
Research Progress of Pyroptosis in Fatty Liver Disease


Abstract
:1. Introduction
2. Pyroptosis
2.1. Inflammasomes
2.2. Caspases
2.3. GSDMs
2.4. Pathways of Pyroptosis
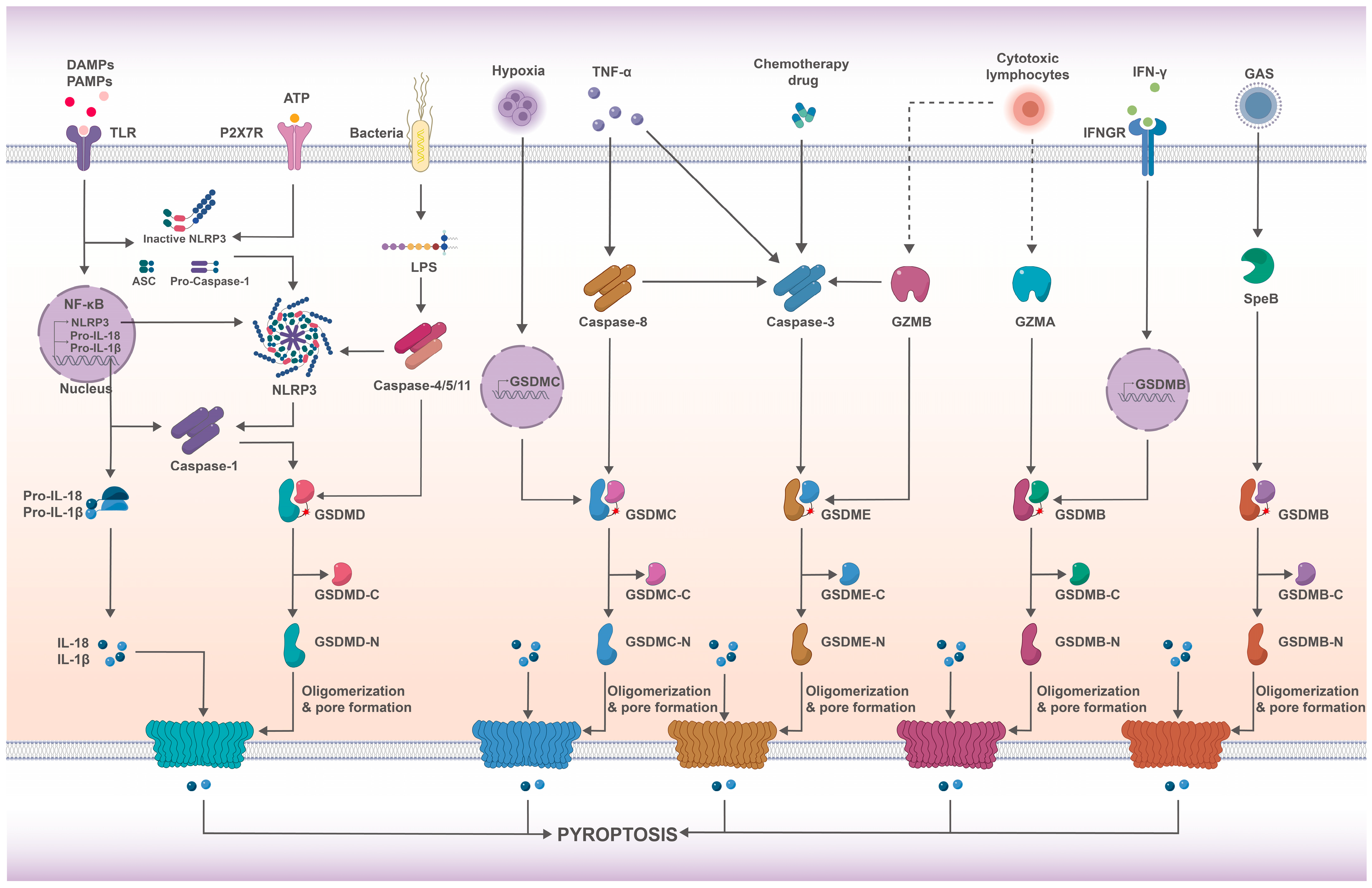
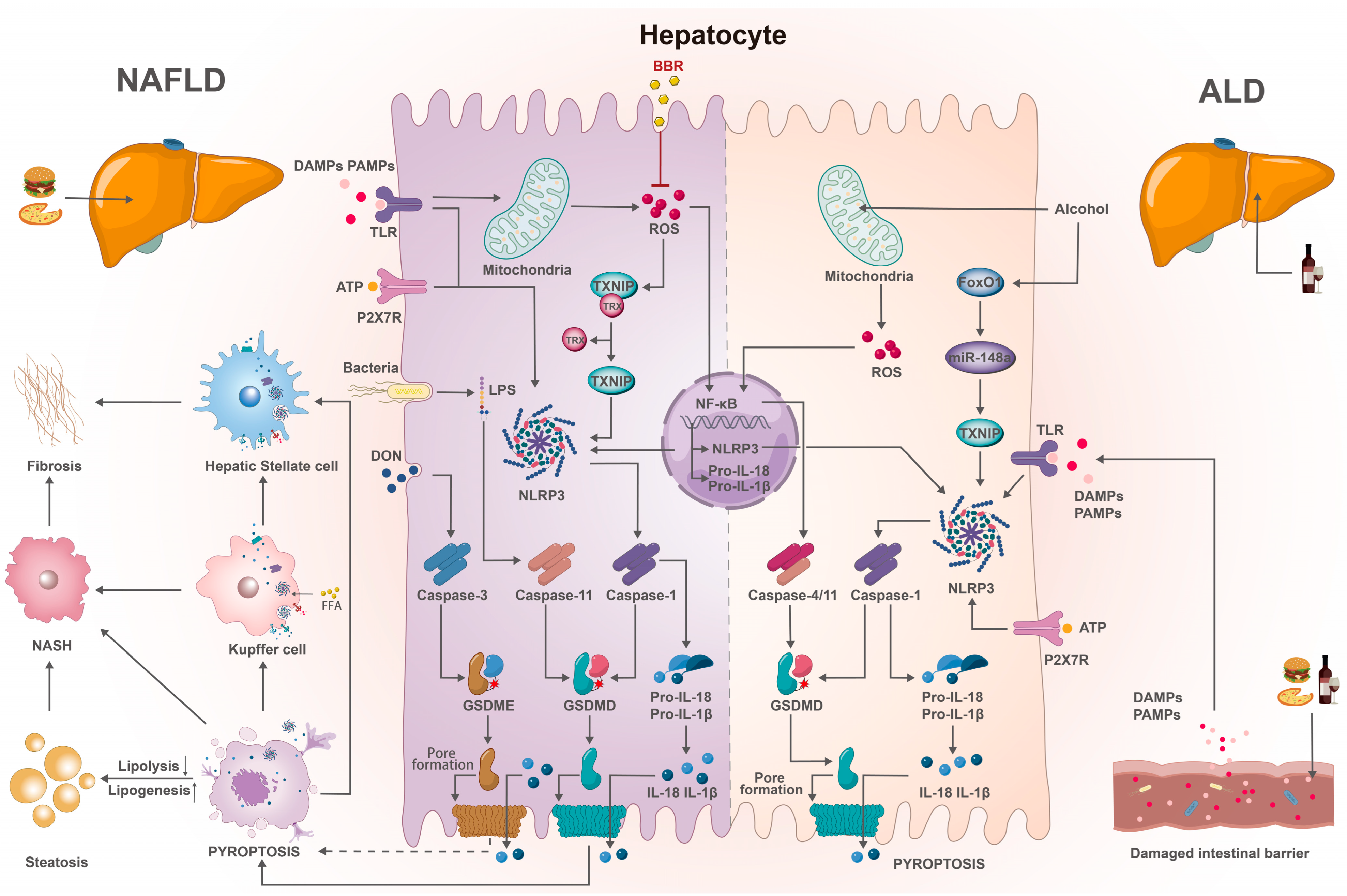
3. Pyroptosis and FLD
3.1. Inflammasomes and FLD
3.2. Caspases and FLD
3.3. GSDMs and FLD
4. Strategies for the Prevention and Treatment of FLD with Pyroptosis
5. Summary and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whalley, S.; Puvanachandra, P.; Desai, A.; Kennedy, H. Hepatology outpatient service provision in secondary care: A study of liver disease incidence and resource costs. Clin. Med. 2007, 7, 119–124. [Google Scholar] [CrossRef]
- Rinella, M.E. Nonalcoholic fatty liver disease: A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.M.; Wong, V.W.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Primers 2015, 1, 15080. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, Y.; Wang, J.; Liu, Y.; Qi, D.; Yao, W.; Jiang, H.; Li, T.; Huang, K.; Zhang, W.; et al. Prevalence and risk factor analysis for the nonalcoholic fatty liver disease in patients with type 2 diabetes mellitus. Medicine 2021, 100, e24940. [Google Scholar] [CrossRef] [PubMed]
- Toshikuni, N. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8393. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Man, S.M.; Karki, R.; Kanneganti, T. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Yang, W.; Liu, L.; Wei, Y.; Fang, C.; Liu, S.; Zhou, F.; Li, Y.; Zhao, G.; Guo, Z.; Luo, Y.; et al. Exercise suppresses nlrp3 inflammasome activation in mice with diet-induced nash: A plausible role of adropin. Lab. Investig. 2021, 101, 369–380. [Google Scholar] [CrossRef]
- Aziz, M.; Jacob, A.; Yang, W.L.; Matsuda, A.; Wang, P. Current trends in inflammatory and immunomodulatory mediators in sepsis. J. Leukoc. Biol. 2013, 93, 329–342. [Google Scholar] [CrossRef]
- Beier, J.I.; Banales, J.M. Pyroptosis: An inflammatory link between nafld and nash with potential therapeutic implications. J. Hepatol. 2018, 68, 643–645. [Google Scholar] [CrossRef]
- Mridha, A.R.; Wree, A.; Robertson, A.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. Nlrp3 inflammasome blockade reduces liver inflammation and fibrosis in experimental nash in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Brennan, M.A.; Cookson, B.T. Salmonella induces macrophage death by caspase-1-dependent necrosis. Mol. Microbiol. 2000, 38, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of gsdmd by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Hamilton, C.; Anand, P.K. Right place, right time: Localisation and assembly of the nlrp3 inflammasome. F1000Res 2019, 8, 676. [Google Scholar] [CrossRef]
- Liston, A.; Masters, S.L. Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat. Rev. Immunol. 2017, 17, 208–214. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev.Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Van Opdenbosch, N.; Lamkanfi, M. Caspases in cell death, inflammation, and disease. Immunity 2019, 50, 1352–1364. [Google Scholar] [CrossRef] [PubMed]
- Kesavardhana, S.S.; Malireddi, R.K.S.R.; Kanneganti, T.T. Caspases in cell death, inflammation, and gasdermin-induced pyroptosis. Annu. Rev. Immunol. 2020, 38, 567–595. [Google Scholar] [CrossRef]
- Porter, A.G.; Janicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Fritsch, M.; Günther, S.D.; Schwarzer, R.; Albert, M.; Schorn, F.; Werthenbach, J.P.; Schiffmann, L.M.; Stair, N.; Stocks, H.; Seeger, J.M.; et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 2019, 575, 683–687. [Google Scholar] [CrossRef]
- Saeki, N.; Kuwahara, Y.; Sasaki, H.; Satoh, H.; Shiroishi, T. Gasdermin (gsdm) localizing to mouse chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm. Genome. 2000, 11, 718–724. [Google Scholar] [CrossRef]
- Feng, S.; Fox, D.; Man, S.M. Mechanisms of gasdermin family members in inflammasome signaling and cell death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef]
- Kuang, S.; Zheng, J.; Yang, H.; Li, S.; Duan, S.; Shen, Y.; Ji, C.; Gan, J.; Xu, X.W.; Li, J. Structure insight of gsdmd reveals the basis of gsdmd autoinhibition in cell pyroptosis. Proc. Natl. Acad. Sci. USA 2017, 114, 10642–10647. [Google Scholar] [CrossRef]
- Shi, P.; Tang, A.; Xian, L.; Hou, S.; Zou, D.; Lv, Y.; Huang, Z.; Wang, Q.; Song, A.; Lin, Z.; et al. Loss of conserved gsdma3 self-regulation causes autophagy and cell death. Biochem. J. 2015, 468, 325–336. [Google Scholar] [CrossRef]
- Aglietti, R.A.; Dueber, E.C. Recent insights into the molecular mechanisms underlying pyroptosis and gasdermin family functions. Trends Immunol. 2017, 38, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin d causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Rogers, C.; Fernandes-Alnemri, T.; Mayes, L.; Alnemri, D.; Cingolani, G.; Alnemri, E.S. Cleavage of dfna5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017, 8, 14128. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme a from cytotoxic lymphocytes cleaves gsdmb to trigger pyroptosis in target cells. Science 2020, 368, eaaz7548. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Zeng, H.; Zhou, Z.; Su, Y.; Cheng, H.; Hou, Y.; She, Y.; Feng, N.; Wang, J.; Shao, F.; et al. Structural mechanisms for regulation of gsdmb pore-forming activity. Nature 2023, 616, 598–605. [Google Scholar] [CrossRef]
- Wang, C.; Shivcharan, S.; Tian, T.; Wright, S.; Ma, D.; Chang, J.; Li, K.; Song, K.; Xu, C.; Rathinam, V.A.; et al. Structural basis for gsdmb pore formation and its targeting by ipah7.8. Nature 2023, 616, 590–597. [Google Scholar] [CrossRef]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhou, B.; Sun, R.Y.; Ai, Y.L.; Cheng, K.; Li, F.N.; Wang, B.R.; Liu, F.J.; Jiang, Z.H.; Wang, W.J.; et al. The metabolite alpha-kg induces gsdmc-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021, 31, 980–997. [Google Scholar] [CrossRef]
- Deng, W.; Bai, Y.; Deng, F.; Pan, Y.; Mei, S.; Zheng, Z.; Min, R.; Wu, Z.; Li, W.; Miao, R.; et al. Streptococcal pyrogenic exotoxin b cleaves gsdma and triggers pyroptosis. Nature 2022, 602, 496–502. [Google Scholar] [CrossRef]
- Larock, D.L.; Johnson, A.F.; Wilde, S.; Sands, J.S.; Monteiro, M.P.; Larock, C.N. Group a streptococcus induces gsdma-dependent pyroptosis in keratinocytes. Nature 2022, 605, 527–531. [Google Scholar] [CrossRef]
- Thornberry, N.A.; Bull, H.G.; Calaycay, J.R.; Chapman, K.T.; Howard, A.D.; Kostura, M.J.; Miller, D.K.; Molineaux, S.M.; Weidner, J.R.; Aunins, J.; et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 1992, 356, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Cerretti, D.P.; Kozlosky, C.J.; Mosley, B.; Nelson, N.; Van Ness, K.; Greenstreet, T.A.; March, C.J.; Kronheim, S.R.; Druck, T.; Cannizzaro, L.A.; et al. Molecular cloning of the interleukin-1 beta converting enzyme. Science 1992, 256, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic lps activates caspase-11: Implications in tlr4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular lps. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynski, A.; et al. Noncanonical inflammasome activation by intracellular lps independent of tlr4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin d for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Ruhl, S.; Broz, P. Caspase-11 activates a canonical nlrp3 inflammasome by promoting k+ efflux. Eur. J. Immunol. 2015, 45, 2927–2936. [Google Scholar] [CrossRef]
- de Gassart, A.; Martinon, F. Pyroptosis: Caspase-11 unlocks the gates of death. Immunity 2015, 43, 835–837. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Munoz-Planillo, R.; Liu, Q.; Nunez, G. Caspase-11 requires the pannexin-1 channel and the purinergic p2x7 pore to mediate pyroptosis and endotoxic shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Miura, M.; Jung, Y.; Zhu, H.; Gagliardini, V.; Shi, L.; Greenberg, A.H.; Yuan, J. Identification and characterization of ich-3, a member of the interleukin-1beta converting enzyme (ice)/ced-3 family and an upstream regulator of ice. J. Biol. Chem. 1996, 271, 20580–20587. [Google Scholar] [CrossRef]
- Sarhan, J.; Liu, B.C.; Muendlein, H.I.; Li, P.; Nilson, R.; Tang, A.Y.; Rongvaux, A.; Bunnell, S.C.; Shao, F.; Green, D.R.; et al. Caspase-8 induces cleavage of gasdermin d to elicit pyroptosis during yersinia infection. Proc. Natl. Acad. Sci. USA 2018, 115, E10888–E10897. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.; You, Y.; Hsu, J.; Nie, L.; Chen, Y.; Wang, Y.; Liu, C.; et al. Pd-l1-mediated gasdermin c expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, Y.; Xia, S.; Kong, Q.; Li, S.; Liu, X.; Junqueira, C.; Meza-Sosa, K.F.; Mok, T.M.Y.; Ansara, J.; et al. Gasdermin e suppresses tumour growth by activating anti-tumour immunity. Nature 2020, 579, 415–420. [Google Scholar] [CrossRef]
- Loveless, R.; Bloomquist, R.; Teng, Y. Pyroptosis at the forefront of anticancer immunity. J. Exp. Clin. Cancer Res. 2021, 40, 264. [Google Scholar] [CrossRef]
- Knorr, J.; Wree, A.; Feldstein, A.E. Pyroptosis in steatohepatitis and liver diseases. J. Mol. Biol. 2022, 434, 167271. [Google Scholar] [CrossRef]
- Wei, H.; Cui, D. Pyroptosis and insulin resistance in metabolic organs. Int. J. Mol. Sci. 2022, 23, 11638. [Google Scholar] [CrossRef]
- Xue, W.; Cui, D.; Qiu, Y. Research progress of pyroptosis in alzheimer’s disease. Front. Molec. Neurosci. 2022, 15, 872471. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Heo, M.J.; Kim, T.H.; You, J.S.; Blaya, D.; Sancho-Bru, P.; Kim, S.G. Alcohol dysregulates mir-148a in hepatocytes through foxo1, facilitating pyroptosis via txnip overexpression. Gut 2019, 68, 708–720. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; Mcgeough, M.D.; Pena, C.A.; Johnson, C.D.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. Nlrp3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatology. 2014, 59, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [PubMed]
- Wree, A.; Mcgeough, M.D.; Peña, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. Nlrp3 inflammasome activation is required for fibrosis development in nafld. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates nlrp3 inflammasomes in mouse kupffer cells through mitochondrial dna release. Cell. Immunol. 2018, 332, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Maroni, L.; Agostinelli, L.; Saccomanno, S.; Pinto, C.; Giordano, D.M.; Rychlicki, C.; De Minicis, S.; Trozzi, L.; Banales, J.M.; Melum, E.; et al. Nlrp3 activation induces il-18 synthesis and affects the epithelial barrier function in reactive cholangiocytes. Am. J. Pathol. 2017, 187, 366–376. [Google Scholar] [CrossRef]
- Koh, E.H.; Yoon, J.E.; Ko, M.S.; Leem, J.; Yun, J.; Hong, C.H.; Cho, Y.K.; Lee, S.E.; Jang, J.E.; Baek, J.Y.; et al. Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut 2021, 70, 1954–1964. [Google Scholar] [CrossRef]
- Iracheta-Vellve, A.; Petrasek, J.; Satishchandran, A.; Gyongyosi, B.; Saha, B.; Kodys, K.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Szabo, G. Inhibition of sterile danger signals, uric acid and atp, prevents inflammasome activation and protects from alcoholic steatohepatitis in mice. J. Hepatol. 2015, 63, 1147–1155. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhao, H.; Lu, J.; Lin, K.; Ni, J.; Wu, G.; Tang, H. Caspase-11–mediated hepatocytic pyroptosis promotes the progression of nonalcoholic steatohepatitis. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 653–664. [Google Scholar] [CrossRef]
- Kai, J.; Yang, X.; Wang, Z.; Wang, F.; Jia, Y.; Wang, S.; Tan, S.; Chen, A.; Shao, J.; Zhang, F.; et al. Oroxylin a promotes pgc-1α/mfn2 signaling to attenuate hepatocyte pyroptosis via blocking mitochondrial ros in alcoholic liver disease. Free. Radic. Biol. Med. 2020, 153, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, C.; Dai, S.; Liu, Y.; Zhang, F.; Peng, C.; Li, Y. Quercetin protects ethanol-induced hepatocyte pyroptosis via scavenging mitochondrial ros and promoting pgc-1α-regulated mitochondrial homeostasis in l02 cells. Oxidative Med. Cell. Longev. 2022, 2022, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Lazaro, R.; Wu, R.; Lee, S.; Zhu, N.L.; Chen, C.L.; French, S.W.; Xu, J.; Machida, K.; Tsukamoto, H. Osteopontin deficiency does not prevent but promotes alcoholic neutrophilic hepatitis in mice. Hepatology. 2015, 61, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Khanova, E.; Wu, R.; Wang, W.; Yan, R.; Chen, Y.; French, S.W.; Llorente, C.; Pan, S.Q.; Yang, Q.; Li, Y.; et al. Pyroptosis by caspase11/4-gasdermin-d pathway in alcoholic hepatitis in mice and patients. Hepatology. 2018, 67, 1737–1753. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Li, J.; Xie, X.; Chen, S.; Huang, Q.; Mu, P.; Jiang, J.; Deng, Y. Deoxynivalenol induces caspase-3/gsdme-dependent pyroptosis and inflammation in mouse liver and heparg cells. Arch. Toxicol. 2022, 96, 3091–3112. [Google Scholar] [CrossRef]
- Wang, X.; Sun, K.; Zhou, Y.; Wang, H.; Zhou, Y.; Liu, S.; Nie, Y.; Li, Y. Nlrp3 inflammasome inhibitor cy-09 reduces hepatic steatosis in experimental nafld mice. Biochem. Biophys. Res. Commun. 2021, 534, 734–739. [Google Scholar] [CrossRef] [PubMed]
- Torres, S.; Brol, M.J.; Magdaleno, F.; Schierwagen, R.; Uschner, F.E.; Klein, S.; Ortiz, C.; Tyc, O.; Bachtler, N.; Stunden, J.; et al. The specific nlrp3 antagonist ifm-514 decreases fibrosis and inflammation in experimental murine non-alcoholic steatohepatitis. Front. Mol. Biosci. 2021, 8, 715765. [Google Scholar] [CrossRef]
- Mai, W.; Xu, Y.; Xu, J.; Zhao, D.; Ye, L.; Yu, G.; Wang, Z.; Lu, Q.; Lin, J.; Yang, T.; et al. Berberine inhibits nod-like receptor family pyrin domain containing 3 inflammasome activation and pyroptosis in nonalcoholic steatohepatitis via the ros/txnip axis. Front. Pharmacol. 2020, 11, 185. [Google Scholar] [CrossRef]
- Yang, G.; Jang, J.H.; Kim, S.W.; Han, S.; Ma, K.; Jang, J.; Kang, H.C.; Cho, Y.; Lee, H.S.; Lee, J.Y. Sweroside prevents non-alcoholic steatohepatitis by suppressing activation of the nlrp3 inflammasome. Int. J. Mol. Sci. 2020, 21, 2790. [Google Scholar] [CrossRef]
- Li, P.; He, K.; Li, J.; Liu, Z.; Gong, J. The role of kupffer cells in hepatic diseases. Mol. Immunol. 2017, 85, 222–229. [Google Scholar] [CrossRef]
- Arsenijevic, A.; Milovanovic, J.; Stojanovic, B.; Djordjevic, D.; Stanojevic, I.; Jankovic, N.; Vojvodic, D.; Arsenijevic, N.; Lukic, M.L.; Milovanovic, M. Gal-3 deficiency suppresses novosphyngobium aromaticivorans inflammasome activation and il-17 driven autoimmune cholangitis in mice. Front. Immunol. 2019, 10, 1309. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of nafld and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef]
- Safari, Z.; Gérard, P. The links between the gut microbiome and non-alcoholic fatty liver disease (nafld). Cell. Mol. Life Sci. 2019, 76, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- de Sant’Ana, L.P.; Ribeiro, D.J.S.; Martins, A.M.A.; Dos Santos, F.N.; Corrêa, R.; Almeida, R.D.N.; Eberlin, M.N.; Maurice, C.F.; Magalhães, K.G. Absence of the caspases 1/11 modulates liver global lipid profile and gut microbiota in high-fat-diet-induced obese mice. Front. Immunol. 2020, 10, 2926. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, P.; An, L.; Sun, N.; Peng, L.; Tang, W.; Ma, D.; Chen, J. Miltirone induces cell death in hepatocellular carcinoma cell through gsdme-dependent pyroptosis. Acta Pharm. Sin. B. 2020, 10, 1397–1413. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, Q.; Ding, C.; Sun, H.; Che, Y.; Huang, H.; Wang, Y.; Wu, J.; Hao, H.; Cao, L. Gasdermin e-derived caspase-3 inhibitors effectively protect mice from acute hepatic failure. Acta Pharmacol. Sin. 2021, 42, 68–76. [Google Scholar] [CrossRef]
- Qu, J.; Yuan, Z.; Wang, G.; Wang, X.; Li, K. The selective nlrp3 inflammasome inhibitor mcc950 alleviates cholestatic liver injury and fibrosis in mice. Int. Immunopharmacol. 2019, 70, 147–155. [Google Scholar] [CrossRef]
- Hajighasem, A.; Farzanegi, P.; Mazaheri, Z. Effects of combined therapy with resveratrol, continuous and interval exercises on apoptosis, oxidative stress, and inflammatory biomarkers in the liver of old rats with non-alcoholic fatty liver disease. Arch. Physiol. Biochem. 2019, 125, 142–149. [Google Scholar] [CrossRef]
- Hashida, R.; Kawaguchi, T.; Bekki, M.; Omoto, M.; Matsuse, H.; Nago, T.; Takano, Y.; Ueno, T.; Koga, H.; George, J.; et al. Aerobic vs. Resistance exercise in non-alcoholic fatty liver disease: A systematic review. J. Hepatol. 2017, 66, 142–152. [Google Scholar] [CrossRef]
- Oh, S.; So, R.; Shida, T.; Matsuo, T.; Kim, B.; Akiyama, K.; Isobe, T.; Okamoto, Y.; Tanaka, K.; Shoda, J. High-intensity aerobic exercise improves both hepatic fat content and stiffness in sedentary obese men with nonalcoholic fatty liver disease. Sci. Rep. 2017, 7, 43029. [Google Scholar] [CrossRef]
- Kar, S.; Shahshahan, H.R.; Hackfort, B.T.; Yadav, S.K.; Yadav, R.; Kambis, T.N.; Lefer, D.J.; Mishra, P.K. Exercise training promotes cardiac hydrogen sulfide biosynthesis and mitigates pyroptosis to prevent high-fat diet-induced diabetic cardiomyopathy. Antioxidants 2019, 8, 638. [Google Scholar] [CrossRef] [PubMed]
- Goh, J.; Goh, K.P.; Abbasi, A. Exercise and adipose tissue macrophages: New frontiers in obesity research? Front. Endocrinol. 2016, 7, 65. [Google Scholar] [CrossRef] [PubMed]
- Kawanishi, N.; Yano, H.; Yokogawa, Y.; Suzuki, K. Exercise training inhibits inflammation in adipose tissue via both suppression of macrophage infiltration and acceleration of phenotypic switching from m1 to m2 macrophages in high-fat-diet-induced obese mice. Exerc. Immunol. Rev. 2010, 16, 105–118. [Google Scholar] [PubMed]
- Lee, J.; Hong, J.; Umetani, M.; Lavoy, E.C.; Kim, J.H.; Park, Y. Vascular protection by exercise in obesity: Inflammasome-associated mechanisms. Med. Sci. Sports. Exerc. 2020, 52, 2538–2545. [Google Scholar] [CrossRef]
- Yu, Y.; Yu, L.; Cheng, N.; Liu, X.; Fang, C.; Liu, S.; Zhu, L. Exercise alleviates the apolipoprotein a5-toll-like receptor 4 axis impairment in mice with high-fat diet-induced non-alcoholic steatohepatitis. Front. Physiol. 2021, 12, 783341. [Google Scholar] [CrossRef]
- Zhang, T.; Li, J.; Liu, C.; Guo, M.; Gao, C.; Zhou, L.; Long, Y.; Xu, Y. Butyrate ameliorates alcoholic fatty liver disease via reducing endotoxemia and inhibiting liver gasdermin d-mediated pyroptosis. Ann. Transl. Med. 2021, 9, 873. [Google Scholar] [CrossRef]
- Luan, J.; Chen, W.; Fan, J.; Wang, S.; Zhang, X.; Zai, W.; Jin, X.; Wang, Y.; Feng, Z.; Zhang, J.; et al. Gsdmd membrane pore is critical for il-1beta release and antagonizing il-1beta by hepatocyte-specific nanobiologics is a promising therapeutics for murine alcoholic steatohepatitis. Biomaterials 2020, 227, 119570. [Google Scholar] [CrossRef]
- Fu, X.; Zhong, Z.; Hu, F.; Zhang, Y.; Li, C.; Yan, P.; Feng, L.; Shen, J.; Huang, B. The protective effects of selenium-enriched spirulina platensis on chronic alcohol-induced liver injury in mice. Food Funct. 2018, 9, 3155–3165. [Google Scholar] [CrossRef]
- Cui, W.; Li, X.; Xue, W.; Wei, H.; Zhou, G.; Qiu, Y.; Cui, D. Exercise affects the formation and recovery of alcoholic liver disease through the il-6–p47phox oxidative–stress axis. Cells 2022, 11, 1305. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Pyroptotic Component | FLD Feature | Experimental Model | Results (Compared with Controls) | Reference |
|---|---|---|---|---|
| NLRP3 | Steatosis | HFD-fed Nlrp3−/−, Asc−/− and Casp1−/− mice | ↓ Steatosis, ↓ weight gain, and ↓ hepatic triglyceride content | [64] |
| Steatosis | CDAA-fed Nlrp3−/− mice | ↓ Steatohepatitis, ↓ liver enlargement, ↓ liver injury, ↓ liver fibrosis, and ↓ activated macrophage infiltration | [65] | |
| Steatosis | Nlrp3 knock-in mice | Severe liver inflammation, hepatic stellate cell activation and collagen deposition | [62] | |
| NASH | MCD-fed Nlrp3−/− mice | ↓ Steatosis, ↓ hepatocyte ballooning, ↓ inflammatory cell infiltration, ↓ expression of NLRP3, ASC and caspase-1 in KCs | [66] | |
| NASH | PA stimulates primary KCs from MCD-fed Nlrp3−/− and WT mice | NLRP3 inflammasome complex formation, the colocalization of NLRP3 with caspase-1, caspase-1 activation and IL-1β secretion | [66] | |
| NASH | MCC950-treated MCD-fed mice and atherogenic diet-fed foz/foz mice | Normal hepatic caspase 1 and IL-1β expression, ↓ ALT/AST, ↓ the severity of NASH pathology and liver fibrosis | [11] | |
| NASH | LPS plus Nig or LPS plus PA primary mouse and human hepatocytes | ↑ Caspase-1- and PI-positive cells, NLRP3 inflammasome proteins release | [63] | |
| NASH | Hepatocyte-specific leucine 351 to proline Nlrp3KICreA mice | ↓ Upregulated fibrosis markers, ↓ collagen deposion and ↓ α-SMA protein expression | [63] | |
| Liver fibrosis | LPS and ATP stimulates cholangiocyte | ↑ IL-18 expression,↓ E-cadherin and Zonulin-1 protein expression | [67] | |
| NLRC4 | NASH | HFHCD-fed mice | ↑ The expression of SMS1, NLRC4 and simple steatosis without significant inflammation and fibrosis | [68] |
| IL-1β | ASH | Lieber-DeCarli ethanol-fed P2rx7-KO and overexpress uricase mice | ASH attenuation, ↓ inflammasome activation and ↓ IL-1β level | [69] |
| Caspase-1 | Liver fibrosis | HFHCD-fed Casp1−/− mice | ↓ Hepatic inflammation and ↓ fibrosis | [68] |
| Caspase-11 | NASH | MCD-fed Casp11−/− mice | ↓ Liver injury, ↓ fibrosis, ↓ inflammation, ↓ GSDMD and IL-1β activation | [70] |
| NLRP3, Caspase-1 | ALD | Lieber-De-Carli diet with binge-fed ALD mice | ↑ Inflammatory infiltration, ↑ intracellular TG and TC, ↑ caspase-1, IL-18, IL-1β, NLRP3 activation | [71] |
| ALD | Ethano and quercetin-treated lL02 cells | ↓ MtDNA production, ↓ NLRP3, ASC, caspase1, IL-18, IL-1β and GSDMD-N expression | [72] | |
| Caspase-11/4 | AH | Hybrid-fed AH mice [73] | ↑ Caspase-11/4, ↑ GSDMD and ↓ IL-1β levels | [74] |
| Caspase-11 | AH | Hybrid-fed AH Casp11−/− mice | ↓ GSDMD level, ↓ hepatocellular death, ↓ liver bacterial load | [74] |
| IL-18 | AH | Hybrid-fed AH IL-18−/− mice | ↑ GSDMD activation, ↑ liver bacterial load and ↑ hepatocyte death | [74] |
| GSDMD | NAFLD | Human liver tissues from NAFLD patients | ↑ GSDMD and GSDMD-N fragment level | [60] |
| NASH | MCD-fed GSDMD−/− mice | ↓ Steatosis, ↓ inflammation, ↑ lipogenic gene expression and↑ lipolytic genes expression | [60] | |
| GSDME | Liver inflammatory injury | DON orally administered mice | Focal steatosis, focal fibrosis and caspase-3, PARP and GSDME activation | [75] |
| Liver inflammatory injury | DON-exposed HepaRG cells | ↓ Cell viability and ↑ Annexin-V/PI double positive cells | [75] | |
| NLRP3 | NAFLD | CY-09-treated HFD-fed mice | ↓ Body weight, ↓ liver lipid droplets and ↓ NAS | [76] |
| NASH | IFM-514 -injected MCD-fed ApoE−/− mice | ↓ IL-1β production, ↓ caspase-1 activation, ↓ NAS and ↓ steatosis | [77] | |
| NASH | BBR-treated MCD Medium-induced AML 12 cells | ↓ Lipid accumulation, ↓ ROS, ↓ lipid peroxides, ↓ NLRP3, caspase-1 and GSDMD-N expression | [78] | |
| NASH | Sweroside-treated MCD-fed mice | ↓ IL-1β, ↓ caspase-1, ↓ hepatic immune cell infiltration, ↓ hepatic triglyceride accumulation, and ↓ liver fibrosis | [79] | |
| NASH | Exercise-intervened HFD-fed or MCD-fed mice | ↓ NLRP3 inflammasome components, ↓ caspase-1 enzymatic activity, ↓ hepatic steatosis, ↓ inflammation, and ↓ fibrosis | [80] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Xue, W.; Wei, H.; Fan, Q.; Li, X.; Qiu, Y.; Cui, D. Research Progress of Pyroptosis in Fatty Liver Disease. Int. J. Mol. Sci. 2023, 24, 13065. https://doi.org/10.3390/ijms241713065
Li R, Xue W, Wei H, Fan Q, Li X, Qiu Y, Cui D. Research Progress of Pyroptosis in Fatty Liver Disease. International Journal of Molecular Sciences. 2023; 24(17):13065. https://doi.org/10.3390/ijms241713065
Chicago/Turabian StyleLi, Rongxuan, Weiyue Xue, Huiting Wei, Qingqing Fan, Xiang Li, Ye Qiu, and Di Cui. 2023. "Research Progress of Pyroptosis in Fatty Liver Disease" International Journal of Molecular Sciences 24, no. 17: 13065. https://doi.org/10.3390/ijms241713065