
A Risk Model for Prognosis and Treatment Response Prediction in Colon Adenocarcinoma Based on Genes Associated with the Characteristics of the Epithelial-Mesenchymal Transition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
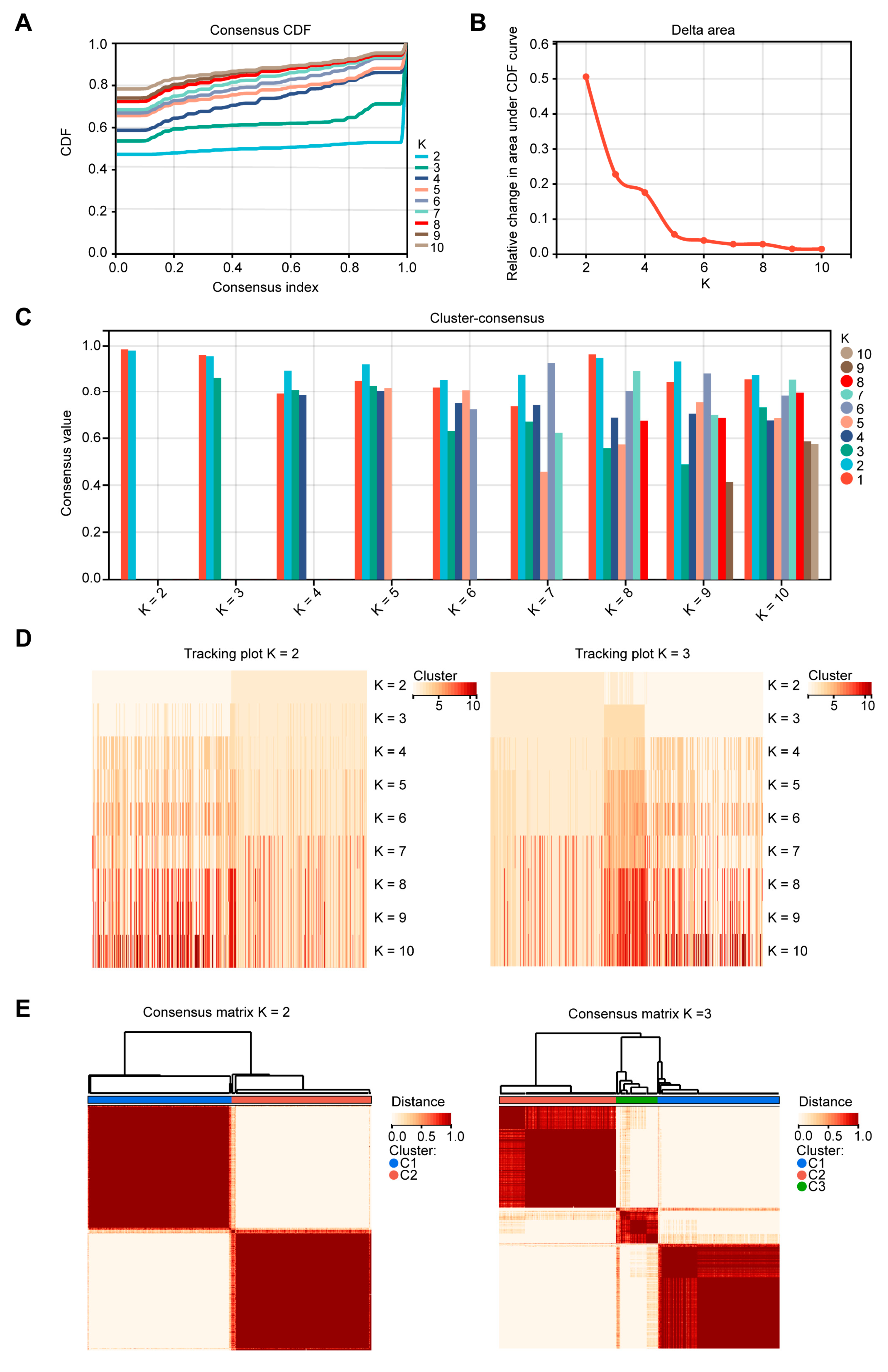
2.1. Patients in Different EMT Subtypes Showed Different Prognoses and EMT Characteristics
2.2. Independent Analysis to Verify the Function of the Three EMT Clusters
2.3. Identification of Modules and Hub Genes Associated with EMT Cluster 2 by WGCNA
2.4. GO and KEGG Analyses of Selected Module Genes and PPI Network Construction
2.5. Construction of the Prognostic Risk Score Model Based on the Hub Genes
2.6. Further Validation of the Prognostic Value of the Risk Score Model
2.7. Immune Infiltrations and Mutation Landscape in the Two Risk Groups
2.8. Risk Model Prediction of Drug Sensitivity and Immunotherapy Response
2.9. GSEA Analysis of Gene Sets Enriched in High- and Low-Risk Groups
3. Discussion
4. Materials and Methods
4.1. Data Collection and Processing
4.2. EMT Signatures Acquisition and Consensus Clustering to Classify COAD Samples into EMT Subtypes
4.3. EMT Marker Collection and Consensus Clustering to Verify the Clustering of 26 EMT Gene Sets
4.4. Construction of a Gene Co-Expression Network by WGCNA
4.5. GO Analysis, KEGG Analysis, and PPI Network Construction
4.6. Prognostic Model Construction Based on EMT Subtype-Associated Hub Genes
4.7. Validation of the Prognostic Model
4.8. TME Analysis
4.9. Gene Mutation Analysis
4.10. Drug Sensitivity Predictions
4.11. Gene Set Enrichment Analysis (GSEA)
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Harilal, S.; Carradori, S.; Mathew, B. A Comprehensive Overview of Colon Cancer—A Grim Reaper of the 21st Century. Curr. Med. Chem. 2021, 28, 2657–2696. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Feng, M.; Bai, L.; Liao, W.; Zhou, K.; Zhang, M.; Wu, Q.; Wen, F.; Lei, W.; Zhang, P.; et al. Comprehensive analysis of EMT-related genes and lncRNAs in the prognosis, immunity, and drug treatment of colorectal cancer. J. Transl. Med. 2021, 19, 391. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Antin, P.; Berx, G.; Blanpain, C.; Brabletz, T.; Bronner, M.; Campbell, K.; Cano, A.; Casanova, J.; Christofori, G.; et al. Guidelines and definitions for research on epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2020, 21, 341–352. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Chanvorachote, P.; Petsri, K.; Thongsom, S. Epithelial to Mesenchymal Transition in Lung Cancer: Potential EMT-Targeting Natural Product-derived Compounds. Anticancer Res. 2022, 42, 4237–4246. [Google Scholar] [CrossRef]
- Wang, L.; Saci, A.; Szabo, P.M.; Chasalow, S.D.; Castillo-Martin, M.; Domingo-Domenech, J.; Siefker-Radtke, A.; Sharma, P.; Sfakianos, J.P.; Gong, Y.; et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat. Commun. 2018, 9, 3503. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Cho, E.S.; Kang, H.E.; Kim, N.H.; Yook, J.I. Therapeutic implications of cancer epithelial-mesenchymal transition (EMT). Arch. Pharm. Res. 2019, 42, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, R.H.; Swier, N.; van Beijnum, J.R.; Nowak-Sliwinska, P. Colorectal Cancer Growth Retardation through Induction of Apoptosis, Using an Optimized Synergistic Cocktail of Axitinib, Erlotinib, and Dasatinib. Cancers 2019, 11, 1878, Erratum in Cancers 2020, 12, 1878. [Google Scholar] [CrossRef]
- Fornasier, G.; Francescon, S.; Baldo, P. An Update of Efficacy and Safety of Cetuximab in Metastatic Colorectal Cancer: A Narrative Review. Adv. Ther. 2018, 35, 1497–1509. [Google Scholar] [CrossRef] [PubMed]
- Taminau, J.; Meganck, S.; Lazar, C.; Steenhoff, D.; Coletta, A.; Molter, C.; Duque, R.; Schaetzen, V.d.; Weiss Solís, D.Y.; Bersini, H.; et al. Unlocking the potential of publicly available microarray data using inSilicoDb and inSilicoMerging R/Bioconductor packages. BMC Bioinform. 2012, 13, 335. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.E.; Li, C.; Rabinovic, A. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics 2007, 8, 118–127. [Google Scholar] [CrossRef]
- Hänzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef]
- Wilkerson, M.D.; Hayes, D.N. ConsensusClusterPlus: A class discovery tool with confidence assessments and item tracking. Bioinformatics 2010, 26, 1572–1573. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; den Hollander, P.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Ay, F.; Kellis, M.; Kahveci, T. SubMAP: Aligning metabolic pathways with subnetwork mappings. J. Comput. Biol. 2011, 18, 219–235. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [PubMed]
- Therneau, T.M.; Grambsch, P.M. The cox model. In Modeling Survival Data: Extending the Cox Model; Springer: Berlin/Heidelberg, Germany, 2000; pp. 39–77. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves using ‘ggplot2’. In R Package, version 0.3; The R Foundation: Ames, IA, USA, 2017. [Google Scholar]
- Wickham, H. Data analysis. In ggplot2; Springer: Berlin/Heidelberg, Germany, 2016; pp. 189–201. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, K.; Shahmoradgoli, M.; Martínez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Maeser, D.; Gruener, R.F.; Huang, R.S. oncoPredict: An R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 2021, 22, bbab260. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.J.; Huang, R.S. Clinical drug response can be predicted using baseline gene expression levels and in vitro drug sensitivity in cell lines. Genome Biol. 2014, 15, R47. [Google Scholar] [CrossRef]
- Geeleher, P.; Cox, N.; Huang, R.S. pRRophetic: An R package for prediction of clinical chemotherapeutic response from tumor gene expression levels. PLoS ONE 2014, 9, e107468. [Google Scholar] [CrossRef]
- Jiang, P.; Gu, S.; Pan, D.; Fu, J.; Sahu, A.; Hu, X.; Li, Z.; Traugh, N.; Bu, X.; Li, B.; et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018, 24, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, H.; Li, T.; Meng, Z.; Zhang, X.; Jiang, S.; Suo, M.; Li, N. A Risk Model for Prognosis and Treatment Response Prediction in Colon Adenocarcinoma Based on Genes Associated with the Characteristics of the Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2023, 24, 13206. https://doi.org/10.3390/ijms241713206
Huang H, Li T, Meng Z, Zhang X, Jiang S, Suo M, Li N. A Risk Model for Prognosis and Treatment Response Prediction in Colon Adenocarcinoma Based on Genes Associated with the Characteristics of the Epithelial-Mesenchymal Transition. International Journal of Molecular Sciences. 2023; 24(17):13206. https://doi.org/10.3390/ijms241713206
Chicago/Turabian StyleHuang, Hongyu, Tianyou Li, Ziqi Meng, Xueqian Zhang, Shanshan Jiang, Mengying Suo, and Na Li. 2023. "A Risk Model for Prognosis and Treatment Response Prediction in Colon Adenocarcinoma Based on Genes Associated with the Characteristics of the Epithelial-Mesenchymal Transition" International Journal of Molecular Sciences 24, no. 17: 13206. https://doi.org/10.3390/ijms241713206