

A Leukemic Target with a Thousand Faces: The Mitochondria

Abstract
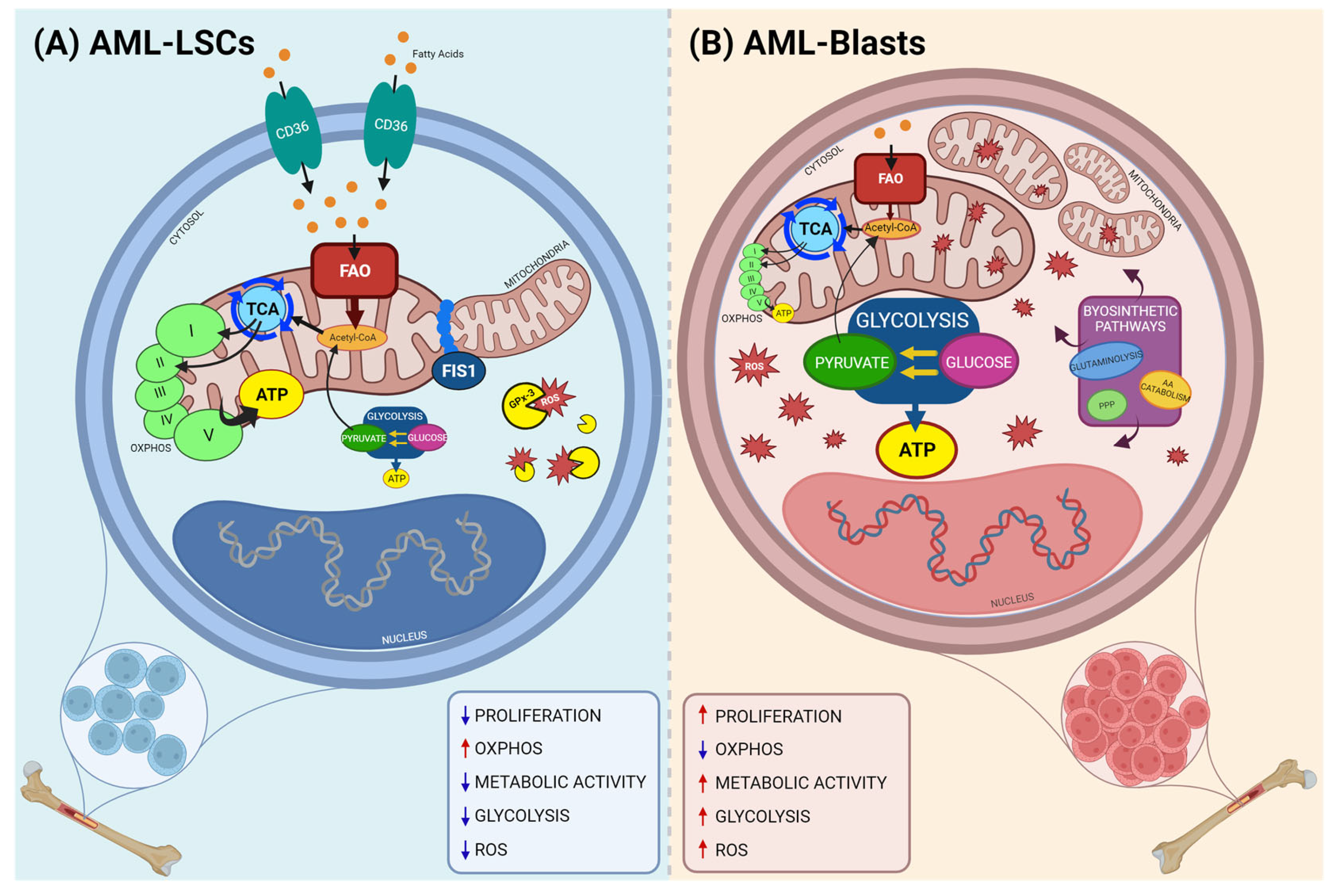
:1. Introduction
2. Altered Metabolic Processes in AML LSCs
3. Mitochondrial Targets and Specific Therapies in AML
3.1. Tricarboxylic Acid (TCA) Cycle Inhibition
3.2. Electron Transport Chain (ETC) Inhibition
3.3. Reactive Oxygen Species (ROS) Regulation
3.4. Amino Acid Metabolism Inhibition
3.5. Mitophagy Inhibition
4. Targeting Bcl-2 Proteins in AML
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- DiNardo, C.D.; Erba, H.P.; Freeman, S.D.; Wei, A.H. Acute myeloid leukaemia. Lancet 2023, 401, 2073–2086. [Google Scholar] [CrossRef]
- Juliusson, G.; Hagberg, O.; Lazarevic, V.L.; Ölander, E.; Antunovic, P.; Cammenga, J.; Wennström, L.; Möllgård, L.; Brune, M.; Jädersten, M.; et al. Improved survival of men 50 to 75 years old with acute myeloid leukemia over a 20-year period. Blood 2019, 134, 1558–1561. [Google Scholar] [CrossRef] [PubMed]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, K.; Maiti, A.; Konopleva, M. Targeting FLT3 Mutation in Acute Myeloid Leukemia: Current Strategies and Future Directions. Cancers 2023, 15, 2312. [Google Scholar] [CrossRef] [PubMed]
- Pommert, L.; Tarlock, K. The evolution of targeted therapy in pediatric AML: Gemtuzumab ozogamicin, FLT3/IDH/BCL2 inhibitors, and other therapies. Hematol. Am. Soc. Hematol. Educ. Program. 2022, 2022, 603–610. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Döhner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef]
- Todisco, E.; Papayannidis, C.; Fracchiolla, N.; Petracci, E.; Zingaretti, C.; Vetro, C.; Martelli, M.P.; Zappasodi, P.; Di Renzo, N.; Gallo, S.; et al. AVALON: The Italian cohort study on real-life efficacy of hypomethylating agents plus venetoclax in newly diagnosed or relapsed/refractory patients with acute myeloid leukemia. Cancer 2023, 129, 992–1004. [Google Scholar] [CrossRef]
- Moujalled, D.M.; Brown, F.C.; Chua, C.C.; Dengler, M.A.; Pomilio, G.; Anstee, N.S.; Litalien, V.; Thompson, E.; Morley, T.; MacRail, S.; et al. Acquired mutations in BAX confer resistance to BH3-mimetic therapy in acute myeloid leukemia. Blood 2023, 141, 634–644. [Google Scholar] [CrossRef]
- Sallman, D.A.; Al Malki, M.M.; Asch, A.S.; Wang, E.S.; Jurcic, J.G.; Bradley, T.J.; Flinn, I.W.; Pollyea, D.A.; Kambhampati, S.; Tanaka, T.N.; et al. Magrolimab in Combination with Azacitidine in Patients with Higher-Risk Myelodysplastic Syndromes: Final Results of a Phase Ib Study. J. Clin. Oncol. 2023, 41, 2815–2826. [Google Scholar] [CrossRef]
- Panina, S.B.; Pei, J.; Kirienko, N.V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 2021, 9, 17. [Google Scholar] [CrossRef]
- Liyanage, S.U.; Hurren, R.; Voisin, V.; Bridon, G.; Wang, X.; Xu, C.; MacLean, N.; Siriwardena, T.P.; Gronda, M.; Yehudai, D.; et al. Leveraging increased cytoplasmic nucleoside kinase activity to target mtDNA and oxidative phosphorylation in AML. Blood 2017, 129, 2657–2666. [Google Scholar] [CrossRef] [PubMed]
- Hanekamp, D.; Cloos, J.; Schuurhuis, G.J. Leukemic stem cells: Identification and clinical application. Int. J. Hematol. 2017, 105, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Culp-Hill, R.; D’Alessandro, A.; Pietras, E.M. Extinguishing the Embers: Targeting AML Metabolism. Trends Mol. Med. 2021, 27, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Glytsou, C.; Zhou, H.; Narang, S.; Reyna, D.E.; Lopez, A.; Sakellaropoulos, T.; Gong, Y.; Kloetgen, A.; Yap, Y.S.; et al. Targeting Mitochondrial Structure Sensitizes Acute Myeloid Leukemia to Venetoclax Treatment. Cancer Discov. 2019, 9, 890–909. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef]
- Wu, S.; Akhtari, M.; Alachkar, H. Characterization of Mutations in the Mitochondrial Encoded Electron Transport Chain Complexes in Acute Myeloid Leukemia. Sci. Rep. 2018, 8, 13301. [Google Scholar] [CrossRef]
- Baccelli, I.; Gareau, Y.; Lehnertz, B.; Gingras, S.; Spinella, J.F.; Corneau, S.; Mayotte, N.; Girard, S.; Frenchette, M.; Blouin-Chagnon, V.; et al. Mubritinib Targets the Electron Transport Chain Complex I and Reveals the Landscape of OXPHOS Dependency in Acute Myeloid Leukemia. Cancer Cell 2019, 36, 84–99.e8. [Google Scholar] [CrossRef]
- Pollyea, D.A.; Stevens, B.M.; Jones, C.L.; Winters, A.; Pei, S.; Minhajuddin, M.; D’Alessandro, A.; Culp-Hill, R.; Riemondy, K.A.; Gillen, A.E.; et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat. Med. 2018, 24, 1859–1866. [Google Scholar] [CrossRef]
- Zecchini, V.; Frezza, C. Metabolic synthetic lethality in cancer therapy. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 723–731. [Google Scholar] [CrossRef]
- Gaude, E.; Frezza, C. Defects in mitochondrial metabolism and cancer. Cancer Metab. 2014, 2, 10. [Google Scholar] [CrossRef]
- Castro, I.; Sampaio-Marques, B.; Ludovico, P. Targeting Metabolic Reprogramming in Acute Myeloid Leukemia. Cells 2019, 8, 967. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.L.; Wang, J.H.; Zhao, A.H.; Xu, X.; Wang, Y.H.; Chen, T.L.; Li, J.M.; Mi, J.Q.; Zhu, Y.M.; Liu, Y.F.; et al. A distinct glucose metabolism signature of acute myeloid leukemia with prognostic value. Blood 2014, 124, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Scadden, D.T. Targeting the Warburg effect for leukemia therapy: Magnitude matters. Mol. Cell Oncol. 2015, 2, e981988. [Google Scholar] [CrossRef]
- Pereira, O.; Teixeira, A.; Sampaio-Marques, B.; Castro, I.; Girão, H.; Ludovico, P. Signalling mechanisms that regulate metabolic profile and autophagy of acute myeloid leukaemia cells. J. Cell Mol. Med. 2018, 22, 4807–4817. [Google Scholar] [CrossRef] [PubMed]
- Poulain, L.; Sujobert, P.; Zylbersztejn, F.; Berreau, S.; Stuani, L.; Lambert, M.; Oalam, T.L.; Chsnais, V.; Birsen, R.; Vergez, F.; et al. High mTORC1 activity drives glycolysis addiction and sensitivity to G6PD inhibition in acute myeloid leukemia cells. Leukemia 2017, 31, 2326–2335. [Google Scholar] [CrossRef]
- Sánchez-Mendoza, S.E.; Rego, E.M. Targeting the mitochondria in acute myeloid leukemia. Appl. Cancer Res. 2017, 37, 22. [Google Scholar] [CrossRef]
- Fulda, S.; Galluzzi, L.; Kroemer, G. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 2010, 9, 447–464. [Google Scholar] [CrossRef]
- Larrue, C.; Saland, E.; Vergez, F.; Serham, N.; Delabesse, E.; Mansat-De Mas, V.; Hospital, M.A.; Tamburini, J.; Manetti, S.; Sarry, J.E.; et al. Antileukemic Activity of 2-Deoxy-d-Glucose through Inhibition of N-Linked Glycosylation in Acute Myeloid Leukemia with FLT3-ITD or c-KIT Mutations. Mol. Cancer Ther. 2015, 14, 2364–2373. [Google Scholar] [CrossRef]
- Samudio, I.; Harmancey, R.; Fiegl, M.; Kantarjian, H.; Konopleva, M.; Korchin, B.; Kaluarachchi, K.; Bornmann, W.; Duvvuri, S.; Taegtmeyer, H.; et al. Pharmacologic inhibition of fatty acid oxidation sensitizes human leukemia cells to apoptosis induction. J. Clin. Investig. 2010, 120, 142–156. [Google Scholar] [CrossRef]
- Kennedy, V.E.; Smith, C.C. FLT3 Mutations in Acute Myeloid Leukemia: Key Concepts and Emerging Controversies. Front. Oncol. 2020, 10, 612880. [Google Scholar] [CrossRef]
- Gregory, M.A.; D’Alessandro, A.; Alvarez-Calderon, F.; Kim, J.; Nemkov, T.; Adane, B.; Rozhok, A.I.; Kumar, A.; Kumar, V.; Pollyea, D.A.; et al. ATM/G6PD-driven redox metabolism promotes FLT3 inhibitor resistance in acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2016, 113, E6669–E6678. [Google Scholar] [CrossRef] [PubMed]
- Oran, B.; Weisdorf, D.J. Survival for older patients with acute myeloid leukemia: A population-based study. Haematologica 2012, 97, 1916–1924. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Peng, M.; Huang, Y.; Zhang, L.; Zhao, X.; Hou, Y. Targeting Mitochondrial Oxidative Phosphorylation Eradicates Acute Myeloid Leukemic Stem Cells. Front. Oncol. 2022, 12, 899502. [Google Scholar] [CrossRef] [PubMed]
- Raffel, S.; Klimmeck, D.; Falcone, M.; Demir, A.; Pouya, A.; Zeisberger, P.; Lutz, C.; Tinelli, M.; Bischel, O.; Bullinger, L.; et al. Quantitative proteomics reveals specific metabolic features of acute myeloid leukemia stem cells. Blood 2020, 136, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.L.; Stevens, B.M.; D’Alessandro, A.; Reisz, J.A.; Culp-Hill, R.; Nemkov, T.; Pei, S.; Khan, N.; Adane, B.; Te, H.; et al. Inhibition of Amino Acid Metabolism Selectively Targets Human Leukemia Stem Cells. Cancer Cell 2018, 34, 724–740.e4. [Google Scholar] [CrossRef]
- De Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial metabolism as a potential therapeutic target in myeloid leukaemia. Leukemia 2022, 36, 1–12. [Google Scholar] [CrossRef]
- Ye, H.; Adane, B.; Khan, N.; Sullivan, T.; Minhajuddin, M.; Gasparetto, M.; Stevens, B.; Pei, S.; Balys, M.; Ashton, J.M.; et al. Leukemic Stem Cells Evade Chemotherapy by Metabolic Adaptation to an Adipose Tissue Niche. Cell Stem Cell 2016, 19, 23–37. [Google Scholar] [CrossRef]
- Shafat, M.S.; Oellerich, T.; Mohr, S.; Robinson, S.D.; Edwards, D.R.; Marlein, C.R.; Piddock, R.E.; Fenech, M.; Zaitseva, L.; Abdul-Aziz, A.; et al. Leukemic blasts program bone marrow adipocytes to generate a protumoral microenvironment. Blood 2017, 129, 1320–1332. [Google Scholar] [CrossRef]
- Skrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell 2011, 20, 674–688. [Google Scholar] [CrossRef]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Pei, S.; Minhajuddin, M.; Adane, B.; Khan, N.; Stevens, B.M.; Mack, S.C.; Lai, S.; Rich, J.N.; Inguva, A.; Shannon, K.M.; et al. AMPK/FIS1-Mediated Mitophagy Is Required for Self-Renewal of Human AML Stem Cells. Cell Stem Cell 2018, 23, 86–100.e6. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed]
- Sillar, J.R.; Germon, Z.P.; DeIuliis, G.N.; Dun, M.D. The Role of Reactive Oxygen Species in Acute Myeloid Leukaemia. Int. J. Mol. Sci. 2019, 20, 6003. [Google Scholar] [CrossRef]
- Herault, O.; Hope, K.J.; Deneault, E.; Mayotte, N.; Changraoui, J.; Wilhelm, B.T.; Cellot, S.; Sauvageau, M.; Andrade-Navarro, M.A.; Hébert, J.; et al. A role for GPx3 in activity of normal and leukemia stem cells. J. Exp. Med. 2012, 209, 895–901. [Google Scholar] [CrossRef]
- Guna, A.; Stevens, T.A.; Inglis, A.J.; Replogle, J.M.; Esantsi, T.K.; Muthukumar, G.; Shaffer, K.C.L.; Wang, M.L.; Pogson, A.G.; Jones, J.J.; et al. MTCH2 is a mitochondrial outer membrane protein insertase. Science 2022, 378, 317–322. [Google Scholar] [CrossRef]
- Khan, D.H.; Mullokandov, M.; Wu, Y.; Gronda, M.; Hurren, R.; Wang, X.; MacLean, N.; Jeyaraju, D.V.; Wei, G.; Laister, R.C.; et al. Mitochondrial Carrier Homolog 2 (MTCH2) regulates the differentiation of AML cells by controlling pyruvate enry into the mitochondria, nuclear localization of pyruvate dehydrogenase complex and H3 and H4 histone acetylation. Blood 2017, 130, 299. [Google Scholar] [CrossRef]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Wirawan, E.; Vanden Berghe, T.; Lippens, S.; Agostinis, P.; Vandenabeele, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef]
- Choubey, V.; Zeb, A.; Kaasik, A. Molecular Mechanisms and Regulation of Mammalian Mitophagy. Cells 2021, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Zhu, P.; Huang, R.; Wang, C.; Sun, L.; Lan, B.; He, Y.; Zhao, H.; Gao, Y. PINK1: The guard of mitochondria. Life Sci. 2020, 259, 118247. [Google Scholar] [CrossRef]
- Gao, A.; Jiang, J.; Xie, F.; Chen, L. Bnip3 in mitophagy: Novel insights and potential therapeutic target for diseases of secondary mitochondrial dysfunction. Clin. Chim. Acta 2020, 506, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Bhujabal, Z.; Birgisdottir, Å.B.; Sjøttem, E.; Brenne, H.B.; Øvervatn, A.; Habisov, S.; Kirkin, V.; Lamark, T.; Johansen, T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep. 2017, 18, 947–961. [Google Scholar] [CrossRef] [PubMed]
- Maynard, R.S.; Hellmich, C.; Bowles, K.M.; Rushworth, S.A. Acute Myeloid Leukaemia Drives Metabolic Changes in the Bone Marrow Niche. Front. Oncol. 2022, 12, 924567. [Google Scholar] [CrossRef]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennet, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef]
- Rakheja, D.; Medeiros, L.J.; Bevan, S.; Chen, W. The emerging role of d-2-hydroxyglutarate as an oncometabolite in hematolymphoid and central nervous system neoplasms. Front. Oncol. 2013, 3, 169. [Google Scholar] [CrossRef]
- Andreozzi, F.; Massaro, F.; Wittnebel, S.; Spilleboudt, C.; Lewalle, P.; Salaroli, A. New Perspectives in Treating Acute Myeloid Leukemia: Driving towards a Patient-Tailored Strategy. Int. J. Mol. Sci. 2022, 23, 3887. [Google Scholar] [CrossRef]
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 22. [Google Scholar] [CrossRef]
- Perini, G.F.; Ribeiro, G.N.; Pinto Neto, J.V.; Campos, L.T.; Hamerschlak, N. BCL-2 as therapeutic target for hematological malignancies. J. Hematol. Oncol. 2018, 11, 65. [Google Scholar] [CrossRef] [PubMed]
- Prieto-Bermejo, R.; Romo-González, M.; Pérez-Fernández, A.; Ijurko, C.; Hernández-Hernández, Á. Reactive oxygen species in haematopoiesis: Leukaemic cells take a walk on the wild side. J. Exp. Clin. Cancer Res. 2018, 37, 125. [Google Scholar] [CrossRef] [PubMed]
- Hole, P.S.; Zabkiewicz, J.; Munje, C.; Newton, Z.; Pearn, L.; White, P.; Marquez, N.; Hills, R.K.; Burnett, A.K.; Tonks, A.; et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood 2013, 122, 3322–3330. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Tonacci, A.; Giordano, L.; Musolino, C.; Gangemi, S. Targeting Redox Regulation as a Therapeutic Opportunity against Acute Leukemia: Pro-Oxidant Strategy or Antioxidant Approach? Antioxidants 2022, 11, 1696. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.H.; Hu, J.; Lo-Coco, F.; Jin, J. The simpler, the better: Oral arsenic for acute promyelocytic leukemia. Blood 2019, 134, 597–605. [Google Scholar] [CrossRef]
- Kumana, C.R.; Mak, R.; Kwong, Y.L.; Gill, H. Resurrection of Oral Arsenic Trioxide for Treating Acute Promyelocytic Leukaemia: A Historical Account from Bedside to Bench to Bedside. Front. Oncol. 2020, 10, 1294. [Google Scholar] [CrossRef]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef]
- Kumar, S.; Yedjou, C.G.; Tchounwou, P.B. Arsenic trioxide induces oxidative stress, DNA damage, and mitochondrial pathway of apoptosis in human leukemia (HL-60) cells. J. Exp. Clin. Cancer Res. 2014, 33, 42. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lopez, M.J.; Mohiuddin, S.S. Biochemistry, Essential Amino Acids; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Zhou, X.; Zheng, M.; Wang, Q.; Aa, J.; Cao, B.; Li, J. Metabolomics analysis identifies lysine and taurine as candidate prognostic biomarkers for AML-M2 patients. Int. J. Hematol. 2020, 111, 761–770. [Google Scholar] [CrossRef]
- Markham, G.D.; Pajares, M.A. Structure-function relationships in methionine adenosyltransferases. Cell Mol. Life Sci. 2009, 66, 636–648. [Google Scholar] [CrossRef]
- Taylor, A.V.; Adao, R.R.; Castro, C.; Domingues, A.F.; Griffin, J.L.; Curti, A.; Pina, C. S-Adenosyl Methionine Synthesis Impacts Maintenance of Acute Myeloid Leukemia Cells with Regulation of Transcriptional Elongation. Blood 2018, 132, 2605. [Google Scholar] [CrossRef]
- Secker, K.A.; Bloechl, B.; Keppeler, H.; Duerr-Stoerzer, S.; Schmid, H.; Schneidawind, D.; Jeong, J.; Hentrich, T.; Schulze-Hentrich, J.M.; Scheidawind, C. MAT2A as Key Regulator and Therapeutic Target in MLLr Leukemogenesis. Cancers 2020, 12, 1342. [Google Scholar] [CrossRef]
- Dhir, A.; Paterson, A.J.; Qiu, S.; Mullen, A.K.; Anderson, N.R.; Bhatia, R. An Epigenetic Screen Identifies Prmt5 as a Target for Inhibition of FLT3-ITD Aml Cell Growth in Combination with Tyrosine Kinase Inhibitors. Blood 2019, 134, 2524. [Google Scholar] [CrossRef]
- Barve, A.; Vega, A.; Shah, P.P.; Ghare, S.; Casson, L.; Wunderlich, M.; Siskind, L.J.; Beverly, L.J. Perturbation of Methionine/S-adenosylmethionine Metabolism as a Novel Vulnerability in MLL Rearranged Leukemia. Cells 2019, 8, 1322. [Google Scholar] [CrossRef]
- Fultang, L.; Gneo, L.; De Santo, C.; Mussai, F.J. Targeting Amino Acid Metabolic Vulnerabilities in Myeloid Malignancies. Front. Oncol. 2021, 11, 674720. [Google Scholar] [CrossRef]
- Finger, E.; Wong, J.; Gray, J.; Rock, D.; Loberg, R.; Fitzpatrick, D.; Smith, R.; Wang, X.; Dos Santos, C.E. Utilization of Metabolomics to Identify Biomarkers in Hematological Malignancies: Role of IDO and the Tryptophan Pathway. Blood 2017, 130 (Suppl. S1), 5100. [Google Scholar] [CrossRef]
- Fox, E.; Oliver, T.; Rowe, M.; Thomas, S.; Zakharia, Y.; Gilman, P.B.; Muller, A.J.; Prendergast, G.C. Indoximod: An Immunometabolic Adjuvant That Empowers T Cell Activity in Cancer. Front. Oncol. 2018, 8, 370. [Google Scholar] [CrossRef]
- Zhou, X.; Cao, B.; Li, J. Targeting Amino Acids to Treat AML. J. Clin. Haematol. 2020, 1, 1–6. [Google Scholar] [CrossRef]
- Matre, P.; Velez, J.; Jacamo, R.; Qi, Y.; Su, X.; Cai, T.; Chan, S.M.; Lodi, A.; Sweeney, S.R.; Ma, H.; et al. Inhibiting glutaminase in acute myeloid leukemia: Metabolic dependency of selected AML subtypes. Oncotarget 2016, 7, 79722–79735. [Google Scholar] [CrossRef]
- Gregory, M.A.; Nemkov, T.; Park, H.J.; Zaberezhnyy, V.; Gehrke, S.; Adane, B.; Jordan, C.t.; Hansen, K.C.; D’Alessandro, A.; DeGregori, J. Targeting Glutamine Metabolism and Redox State for Leukemia Therapy. Clin. Cancer Res. 2019, 25, 4079–4090. [Google Scholar] [CrossRef] [PubMed]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, P.C.; Ochoa, A.C.; Al-Khami, A.A. Arginine Metabolism in Myeloid Cells Shapes Innate and Adaptive Immunity. Front. Immunol. 2017, 8, 93. [Google Scholar] [CrossRef]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, Y.; Li, B.; Shen, K.; Li, Q.; Ni, Y.; Huang, L. Mitophagy in carcinogenesis, drug resistance and anticancer therapeutics. Cancer Cell Int. 2021, 21, 350. [Google Scholar] [CrossRef]
- Kong, Y.L.; Huang, Y.; Wu, J.Z.; Cao, X.; Liang, J.H.; Xia, Y.; Wu, W.; Cao, L.; Zhu, H.Y.; Wang, L.; et al. Expression of autophagy related genes in chronic lymphocytic leukemia is associated with disease course. Leuk. Res. 2018, 66, 8–14. [Google Scholar] [CrossRef]
- Seo, W.; Silwal, P.; Song, I.C.; Jo, E.K. The dual role of autophagy in acute myeloid leukemia. J. Hematol. Oncol. 2022, 15, 51. [Google Scholar] [CrossRef]
- Du, W.; Xu, A.; Huang, Y.; Cao, J.; Zhu, H.; Yang, B.; Shao, X.; He, Q.; Ying, M. The role of autophagy in targeted therapy for acute myeloid leukemia. Autophagy 2021, 17, 2665–2679. [Google Scholar] [CrossRef]
- Fay, H.R.S.; Dykstra, K.M.; Johnson, M.; Cronin, T.L.; Lutgen-Dunckley, L.; Martens, B.L.; Moberg, J.R.; Guzman, M.L.; Wang, E. Mitophagy Plays a Key Role in the Anti-Leukemic Activity of Autophagy Inhibitors Under Hypoxia in Acute Myeloid Leukemia. Blood 2019, 134 (Suppl. S1), 1278. [Google Scholar] [CrossRef]
- Dykstra, K.M.; Fay, H.R.S.; Massey, A.C.; Yang, N.; Johnson, M.; Portwood, S.; Guzman, M.L.; Wang, E.S. Inhibiting autophagy targets human leukemic stem cells and hypoxic AML blasts by disrupting mitochondrial homeostasis. Blood Adv. 2021, 5, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.; Yin, J.; Wang, C.; Yang, M.; Gu, J.; He, M.; Xu, H.; Fu, W.; Zhang, W.; et al. A mitophagy inhibitor targeting p62 attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Lett. 2021, 510, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Grønningsæter, I.S.; Reikvam, H.; Aasebø, E.; Bartaula-Brevik, S.; Hernandez-Valladares, M.; Selheim, F.; Berven, F.S.; Tvedt, T.H.; Bruserud, Ø.; Hatfield, K.J. Effects of the Autophagy-Inhibiting Agent Chloroquine on Acute Myeloid Leukemia Cells; Characterization of Patient Heterogeneity. J. Pers. Med. 2021, 11, 779. [Google Scholar] [CrossRef] [PubMed]
- Karami, H.; Baradaran, B.; Esfahani, A.; Sakhinia, M.; Sakhinia, E. Therapeutic Effects of Myeloid Cell Leukemia-1 siRNA on Human Acute Myeloid Leukemia Cells. Adv. Pharm. Bull. 2014, 4, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Sato, K.; Uchida, Y.; Kato, N.; Aikawa, A.; Ogata, K.; Kamimura, H.; Uemura, H.; Yoshimitsu, M.; Ishitsuka, K.; et al. The small molecule STF-62247 induces apoptotic and autophagic cell death in leukemic cells. Oncotarget 2018, 9, 27645–27655. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, L.; Shen, K.; Jin, H.; Li, H.; Cheng, Y.; Ma, X. Efficacy and Safety of Bcl-2 Inhibitor Venetoclax in Hematological Malignancy: A Systematic Review and Meta-Analysis of Clinical Trials. Front. Pharmacol. 2019, 10, 697. [Google Scholar] [CrossRef]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Stojanov, P.; McKenna, A.; Stevenson, K.; Lawrence, M.S.; Sougnez, C.; Stewart, C.; Sivachenko, A.; Wang, L.; et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 2013, 152, 714–726. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Rodriguez-Blanco, G.; Sumpton, D.; Cloix, C.; Mullin, M.; Mackay, G.M.; O’Neill, K.; Lemgruber, L.; Luo, X.; Tait, S.W.G. Venetoclax causes metabolic reprogramming independent of BCL-2 inhibition. Cell Death Dis. 2020, 11, 616. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.R.; Yunis, J.; Nowell, P.C.; Croce, C.M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Zhou, J.D.; Zhang, T.J.; Xu, Z.J.; Gu, Y.; Ma, J.C.; Li, X.X.; Guo, H.; Wen, X.M.; Zhang, W.; Yang, L.; et al. BCL2 overexpression: Clinical implication and biological insights in acute myeloid leukemia. Diagn. Pathol. 2019, 14, 68. [Google Scholar] [CrossRef] [PubMed]
- Stilgenbauer, S.; Eichhorst, B.; Schetelig, J.; Coutre, S.; Seymour, S.F.; Munir, T.; Puvvada, S.D.; Wendtner, C.M.; Roberts, A.W.; Jurczak, W.; et al. Venetoclax in relapsed or refractory chronic lymphocytic leukaemia with 17p deletion: A multicentre, open-label, phase 2 study. Lancet Oncol. 2016, 17, 768–778. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.F.; Kipps, T.J.; Eichhorst, B.; Hillmen, P.; D’Rozario, J.; Assouline, S.; Owen, C.; Gerecitano, J.; Robak, T.; De la Serna, J.; et al. Venetoclax-Rituximab in Relapsed or Refractory Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2018, 378, 1107–1120. [Google Scholar] [CrossRef] [PubMed]
- Tiribelli, M.; Michelutti, A.; Cavallin, M.; Di Giusto, S.; Simeone, E.; Fanin, R.; Damiani, D. BCL-2 Expression in AML Patients over 65 Years: Impact on Outcomes across Different Therapeutic Strategies. J. Clin. Med. 2021, 10, 5096. [Google Scholar] [CrossRef]
- Tzifi, F.; Economopoulou, C.; Gourgiotis, D.; Ardavanis, A.; Papageorgiou, S.; Scorilas, A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv. Hematol. 2012, 2012, 524308. [Google Scholar] [CrossRef]
- Andreeff, M.; Jiang, S.; Zhang, X.; Konopleva, M.; Estrov, Z.; Snell, V.E.; Xie, Z.; Okcu, M.F.; Sanchez-Williams, G.; Dong, J.; et al. Expression of Bcl-2-related genes in normal and AML progenitors: Changes induced by chemotherapy and retinoic acid. Leukemia 1999, 13, 1881–1892. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Pratz, K.; Pullarkat, V.; Jonas, B.A.; Arellano, M.; Becker, P.S.; Frankfurt, O.; Konopleva, M.; Wei, A.H.; Kantarjian, H.M.; et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019, 133, 7–17. [Google Scholar] [CrossRef]
- Brancati, S.; Gozzo, L.; Romano, G.L.; Vetro, C.; Dulcamare, I.; Maugeri, C.; Parisi, M.; Longo, L.; Vitale, D.C.; Di Raimondo, F.; et al. Venetoclax in Relapsed/Refractory Acute Myeloid Leukemia: Are Supporting Evidences Enough? Cancers 2021, 14, 22. [Google Scholar] [CrossRef]
- Tenold, M.E.; Moskoff, B.N.; Benjamin, D.J.; Hoeg, R.T.; Rosenberg, A.S.; Abedi, M.; Tuscano, J.M.; Jonas, B.A. Outcomes of Adults with Relapsed/Refractory Acute Myeloid Leukemia Treated with Venetoclax Plus Hypomethylating Agents at a Comprehensive Cancer Center. Front. Oncol. 2021, 11, 649209. [Google Scholar] [CrossRef]
- Van Gils, N.; Denkers, F.; Smit, L. Escape from Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia. Front. Oncol. 2021, 11, 659253. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Maiti, A.; Loghavi, S.; Pourebrahim, R.; Kadia, T.M.; Rausch, C.R.; Furudate, K.; Daver, N.G.; Alvarado, E.; Ohanian, M.; et al. Outcomes of TP53-mutant acute myeloid leukemia with decitabine and venetoclax. Cancer 2021, 127, 3772–3781. [Google Scholar] [CrossRef] [PubMed]
- Singh Mali, R.; Zhang, Q.; DeFilippis, R.A.; Cavazos, A.; Kuruvilla, V.M.; Raman, J.; Mody, V.; Choo, E.F.; Dail, M.; Shah, N.P.; et al. Venetoclax combines synergistically with FLT3 inhibition to effectively target leukemic cells in FLT3-ITD+ acute myeloid leukemia models. Haematologica 2021, 106, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Singh Mali, R.; Lasater, E.A.; Dyle, K.; Malla, R.; Boghaert, E.; Souers, A.; Leverson, J.D.; Sampath, D. FLT3-ITD Activation Mediates Resistance to the BCL-2 Selective Antagonist, Venetoclax, in FLT3-ITD Mutant AML Models. Blood 2017, 130, 1348. [Google Scholar] [CrossRef]
- Zhang, Q.; Riley-Gillis, B.; Han, L.; Jia, Y.; Lodi, A.; Zhang, H.; Ganesan, S.; Pan, R.; Konoplev, S.N.; Sweeney, S.R.; et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct. Target. Ther. 2022, 7, 51. [Google Scholar] [CrossRef]
- Tausch, E.; Close, W.; Dolnik, A.; Bloehdorn, J.; Chyla, B.; Bullinger, L.; Döhner, H.; Mertens, D.; Stilgenbauer, S. Venetoclax resistance and acquired BCL2 mutations in chronic lymphocytic leukemia. Haematologica 2019, 104, e434–e437. [Google Scholar] [CrossRef]
- Punnoose, E.A.; Leverson, J.D.; Peale, F.; Boghaert, E.R.; Belmont, L.D.; Tan, N.; Young, A.; Mitten, M.; Ingalla, E.; Darbonne, W.C.; et al. Expression Profile of BCL-2, BCL-XL, and MCL-1 Predicts Pharmacological Response to the BCL-2 Selective Antagonist Venetoclax in Multiple Myeloma Models. Mol. Cancer Ther. 2016, 15, 1132–1144. [Google Scholar] [CrossRef]
- Przedborski, M.; Sharon, D.; Cathelin, S.; Chan, S.; Kohandel, M. An integrative systems biology approach to overcome venetoclax resistance in acute myeloid leukemia. PLoS Comput. Biol. 2022, 18, e1010439. [Google Scholar] [CrossRef]
- Guièze, R.; Liu, V.M.; Rosebrock, D.; Jourdain, A.A.; Hernandez-Sanchez, M.; Martinez-Zurita, A.; Sun, J.; Ten Hacken, E.; Baranowski, K.; Thompson, P.A.; et al. Mitochondrial Reprogramming Underlies Resistance to BCL-2 Inhibition in Lymphoid Malignancies. Cancer Cell 2019, 36, 369–384.e13. [Google Scholar] [CrossRef]
- Bolomsky, A.; Vogler, M.; Köse, M.C.; Heckman, C.A.; Ehx, G.; Ludwig, H.; Caers, J. MCL-1 inhibitors, fast-lane development of a new class of anti-cancer agents. J. Hematol. Oncol. 2020, 13, 173. [Google Scholar] [CrossRef]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, H.E.; Fischer, M.A.; Lee, T.; Gorska, A.E.; Arrate, M.P.; Fuller, L.; Boyd, K.L.; Strickland, S.A.; Sensintaffar, J.; Hogdal, L.J.; et al. A Novel MCL1 Inhibitor Combined with Venetoclax Rescues Venetoclax-Resistant Acute Myelogenous Leukemia. Cancer Discov. 2018, 8, 1566–1581. [Google Scholar] [CrossRef] [PubMed]
- Gavriatopoulou, M.; Paschou, S.A.; Ntanasis-Stathopoulos, I.; Dimopoulos, M.A. Metabolic Disorders in Multiple Myeloma. Int. J. Mol. Sci. 2021, 22, 11430. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Y.; Yu, L.; Yang, L. Mechanisms of venetoclax resistance and solutions. Front. Oncol. 2022, 12, 1005659. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Name | Code | Mechanism | Status |
|---|---|---|---|
| Arsenic Trioxide + All-trans Retinoic Acid | ATO/ATRA | Stimulation of promyelocytic cells differentiation | FDA approved |
| Bafilomycin A1 (Baf A1) | CAS 88899-55-2 | Inhibition of autophagic flux | FDA approved |
| Chloroquine (CQ) | P01BA01 | Inhibition of autophagosome-lysosome fusion | FDA approved |
| Chloroquine + Venetoclax | Induction of cancer cell death | Preclinical studies | |
| DZ2002 | DZ2002 | SAHH inhibitor | |
| Enasidenib | AG-221 | IDH2 inhibitor | FDA approved |
| Hydroxychloroquine + Azacytidine | NCT01682516 | DNA methyltransferase inhibition and autophagy inhibition | Phase I/II clinical trial |
| Indoximod | NLG-8189 | IDO inhibitor | |
| Indoximod + Idarubicin + Cytarabine | NCT02835729 | IDO inhibition and DNA topoisomerase and DNA polymerase inhibition | Phase I clinical trial |
| Ivosidenib | AG-120 | IDH1 inhibitor | FDA approved |
| Linrodostat | BMS-986205 | IDO1 inhibitor | Phase III clinical trial |
| Linrodostat + Nivulimab | NCT02658890 | IDO inhibition | Phase I/II clinical trial |
| LLY-283 | LLY-283 | PRMT5 inhibitor | Preclinical studies |
| Mubritinib | TAK-165 | HER2 inhibitor | FDA approved |
| Numidargistat | CB-1158 (NCT02903914) | Arginase inhibitor | Phase I clinical trial |
| PF-9366 | PF-9366 | Mat2A inhibitor | Preclinical studies |
| Quizartinib | AC-220 | FLT3 inhibitor | Under approval |
| Spautin-1 | Spautin-1 | Autophagy inhibitor | Phase I/II clinical trial |
| Telaglenastat | CB-839 | GLSs inhibitor | |
| Venetoclax | L01XX52 | Bcl-2 inhibitor | FDA approved |
| Venetoclax + Azacytydine | NCT02993523 | Inhibition of SDH glutathionylation | Phase III clinical trial |
| XRK3F2 | XRK3F2 | P62–ZZ inhibitor | Preclinical studies |
| Zalcitabine | ddC | Mitochondrial DNA polymerase inhibitor | FDA approved |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maffeo, B.; Panuzzo, C.; Moraca, A.; Cilloni, D. A Leukemic Target with a Thousand Faces: The Mitochondria. Int. J. Mol. Sci. 2023, 24, 13069. https://doi.org/10.3390/ijms241713069
Maffeo B, Panuzzo C, Moraca A, Cilloni D. A Leukemic Target with a Thousand Faces: The Mitochondria. International Journal of Molecular Sciences. 2023; 24(17):13069. https://doi.org/10.3390/ijms241713069
Chicago/Turabian StyleMaffeo, Beatrice, Cristina Panuzzo, Amedeo Moraca, and Daniela Cilloni. 2023. "A Leukemic Target with a Thousand Faces: The Mitochondria" International Journal of Molecular Sciences 24, no. 17: 13069. https://doi.org/10.3390/ijms241713069