Immunol. Res. 2021, 9, 62–74. [Google Scholar] [CrossRef]
- Guedan, S.; Posey, A.D., Jr.; Shaw, C.; Wing, A.; Da, T.; Patel, P.R.; McGettigan, S.E.; Casado-Medrano, V.; Kawalekar, O.U.; Uribe-Herranz, M.; et al. Enhancing CAR T cell persistence through ICOS and 4-1BB costimulation. JCI Insight 2018, 3, e96976. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, D.; Arshad, Z.; Smith, J.; Stanic, T.; Holländer, G.; Brindley, D. CAR-T cells: A systematic review and mixed methods analysis of the clinical trial landscape. Mol. Ther. 2018, 26, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, W.; Li, Y.; Yi, Y.; Yu, Z.; Liu, X.; Zhang, L.; Zheng, Y.; Niu, T. A systematic review on performance analysis of critical time points in multiple myeloma treated by CAR-T cell immunotherapy. Int. Immunopharmacol. 2023, 114, 109592. [Google Scholar] [CrossRef] [PubMed]
- González-Amaro, R.; Cortes, J.R.; Sánchez-Madrid, F.; Martín, P. Is CD69 an effective brake to control inflammatory diseases? Trends Mol. Med. 2013, 19, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Fernández, M.; de la Fuente, H.; Martín, P.; Cibrián, D.; Sánchez-Madrid, F. Unraveling CD69 signaling pathways, ligands and laterally associated molecules. EXCLI J. 2023, 22, 334. [Google Scholar]
- Qiao, G.; Chen, M.; Mohammadpour, H.; MacDonald, C.R.; Bucsek, M.J.; Hylander, B.L.; Barbi, J.J.; Repasky, E.A. Chronic Adrenergic Stress Contributes to Metabolic Dysfunction and an Exhausted Phenotype in T Cells in the Tumor Microenvironment. Cancer Immunol. Res. 2021, 9, 651–664. [Google Scholar] [CrossRef] [PubMed]
- Devi, S.; Alexandre, Y.O.; Loi, J.K.; Gillis, R.; Ghazanfari, N.; Creed, S.J.; Holz, L.E.; Shackleford, D.; Mackay, L.K.; Heath, W.R.; et al. Adrenergic regulation of the vasculature impairs leukocyte interstitial migration and suppresses immune responses. Immunity 2021, 54, 1219–1230. [Google Scholar] [CrossRef]
- Steinman, R.M.; Hemmi, H. Dendritic cells: Translating innate to adaptive immunity. In From Innate Immunity to Immunological Memory; Springer: Berlin/Heidelberg, Germany, 2006; Volume 311, pp. 17–58. [Google Scholar] [CrossRef]
- Patel, S.; Burga, R.A.; Powell, A.B.; Chorvinsky, E.A.; Hoq, N.; McCormack, S.E.; Van Pelt, S.N.; Hanley, P.J.; Cruz, C.R.Y. Beyond CAR T cells: Other cell-based immunotherapeutic strategies against cancer. Front. Oncol. 2019, 9, 196. [Google Scholar] [CrossRef]
- Hervé, J.; Dubreil, L.; Tardif, V.; Terme, M.; Pogu, S.; Anegon, I.; Rozec, B.; Gauthier, C.; Bach, J.M.; Blancou, P. β2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J. Immunol. 2013, 190, 3163–3171. [Google Scholar] [CrossRef]
- Nissen, M. The Impact of β-Adrenergic Signalling on Immune Function and Immunotherapy in Lymphoma. Ph.D. Thesis, The University of Queensland, Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Daher, C.; Vimeux, L.; Stoeva, R.; Peranzoni, E.; Bismuth, G.; Wieduwild, E.; Lucas, B.; Donnadieu, E.; Bercovici, N.; Trautmann, A.; et al. Blockade of β-Adrenergic Receptors Improves CD8+ T-cell Priming and Cancer Vaccine Efficacyβ-Blockers Boost T-cell Priming and Cancer Vaccine Efficacy. Cancer Immunol. Res. 2019, 7, 1849–1863. [Google Scholar] [CrossRef]
- Egri, N.; de Landazuri, I.O.; San Bartolome, C.; Ortega, J.R.; Espanol-Rego, M.; Juan, M. CART manufacturing process and reasons for academy-pharma collaboration. Immunol. Lett. 2020, 217, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Won, A.; Refaeli, Y.; Van Parijs, L. IL-2 and related cytokines can promote T cell survival by activating AKT. J. Immunol. 2002, 168, 597–603. [Google Scholar] [CrossRef]
- Feldman, R.D.; Hunninghake, G.; McArdle, W. Beta-adrenergic-receptor-mediated suppression of interleukin 2 receptors in human lymphocytes. J. Immunol. 1987, 139, 3355–3359. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Medina, B.E.; Cadena-Medina, D.A.; Esparza, E.; Arrieta, A.J.; Kirken, R.A. Isoproterenol-induced beta-2 adrenergic receptor activation negatively regulates interleukin-2 signaling. Biochem. J. 2018, 475, 2907–2923. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Yang, R.; Tu, J.; Xi, Y.; Yang, S.; Lv, L.; Zhai, X.; Zhu, Y.; Dong, D.; Tao, X. Metabolic reprogramming of immune cells in pancreatic cancer progression. Biomed. Pharmacother. 2023, 157, 113992. [Google Scholar] [CrossRef]
- Cao, Y.; Rathmell, J.C.; Macintyre, A.N. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE 2014, 9, e104104. [Google Scholar] [CrossRef]
- Corcoran, S.E.; O’Neill, L.A. HIF1α and metabolic reprogramming in inflammation. J. Clin. Investig. 2016, 126, 3699–3707. [Google Scholar] [CrossRef] [PubMed]
- Kokolus, K.M.; Capitano, M.L.; Lee, C.T.; Eng, J.W.L.; Waight, J.D.; Hylander, B.L.; Sexton, S.; Hong, C.C.; Gordon, C.J.; Abrams, S.I.; et al. Baseline tumor growth and immune control in laboratory mice are significantly influenced by subthermoneutral housing temperature. Proc. Natl. Acad. Sci. USA 2013, 110, 20176–20181. [Google Scholar] [CrossRef]
- Chen, Z.; Vaeth, M.; Eckstein, M.; Delgobo, M.; Ramos, G.; Frantz, S.; Hofmann, U.; Gladow, N. Characterization of the effect of the GLUT-1 inhibitor BAY-876 on T cells and macrophages. Eur. J. Pharmacol. 2023, 945, 175552. [Google Scholar] [CrossRef]
- Park, S.Y.; Kang, J.H.; Jeong, K.J.; Lee, J.; Han, J.W.; Choi, W.S.; Kim, Y.K.; Kang, J.; Park, C.G.; Lee, H.Y. Retracted: Norepinephrine induces VEGF expression and angiogenesis by a hypoxia-inducible factor-1α protein-dependent mechanism. Int. J. Cancer 2011, 128, 2306–2316. [Google Scholar] [CrossRef]
- Chang, C.H.; Curtis, J.D.; Maggi Jr, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; Van Der Windt, G.J.; Blagih, J.; Qiu, J. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Menk, A.V.; Moreci, R.S.; Whetstone, R.D.; Dadey, R.E.; Watkins, S.C.; Ferris, R.L.; Delgoffe, G.M. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 2016, 45, 374–388. [Google Scholar] [CrossRef]
- Jensen, A.W.P.; Carnaz Simões, A.M.; thor Straten, P.; Holmen Olofsson, G. Adrenergic signaling in immunotherapy of cancer: Friend or foe? Cancers 2021, 13, 394. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef] [PubMed]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8+ T cell exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Yu, Y.R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.C.; et al. Disturbed mitochondrial dynamics in CD8+ TILs reinforce T cell exhaustion. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Franchina, D.G.; Dostert, C.; Brenner, D. Reactive oxygen species: Involvement in T cell signaling and metabolism. Trends Immunol. 2018, 39, 489–502. [Google Scholar] [CrossRef]
- Ligtenberg, M.A.; Mougiakakos, D.; Mukhopadhyay, M.; Witt, K.; Lladser, A.; Chmielewski, M.; Riet, T.; Abken, H.; Kiessling, R. Coexpressed catalase protects chimeric antigen receptor–redirected T cells as well as bystander cells from oxidative stress–induced loss of antitumor activity. J. Immunol. 2016, 196, 759–766. [Google Scholar] [CrossRef]
- Chen, X.; Song, M.; Zhang, B.; Zhang, Y. Reactive oxygen species regulate T cell immune response in the tumor microenvironment. Oxid. Med. Cell. Longev. 2016, 2016, 1580967. [Google Scholar] [CrossRef]
- Case, A.J.; Roessner, C.T.; Tian, J.; Zimmerman, M.C. Mitochondrial superoxide signaling contributes to norepinephrine-mediated T-lymphocyte cytokine profiles. PLoS ONE 2016, 11, e0164609. [Google Scholar] [CrossRef]
- Ogbodo, E.J.; Ruff, M.W.; Sakemura, R.L.; ManriquezRoman, C.; Huynh, T.; Girsch, J.H.; Sirpilla, O.L.; Yun, K.; Stewart, C.M.; Can, I.; et al. Simultaneous targeting of EphA3 on glioblastoma and tumor microenvironment to overcome resistance to CART cell therapy in brain cancer. Cancer Res. 2023, 83, 5017. [Google Scholar] [CrossRef]
- He, J.; Munir, F.; Ragoonanan, D.; Zaky, W.; Khazal, S.J.; Tewari, P.; Fueyo, J.; Gomez-Manzano, C.; Jiang, H. Combining CAR T Cell Therapy and Oncolytic Virotherapy for Pediatric Solid Tumors: A Promising Option. Immuno 2023, 3, 37–58. [Google Scholar] [CrossRef]
- Dünser, M.W.; Hasibeder, W.R. Sympathetic overstimulation during critical illness: Adverse effects of adrenergic stress. J. Intensive Care Med. 2009, 24, 293–316. [Google Scholar] [CrossRef]
- Qin, J.F.; Jin, F.J.; Li, N.; Guan, H.T.; Lan, L.; Ni, H.; Wang, Y. Adrenergic receptor β2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015, 48, 295. [Google Scholar] [CrossRef] [PubMed]
- Oh, M.S.; Guzner, A.; Wainwright, D.A.; Mohindra, N.A.; Chae, Y.K.; Behdad, A.; Villaflor, V.M. The impact of beta blockers on survival outcomes in patients with non–small-cell lung cancer treated with immune checkpoint inhibitors. Clin. Lung Cancer 2021, 22, e57–e62. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farooq, M.A.; Ajmal, I.; Hui, X.; Chen, Y.; Ren, Y.; Jiang, W. β2-Adrenergic Receptor Mediated Inhibition of T Cell Function and Its Implications for CAR-T Cell Therapy. Int. J. Mol. Sci. 2023, 24, 12837. https://doi.org/10.3390/ijms241612837
Farooq MA, Ajmal I, Hui X, Chen Y, Ren Y, Jiang W. β2-Adrenergic Receptor Mediated Inhibition of T Cell Function and Its Implications for CAR-T Cell Therapy. International Journal of Molecular Sciences. 2023; 24(16):12837. https://doi.org/10.3390/ijms241612837
Chicago/Turabian StyleFarooq, Muhammad Asad, Iqra Ajmal, Xinhui Hui, Yiran Chen, Yaojun Ren, and Wenzheng Jiang. 2023. "β2-Adrenergic Receptor Mediated Inhibition of T Cell Function and Its Implications for CAR-T Cell Therapy" International Journal of Molecular Sciences 24, no. 16: 12837. https://doi.org/10.3390/ijms241612837
APA StyleFarooq, M. A., Ajmal, I., Hui, X., Chen, Y., Ren, Y., & Jiang, W. (2023). β2-Adrenergic Receptor Mediated Inhibition of T Cell Function and Its Implications for CAR-T Cell Therapy. International Journal of Molecular Sciences, 24(16), 12837. https://doi.org/10.3390/ijms241612837