

Using cfDNA and ctDNA as Oncologic Markers: A Path to Clinical Validation
 ,
, 
Abstract
:1. Introduction
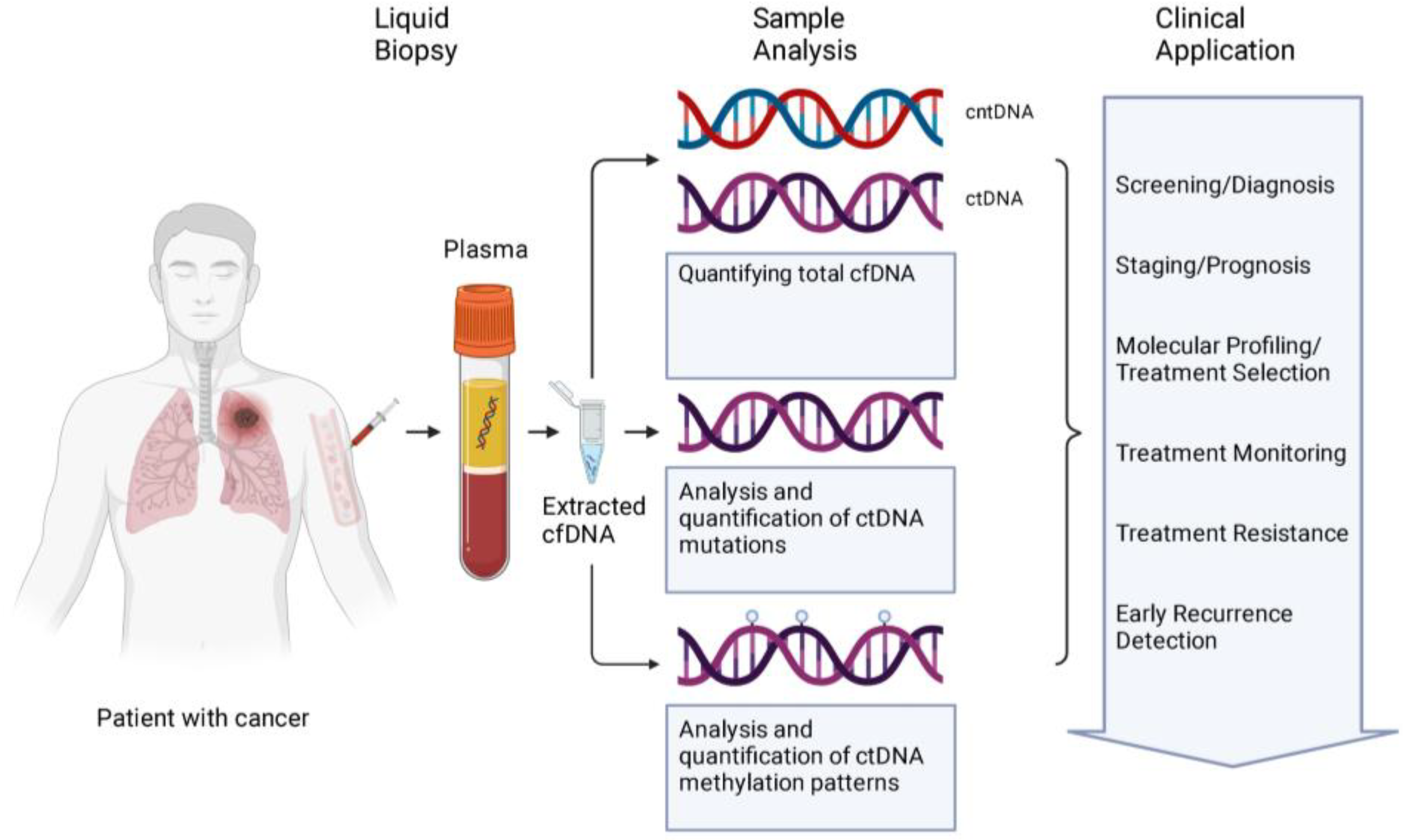
2. Circulating Free DNA
3. Total cfDNA Concentration
4. ctDNA
4.1. Techniques
4.2. Early Detection/Screening
4.3. Treatment Selection/Companion Diagnostics
4.3.1. BRCA
4.3.2. EGFR
4.3.3. Other
4.4. Minimal Residual Disease
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cancer. World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cancer (accessed on 15 March 2023).
- Incidence and Relative Survival by Stage at Diagnosis for Common Cancers. Centers for Disease Control and Prevention. Published 10 November 2021. Available online: https://www.cdc.gov/cancer/uscs/about/data-briefs/no25-incidence-relative-survival-stage-diagnosis.htm (accessed on 15 March 2023).
- Reddy, S.R.; Broder, M.S.; Chang, E.; Paydar, C.; Chung, K.C.; Kansal, A.R. Cost of cancer management by stage at diagnosis among Medicare beneficiaries. Curr. Med. Res. Opin. 2022, 38, 1285–1294. [Google Scholar] [CrossRef] [PubMed]
- Halabi, S.; Owzar, K. The importance of identifying and validating prognostic factors in oncology. Semin. Oncol. 2010, 37, e9–e18. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, V.; Tarazona, N.; Cejalvo, J.M.; Lombardi, P.; Huerta, M.; Roselló, S.; Fleitas, T.; Roda, D.; Cervantes, A. Personalized Medicine: Recent Progress in Cancer Therapy. Cancers 2020, 12, 1009. [Google Scholar] [CrossRef] [PubMed]
- A & B Recommendations. A and B Recommendations|United States Preventive Services Taskforce. Available online: https://www.uspreventiveservicestaskforce.org/uspstf/recommendation-topics/uspstf-a-and-b-recommendations. (accessed on 15 March 2023).
- Recommendation: Breast Cancer: Screening|United States Preventive Services Taskforce. Available online: https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/breast-cancer-screening#tab1 (accessed on 15 March 2023).
- Recommendation: Cervical Cancer Screening|United States Preventive Services Taskforce. Published 21 August 2018. Available online: https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/cervical-cancer-screening (accessed on 15 March 2023).
- Recommendation: Colorectal Cancer Screening|United States Preventive Services Taskforce. Published 18 May 2021. Available online: https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/colorectal-cancer-screening (accessed on 15 March 2023).
- Gilson, P.; Merlin, J.L.; Harlé, A. Deciphering Tumour Heterogeneity: From Tissue to Liquid Biopsy. Cancers 2022, 14, 1384. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, A.; Kelly, P.; Turkington, R.C.; Jones, C.; Coleman, H.G.; McCain, R.S. Pancreatic cancer: A review of clinical diagnosis, epidemiology, treatment and outcomes. In World Journal of Gastroenterology; Baishideng Publishing Group Co.: Pleasanton, CA, USA, 2018; Volume 24, pp. 4846–4861. [Google Scholar] [CrossRef]
- Rogers, T.K. Minimising diagnostic delay in lung cancer. Thorax 2019, 74, 319. [Google Scholar] [CrossRef] [PubMed]
- Doubeni, C.A.; Doubeni, A.R.; Myers, A.E. Diagnosis and Management of Ovarian Cancer. Am. Fam. Physician 2016, 93, 937–944. [Google Scholar]
- Pantel, K.; Alix-Panabières, C. Circulating tumour cells in cancer patients: Challenges and perspectives. Trends Mol. Med. 2010, 16, 398–406. [Google Scholar] [CrossRef]
- Killock, D. CancerSEEK and destroy—A blood test for early cancer detection. Nat. Rev. Clin. Oncol. 2018, 15, 133. [Google Scholar] [CrossRef]
- Nakamura, Y.; Taniguchi, H.; Ikeda, M.; Bando, H.; Kato, K.; Morizane, C.; Esaki, T.; Komatsu, Y.; Kawamoto, Y.; Takahashi, N. Clinical utility of circulating tumor DNA sequencing in advanced gastrointestinal cancer: SCRUM-Japan GI-SCREEN and GOZILA studies. Nat. Med. 2020, 26, 1859–1864. [Google Scholar] [CrossRef]
- Sama, S.; Le, T.; Ullah, A.; Elhelf, I.A.; Kavuri, S.K.; Karim, N.A. The Role of Serial Liquid Biopsy in the Management of Metastatic Non-Small Cell Lung Cancer (NSCLC). Clin. Pract. 2022, 12, 419–424. [Google Scholar] [CrossRef]
- Liquid Biopsy: What It Is & Procedure Details. Cleveland Clinic. Available online: https://my.clevelandclinic.org/health/diagnostics/23992-liquid-biopsy (accessed on 15 March 2023).
- Giacona, M.B.; Ruben, G.C.; Iczkowski, K.A.; Roos, T.B.; Porter, D.M.; Sorenson, G.D. Cell-Free DNA in Human Blood Plasma: Length Measurements in Patients with Pancreatic Cancer and Healthy Controls. Pancreas 1998, 17, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Alcaide, M.; Cheung, M.; Hillman, J.; Rassekh, S.R.; Deyell, R.J.; Batist, G.; Karsan, A.; Wyatt, A.W.; Johnson, N.; Scott, D.W.; et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Sci. Rep. 2020, 10, 12564. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.-L. Soluble Normal and Mutated DNA Sequences from Single-Copy Genes in Human Blood. Volume 3. Available online: http://aacrjournals.org/cebp/article-pdf/3/1/67/2287024/67.pdf (accessed on 15 March 2023).
- Streleckiene, G.; Reid, H.M.; Arnold, N.; Bauerschlag, D.; Forster, M. Quantifying cell free DNA in urine: Comparison between commercial kits, impact of gender and inter-individual variation. BioTechniques 2018, 64, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Escudero, L.; Martínez-Ricarte, F.; Seoane, J. ctDNA-Based Liquid Biopsy of Cerebrospinal Fluid in Brain Cancer. Cancers 2021, 13, 1989. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, N.; Kamataki, A.; Yamaki, J.; Homma, Y. Characterization of circulating DNA in healthy human plasma. Clin. Chim. Acta 2008, 387, 55–58. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kawane, K.; Fukuyama, H.; Kondoh, G.; Takeda, J.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. Requirement of DNase II for Definitive Erythropoiesis in the Mouse Fetal Liver. Science 2001, 292, 1546–1549. [Google Scholar] [CrossRef]
- Moss, J.; Magenheim, J.; Neiman, D.; Zemmour, H.; Loyfer, N.; Korach, A.; Samet, Y.; Maoz, M.; Druid, H.; Arner, P.; et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat. Commun. 2018, 9, 5068. [Google Scholar] [CrossRef]
- Hummel, E.M.; Hessas, E.; Müller, S.; Beiter, T.; Fisch, M.; Eibl, A.; Wolf, O.T.; Giebel, B.; Platen, P.; Kumsta, R.; et al. Cell-free DNA release under psychosocial and physical stress conditions. Transl. Psychiatry 2018, 8, 236. [Google Scholar] [CrossRef]
- Duvvuri, B.; Lood, C. Cell-free DNA as a biomarker in autoimmune rheumatic diseases. In Frontiers in Immunology; Frontiers Media S.A.: Lausanne, Switzerland, 2019; Volume 10. [Google Scholar] [CrossRef]
- Saukkonen, K.; Lakkisto, P.; Pettilä, V.; Varpula, M.; Karlsson, S.; Ruokonen, E.; Pulkki, K.; for the Finnsepsis Study Group. Cell-Free Plasma DNA as a Predictor of Outcome in Severe Sepsis and Septic Shock. Clin. Chem. 2008, 54, 1000–1007. [Google Scholar] [CrossRef]
- Chang, C.P.-Y.; Chia, R.-H.; Wu, T.-L.; Tsao, K.-C.; Sun, C.-F.; Wu, J.T. Elevated cell-free serum DNA detected in patients with myocardial infarction. Clin. Chim. Acta 2003, 327, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Chan, C.W.M.; Chan, K.C.A.; Cheng, S.H.; Wong, J.; Wong, V.W.-S.; Wong, G.L.H.; Chan, S.L.; Mok, T.S.K.; Chan, H.L.Y.; et al. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, E1317–E1325. [Google Scholar] [CrossRef]
- Jahr, S.; Hentze, H.; Englisch, S.; Hardt, D.; Fackelmayer, F.O.; Hesch, R.-D.; Knippers, R. DNA Fragments in the Blood Plasma of Cancer Patients: Quantitations and Evidence for Their Origin from Apoptotic and Necrotic Cells1. Cancer Res. 2001, 61, 1659–1665. [Google Scholar]
- Stroun, M.; Lyautey, J.; Lederrey, C.; Olson-Sand, A.; Anker, P. About the possible origin and mechanism of circulating DNA: Apoptosis and active DNA release. Clin. Chim. Acta 2001, 313, 139–142. [Google Scholar] [CrossRef]
- Zhang, R.; Shao, F.; Wu, X.; Ying, K. Value of quantitative analysis of circulating cell free DNA as a screening tool for lung cancer: A meta-analysis. Lung Cancer 2010, 69, 225–231. [Google Scholar] [CrossRef]
- Feng, J.; Gang, F.; Li, X.; Jin, T.; Houbao, H.; Yu, C.; Guorong, L. Plasma cell-free DNA and its DNA integrity as biomarker to distinguish prostate cancer from benign prostatic hyperplasia in patients with increased serum prostate-specific antigen. Int. Urol. Nephrol. 2013, 45, 1023–1028. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, O.; Bigosch, C.; Huegli, B.; Jermann, M.; Marx, A.; Chassé, E.; Ratschiller, D.; Weder, W.; Joerger, M.; Betticher, D.C.; et al. Circulating Deoxyribonucleic Acid As Prognostic Marker in Non–Small-Cell Lung Cancer Patients Undergoing Chemotherapy. J. Clin. Oncol. 2004, 22, 4157–4164. [Google Scholar] [CrossRef]
- Mouliere, F.; el Messaoudi, S.; Pang, D.; Dritschilo, A.; Thierry, A.R. Multi-marker analysis of circulating cell-free DNA toward personalized medicine for colorectal cancer. Mol. Oncol. 2014, 8, 927–941. [Google Scholar] [CrossRef]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Lee, T.-H.; Montalvo, L.; Chrebtow, V.; Busch, M.P. Quantitation of genomic DNA in plasma and serum samples: Higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001, 41, 276–282. [Google Scholar] [CrossRef]
- Pittella-Silva, F.; Chin, Y.M.; Chan, H.T.; Nagayama, S.; Miyauchi, E.; Low, S.-K.; Nakamura, Y. Plasma or Serum: Which Is Preferable for Mutation Detection in Liquid Biopsy? Clin. Chem. 2020, 66, 946–957. [Google Scholar] [CrossRef]
- Frattini, M.; Gallino, G.; Signoroni, S.; Balestra, D.; Battaglia, L.; Sozzi, G.; Leo, E.; Pilotti, S.; Pierotti, M.A. Quantitative Analysis of Plasma DNA in Colorectal Cancer Patients. Ann. N. Y. Acad. Sci. 2006, 1075, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Allen, D.; Butt, A.; Cahill, D.; Wheeler, M.; Popert, R.; Swaminathan, R. Role of cell-free plasma DNA as a diagnostic marker for prostate cancer. Ann. N. Y. Acad. Sci. 2004, 1022, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Bettegowda, C.; Sausen, M.; Leary, R.J.; Kinde, I.; Wang, Y.; Agrawal, N.; Bartlett, B.R.; Wang, H.; Luber, B.; Alani, R.M.; et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl. Med. 2014, 6, 224ra24. [Google Scholar] [CrossRef] [PubMed]
- Petit, J.; Carroll, G.; Gould, T.; Pockney, P.; Dun, M.; Scott, R.J. Cell-Free DNA as a Diagnostic Blood-Based Biomarker for Colorectal Cancer: A Systematic Review. J. Surg. Res. 2019, 236, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Bronkhorst, A.J.; Ungerer, V.; Holdenrieder, S. Comparison of methods for the quantification of cell-free DNA isolated from cell culture supernatant. Tumor Biol. 2019, 41, 1010428319866369. [Google Scholar] [CrossRef]
- Chen, E.; Cario, C.L.; Leong, L.; Lopez, K.; Márquez, C.P.; Chu, C.; Li, P.S.; Oropeza, E.; Tenggara, I.; Cowan, J.; et al. Cell-free DNA concentration and fragment size as a biomarker for prostate cancer. Sci. Rep. 2021, 11, 5040. [Google Scholar] [CrossRef]
- Stebbing, J.; Takis, P.G.; Sands, C.J.; Maslen, L.; Lewis, M.R.; Gleason, K.; Page, K.; Guttery, D.; Fernandez-Garcia, D.; Primrose, L.; et al. Comparison of phenomics and cfDNA in a large breast screening population: The Breast Screening and Monitoring Study (BSMS). Oncogene 2023, 42, 825–832. [Google Scholar] [CrossRef]
- Lan, Y.-T.; Chen, M.-H.; Fang, W.-L.; Hsieh, C.-C.; Lin, C.-H.; Jhang, F.-Y.; Yang, S.-H.; Lin, J.-K.; Chen, W.-S.; Jiang, J.-K.; et al. Clinical relevance of cell-free DNA in gastrointestinal tract malignancy. Oncotarget 2016, 8, 3009–3017. [Google Scholar] [CrossRef]
- Khorana, A.A.; Mangu, P.B.; Berlin, J.; Engebretson, A.; Hong, T.S.; Maitra, A.; Mohile, S.G.; Mumber, M.; Schulick, R.; Shapiro, M.; et al. Potentially Curable Pancreatic Cancer: American Society of Clinical Oncology Clinical Practice Guideline Update. J. Clin. Oncol. 2017, 35, 2324–2328. [Google Scholar] [CrossRef]
- Bergquist, J.R.; Puig, C.A.; Shubert, C.; Groeschl, R.T.; Habermann, E.; Kendrick, M.L.; Nagorney, D.M.; Smoot, R.L.; Farnell, M.B.; Truty, M.J. Carbohydrate Antigen 19-9 Elevation in Anatomically Resectable, Early Stage Pancreatic Cancer Is Independently Associated with Decreased Overall Survival and an Indication for Neoadjuvant Therapy: A National Cancer Database Study. J. Am. Coll. Surg. 2016, 223, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Luo, G.; Lu, R.; Shi, W.; Cheng, H.; Lu, Y.; Jin, K.; Yang, C.; Wang, Z.; Long, J.; et al. Distribution of Lewis and Secretor polymorphisms and corresponding CA19-9 antigen expression in a Chinese population. FEBS Open Bio 2017, 7, 1660–1671. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-H.; Demissie, K.; Shih, W.; Mehta, A.R.; Stein, M.N.; Roberts, C.B.; DiPaola, R.S.; Lu-Yao, G.L. Contemporary Risk Profile of Prostate Cancer in the United States. J. Natl. Cancer Inst. 2009, 101, 1280–1283. [Google Scholar] [CrossRef] [PubMed]
- Dess, R.T.; Sun, Y.; Jackson, W.C.; Jairath, N.K.; Kishan, A.U.; Wallington, D.G.; Mahal, B.A.; Stish, B.J.; Zumsteg, Z.S.; Den, R.B.; et al. Association of Presalvage Radiotherapy PSA Levels After Prostatectomy with Outcomes of Long-term Antiandrogen Therapy in Men With Prostate Cancer. JAMA Oncol. 2020, 6, 735–743. [Google Scholar] [CrossRef]
- Ilic, D.; Djulbegovic, M.; Jung, J.H.; Hwang, E.C.; Zhou, Q.; Cleves, A.; Agoritsas, T.; Dahm, P. Prostate cancer screening with prostate-specific antigen (PSA) test: A systematic review and meta-analysis. BMJ 2018, 362, k3519. [Google Scholar] [CrossRef]
- Harris, L.; Fritsche, H.; Mennel, R.; Norton, L.; Ravdin, P.; Taube, S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C. American Society of Clinical Oncology 2007 Update of Recommendations for the Use of Tumor Markers in Breast Cancer. J. Clin. Oncol. 2007, 25, 5287–5312. [Google Scholar] [CrossRef]
- Locker, G.Y.; Hamilton, S.; Harris, J.; Jessup, J.M.; Kemeny, N.; Macdonald, J.S.; Somerfield, M.R.; Hayes, D.F.; Bast, R.C. ASCO 2006 Update of Recommendations for the Use of Tumor Markers in Gastrointestinal Cancer. J. Clin. Oncol. 2006, 24, 5313–5327. [Google Scholar] [CrossRef]
- Huang, C.-J.; Huang, W.-Y.; Chen, C.-Y.; Chao, Y.-J.; Chiang, N.-J.; Shan, Y.-S. Cancer-cell-derived cell-free DNA can predict distant metastasis earlier in pancreatic cancer: A prospective cohort study. Ther. Adv. Med. Oncol. 2022, 14, 17588359221106558. [Google Scholar] [CrossRef]
- Bunduc, S.; Gede, N.; Váncsa, S.; Lillik, V.; Kiss, S.; Dembrovszky, F.; Eróss, B.; Szakács, Z.; Gheorghe, C.; Mikó, A.; et al. Prognostic role of cell-free DNA biomarkers in pancreatic adenocarcinoma: A systematic review and meta–analysis. Crit. Rev. Oncol./Hematol. 2022, 169, 103548. [Google Scholar] [CrossRef]
- Liu, H.; Gao, Y.; Vafaei, S.; Gu, X.; Zhong, X. The Prognostic Value of Plasma Cell-Free DNA Concentration in the Prostate Cancer: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 599602. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Garcia, D.; Hills, A.; Page, K.; Hastings, R.K.; Toghill, B.; Goddard, K.S.; Ion, C.; Ogle, O.; Boydell, A.R.; Gleason, K.; et al. Plasma cell-free DNA (cfDNA) as a predictive and prognostic marker in patients with metastatic breast cancer. Breast Cancer Res. 2019, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, C.; Wan, S.; Mu, Z.; Zhang, Z.; Abu-Khalaf, M.M.; Fellin, F.M.; Silver, D.P.; Neupane, M.; Jaslow, R.J.; et al. Association of clinical outcomes in metastatic breast cancer patients with circulating tumour cell and circulating cell-free DNA. Eur. J. Cancer 2018, 106, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.-L.G.; Boysen, A.K.; Pallisgård, N.; Johansen, J.S.; Tabernero, J.; Sørensen, M.M.; Jensen, B.V.; Hansen, T.F.; Sefrioui, D.; Andersen, R.F.; et al. Cell-Free DNA in Metastatic Colorectal Cancer: A Systematic Review and Meta-Analysis. Oncol. 2017, 22, 1049–1055. [Google Scholar] [CrossRef] [PubMed]
- Pastor, B.; André, T.; Henriques, J.; Trouilloud, I.; Tournigand, C.; Jary, M.; Mazard, T.; Louvet, C.; Azan, S.; Bauer, A.; et al. Monitoring levels of circulating cell-free DNA in patients with metastatic colorectal cancer as a potential biomarker of responses to regorafenib treatment. Mol. Oncol. 2021, 15, 2401–2411. [Google Scholar] [CrossRef]
- Ai, B.; Liu, H.; Huang, Y.; Peng, P. Circulating cell-free DNA as a prognostic and predictive biomarker in non-small cell lung cancer. Oncotarget 2016, 7, 44583–44595. [Google Scholar] [CrossRef]
- Hyun, M.H.; Sung, J.S.; Kang, E.J.; Choi, Y.J.; Park, K.H.; Shin, S.W.; Lee, S.Y.; Kim, Y.H. Quantification of circulating cell-free DNA to predict patient survival in non-small-cell lung cancer. Oncotarget 2017, 8, 94417–94430. [Google Scholar] [CrossRef]
- Ma, G.; Wang, J.; Huang, H.; Han, X.; Xu, J.; Veeramootoo, J.S.; Xia, T.; Wang, S. Identification of the plasma total cfDNA level before and after chemotherapy as an indicator of the neoadjuvant chemotherapy response in locally advanced breast cancer. Cancer Med. 2020, 9, 2271–2282. [Google Scholar] [CrossRef]
- Lehner, J.; Stötzer, O.J.; Fersching, D.; Nagel, D.; Holdenrieder, S. Circulating plasma DNA and DNA integrity in breast cancer patients undergoing neoadjuvant chemotherapy. Clin. Chim. Acta 2013, 425, 206–211. [Google Scholar] [CrossRef]
- Tissot, C.; Toffart, A.-C.; Villar, S.; Souquet, P.-J.; Merle, P.; Moro-Sibilot, D.; Pérol, M.; Zavadil, J.; Brambilla, C.; Olivier, M.; et al. Circulating free DNA concentration is an independent prognostic biomarker in lung cancer. Eur. Respir. J. 2015, 46, 1773. [Google Scholar] [CrossRef]
- Kumar, S.; Guleria, R.; Singh, V.; Bharti, A.C.; Mohan, A.; Das, B.C. Plasma DNA level in predicting therapeutic efficacy in advanced nonsmall cell lung cancer. Eur. Respir. J. 2010, 36, 885. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.; Xia, W.; Ding, Q.; Shu, Y.; Xu, T.; Geng, Y.; Lu, Y.; Chen, D.; Xu, J.; Wang, F.; et al. Can plasma DNA monitoring be employed in personalized chemotherapy for patients with advanced lung cancer? Biomed. Pharmacother. 2012, 66, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Gristina, V.; Barraco, N.; La Mantia, M.; Castellana, L.; Insalaco, L.; Bono, M.; Perez, A.; Sardo, D.; Inguglia, S.; Iacono, F.; et al. Clinical Potential of Circulating Cell-Free DNA (cfDNA) for Longitudinally Monitoring Clinical Outcomes in the First-Line Setting of Non-Small-Cell Lung Cancer (NSCLC): A Real-World Prospective Study. Cancers 2022, 14, 6013. [Google Scholar] [CrossRef]
- Kuo, Y.B.; Chen, J.S.; Fan, C.W.; Li, Y.S.; Chan, E.C. Comparison of KRAS mutation analysis of primary tumors and matched circulating cell-free DNA in plasmas of patients with colorectal cancer. Clin. Chim. Acta. 2014, 433, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Chi, K.N.; Barnicle, A.; Sibilla, C.; Lai, Z.; Corcoran, C.; Williams, J.A.; Barrett, J.C.; Adelman, C.A.; Qiu, P.; Easter, A.; et al. Concordance of BRCA1, BRCA2 (BRCA), and ATM mutations identified in matched tumor tissue and circulating tumor DNA (ctDNA) in men with metastatic castration-resistant prostate cancer (mCRPC) screened in the PROfound study. J. Clin. Oncol. 2021, 39, 26. [Google Scholar] [CrossRef]
- Vandekerkhove, G.; Lavoie, J.-M.; Annala, M.; Sundahl, N.; Sano, T.; Struss, W.J.; Todenhöfer, T.; Ost, P.; Chi, K.N.; Black, P.C.; et al. Genomic concordance between profiling of circulating tumor DNA (ctDNA) and matched tissue in metastatic urothelial carcinoma. J. Clin. Oncol. 2019, 37, 457. [Google Scholar] [CrossRef]
- Chae, Y.K.; Davis, A.A.; Jain, S.; Santa-Maria, C.; Flaum, L.; Beaubier, N.; Platanias, L.C.; Gradishar, W.; Giles, F.J.; Cristofanilli, M. Concordance of Genomic Alterations by Next-Generation Sequencing in Tumor Tissue versus Circulating Tumor DNA in Breast Cancer. Mol. Cancer Ther. 2017, 16, 1412–1420. [Google Scholar] [CrossRef]
- Xu, B.; Shan, G.; Wu, Q.; Li, W.; Wang, H.; Li, H.; Yang, Y.; Long, Q.; Zhao, P. Concordance of Genomic Alterations between Circulating Tumor DNA and Matched Tumor Tissue in Chinese Patients with Breast Cancer. J. Oncol. 2020, 2020, 4259293. [Google Scholar] [CrossRef]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’connell, A.; Feeney, N.; Mach, S.L.; Jänne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection of EGFR and KRAS Mutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef]
- Hindson, B.J.; Ness, K.D.; Masquelier, D.A.; Belgrader, P.; Heredia, N.J.; Makarewicz, A.J.; Bright, I.J.; Lucero, M.Y.; Hid-dessen, A.L.; Legler, T.C.; et al. High-Throughput Droplet Digital PCR System for Absolute Quantitation of DNA Copy Number. Anal. Chem. 2011, 83, 8604–8610. [Google Scholar] [CrossRef]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; Dressman, D.; He, Y.; Shen, D.; Szabo, S.; Diaz, L.A.; Goodman, S.N.; David, K.A.; Juhl, H.; et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc. Natl. Acad. Sci. USA 2005, 102, 16368–16373. [Google Scholar] [CrossRef]
- Heitzer, E.; Auer, M.; Hoffmann, E.M.; Pichler, M.; Gasch, C.; Ulz, P.; Lax, S.; Waldispuehl-Geigl, J.; Mauermann, O.; Mohan, S.; et al. Establishment of tumor-specific copy number alterations from plasma DNA of patients with cancer. Int. J. Cancer 2013, 133, 346–356. [Google Scholar] [CrossRef] [PubMed]
- Carrera, P.; Righetti, P.G.; Gelfi, C.; Ferrari, M. Amplification Refractory Mutation System Analysis of Point Mutations by Capillary Electrophoresis. In Capillary Electrophoresis of Nucleic Acids: Volume II: Practical Applications of Capillary Electrophoresis; Mitchelson, K.R., Cheng, J., Eds.; Humana Press: Totowa, NJ, USA, 2001; pp. 95–108. [Google Scholar] [CrossRef]
- Spindler, K.-L.G.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative Cell-Free DNA, KRAS, and BRAF Mutations in Plasma from Patients with Metastatic Colorectal Cancer during Treatment with Cetuximab and Irinotecan. Clin. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef]
- Stadler, J.; Eder, J.; Pratscher, B.; Brandt, S.; Schneller, D.; Müllegger, R.; Vogl, C.; Trautinger, F.; Brem, G.; Burgstaller, J.P. SNPase-ARMS qPCR: Ultrasensitive Mutation-Based Detection of Cell-Free Tumor DNA in Melanoma Patients. PLoS ONE 2015, 10, e0142273. [Google Scholar] [CrossRef]
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui DW, Y.; Kaper, F.; Dawson, S.-J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive Identification and Monitoring of Cancer Mutations by Targeted Deep Sequencing of Plasma DNA. Sci. Transl. Med. 2012, 4, ra68–ra136. [Google Scholar] [CrossRef] [PubMed]
- Ultra-Sensitive NGS-Based Liquid Biopsy Technology in Companion Diagnostics. 2021. Available online: https://www.amp.org/AMP/assets/File/education/ocw/QIAGEN_Sysmex_Inostics_Partnership_08252021.pdf?pass=99 (accessed on 15 May 2023).
- Kinde, I.; Wu, J.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B. Detection and quantification of rare mutations with massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 9530–9535. [Google Scholar] [CrossRef]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Leary, R.J.; Kinde, I.; Diehl, F.; Schmidt, K.; Clouser, C.; Duncan, C.; Antipova, A.; Lee, C.; McKernan, K.; De La Vega, F.M.; et al. Development of Personalized Tumor Biomarkers Using Massively Parallel Sequencing. Sci. Transl. Med. 2010, 2, 20ra14. [Google Scholar] [CrossRef]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in Cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; CCGA Consortium. Sensitive and specific multi-cancer detection and localization using methylation signatures in cell-free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Desai, M.; Shchegrov, S.R.; Chai, S.; Zhou, Y.; Nguyen, T.; Cho, Y.; Melton, C.; Scott, E.; Roychowdhury-Saha, M.; Chang, P.-Y.; et al. Abstract LB297: Analytical validation of a tissue-free, multi-cancer, post-diagnosis cancer research test that uses cell-free DNA methylation profiling. Cancer Res. 2023, 83, LB297. [Google Scholar] [CrossRef]
- Liang, N.; Li, B.; Jia, Z.; Wang, C.; Wu, P.; Zheng, T.; Wang, Y.; Qiu, F.; Wu, Y.; Su, J.; et al. Ultrasensitive detection of circulating tumour DNA via deep methylation sequencing aided by machine learning. Nat. Biomed. Eng. 2021, 5, 586–599. [Google Scholar] [CrossRef]
- Li, B.; Su, J.; Zhang, G.; Xu, J.; Peng, J.; Zhou, Y.; Qiu, F.; Fang, S.; Wen, X.; Wang, G.; et al. Abstract 5116: Analytical performance of ELSA-seq, a blood-based test for early detection of multiple cancers. Cancer Res. 2022, 82, 5116. [Google Scholar] [CrossRef]
- Chen, X.; Gole, J.; Gore, A.; He, Q.; Lu, M.; Min, J.; Yuan, Z.; Yang, X.; Jiang, Y.; Zhang, T.; et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat. Commun. 2020, 11, 3475. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.Y.; Burgener, J.M.; Bratman, S.V.; De Carvalho, D.D. Preparation of cfMeDIP-seq libraries for methylome profiling of plasma cell-free DNA. Nat. Protoc. 2019, 14, 2749–2780. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.; Wu, Y.-L.; Lee, J.S.; Yu, C.-J.; Sriuranpong, V.; Sandoval-Tan, J.; Ladrera, G.; Thongprasert, S.; Srimuninnimit, V.; Liao, M.; et al. Detection and Dynamic Changes of EGFR Mutations from Circulating Tumor DNA as a Predictor of Survival Outcomes in NSCLC Patients Treated with First-line Intercalated Erlotinib and Chemotherapy. Clin. Cancer Res. 2015, 21, 3196–3203. [Google Scholar] [CrossRef]
- Marsico, G.; Sharma, G.; Perry, M.; Hackinger, S.; Forshew, T.; Howarth, K.; Platt, J.; Rosenfeld, N.; Osborne, R. Abstract 3097: Analytical development of the RaDaRTM assay, a highly sensitive and specific assay for the monitoring of minimal residual disease. Cancer Res. 2020, 80, 3097. [Google Scholar] [CrossRef]
- Coombes, R.C.; Page, K.; Salari, R.; Hastings, R.K.; Armstrong, A.C.; Ahmed, S.; Ali, S.; Cleator, S.J.; Kenny, L.M.; Stebbing, J.; et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin. Cancer Res. 2019, 25, 4255–4263. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [PubMed]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P210040 (accessed on 15 May 2023).
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P140020 (accessed on 15 May 2023).
- Devices@FDA. Available online: https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm?db=pma&id=320540 (accessed on 15 May 2023).
- Devices@FDA. Available online: https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm?db=pma&id=320651 (accessed on 15 May 2023).
- Devices@FDA. Available online: https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm?db=pma&id=452325 (accessed on 15 May 2023).
- Devices@FDA. Available online: https://www.accessdata.fda.gov/scripts/cdrh/devicesatfda/index.cfm?db=pma&id=454228 (accessed on 15 May 2023).
- CancerSEEK. Early Detection Research Network. Available online: https://edrn.nci.nih.gov/data-and-resources/biomarkers/cancerseek/ (accessed on 15 May 2023).
- GRAIL Announces Significant Progress with Multi-Cancer Early Detection Test Including FDA Breakthrough Device Designation. GRAIL. Available online: https://grail.com/press-releases/grail-announces-significant-progress-with-multi-cancer-early-detection-test-including-fda-breakthrough-device-designation/ (accessed on 15 May 2023).
- FDA Grants Breakthrough Device Designation for Inivatas Radar Assay. Inivata. Available online: https://www.inivata.com/fda-grants-breakthrough-device-designation-for-inivatas-radar-assay/ (accessed on 15 May 2023).
- Nilson, E. Bluestar Genomics Receives FDA Breakthrough Device Designation for First-of-Its-Kind Pancreatic Cancer Screening Test. ClearNote Health. Available online: https://www.clearnotehealth.com/press-release/bluestar-genomics-receives-fda-breakthrough-device-designation-for-first-of-its-kind-pancreatic-cancer-screening-test/ (accessed on 15 May 2023).
- FDA Grants Two New Breakthrough Device Designations for Natera’s SignateraTM MRD Test. Natera. Available online: https://www.natera.com/company/news/fda-grants-two-new-breakthrough-device-designations-for-nateras-signatera-mrd-test-2/ (accessed on 15 May 2023).
- Predicine’s Liquid Biopsy Next-Generation Sequencing (NGS) Assay is Granted Breakthrough Device Designation by U.S. Food and Drug Administration—Predicine|Advancing Precision Cancer Therapies. Precidine. Available online: https://www.predicine.com/2022/09/20/predicines-liquid-biopsy-next-generation-sequencing-ngs-assay-is-granted-breakthrough-device-designation-by-u-s-food-and-drug-administration/ (accessed on 15 May 2023).
- Burning Rock Received FDA Breakthrough Device Designation for its OverC Multi-Cancer Detection Blood Test. Burning Rock. Available online: https://ir.brbiotech.com/news-releases/news-release-details/burning-rock-received-fda-breakthrough-device-designation-its (accessed on 15 May 2023).
- Potter, N.T.; Hurban, P.; White, M.N.; Whitlock, K.D.; Lofton-Day, C.E.; Tetzner, R.; Koenig, T.; Quigley, N.B.; Weiss, G. Validation of a Real-Time PCR–Based Qualitative Assay for the Detection of Methylated SEPT9 DNA in Human Plasma. Clin. Chem. 2014, 60, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.A.; Barclay, R.L.; Mergener, K.; Weiss, G.; König, T.; Beck, J.; Potter, N.T. Plasma Septin9 versus fecal immunochemical testing for colorectal cancer screening: A prospective multicenter study. PLoS ONE 2014, 9, e98238. [Google Scholar] [CrossRef]
- Haan, D.; Bergamaschi, A.; Guler, G.D.; Friedl, V.; Ning, Y.; Reggiardo, R.; Kesling, M.; Collins, M.; Gibb, B.; Pitea, A.; et al. Validation of a Pancreatic Cancer Detection Test in New-Onset Diabetes Using Cell-Free DNA 5-Hydroxymethylation Signatures. MedRxiv 2021. [Google Scholar] [CrossRef]
- Lennon, A.M.; Buchanan, A.H.; Kinde, I.; Warren, A.; Honushefsky, A.; Cohain, A.T.; Ledbetter, D.H.; Sanfilippo, F.; Sheridan, K.; Rosica, D.; et al. Feasibility of blood testing combined with PET-CT to screen for cancer and guide intervention. Science 2020, 369, eabb9601. [Google Scholar] [CrossRef]
- Liu, M.C.; Klein, E.; Hubbell, E.; Maddala, T.; Aravanis, A.M.; Beausang, J.F.; Filippova, D.; Gross, S.; Jamshidi, A.; Kurtzman, K.; et al. 50O—Plasma cell-free DNA (cfDNA) assays for early multi-cancer detection: The circulating cell-free genome atlas (CCGA) study. Ann. Oncol. 2018, 29, viii14. [Google Scholar] [CrossRef]
- Klein, E.A.; Richards, D.; Cohn, A.; Tummala, M.; Lapham, R.; Cosgrove, D.; Chung, G.; Clement, J.; Gao, J.; Hunkapiller, N.; et al. Clinical validation of a targeted methylation-based multi-cancer early detection test using an independent validation set. Ann. Oncol. 2021, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Wang, C.; Yang, X.; Fang, S.; Zhang, Y.; Wang, G.; Liu, F.; Wen, X.; Zhao, J.; Zhou, G.; et al. 909P A multi-cancer early detection model based on liquid biopsy of multi-omics biomarkers: A proof of concept study (PROMISE study). Ann. Oncol. 2022, 33, S963–S964. [Google Scholar] [CrossRef]
- Gao, Q.; Li, B.; Cai, S.; Xu, J.; Wang, C.; Su, J.; Fang, S.; Qiu, F.; Wen, X.; Zhang, Y.; et al. Early detection and localization of multiple cancers using a blood-based methylation assay (ELSA-seq). J. Clin. Oncol. 2021, 39, 459. [Google Scholar] [CrossRef]
- Gao, Q.; Li, B.; Xu, J.; Fang, S.; Qiu, F.; Su, J.; Wang, G.; Cai, S.; Zhao, H.; Zhang, W.; et al. Analysis of epigenomic signatures in cell-free DNA (cfDNA) from cancer patients and high-risk controls: A blinded test cohort of THUNDER-II study. J. Clin. Oncol. 2021, 39, e22518. [Google Scholar] [CrossRef]
- Gao, Q.; Zhang, Y.; Xu, J.; Wang, G.; Zhao, J.; Wen, X.; Li, B.; Zhang, Z.; Cai, S.; Fan, J.; et al. Clinical validation of a multicancer detection blood test by circulating cell-free DNA (cfDNA) methylation sequencing: The THUNDER study. J. Clin. Oncol. 2022, 40, 10544. [Google Scholar] [CrossRef]
- Liles, E.G.; Coronado, G.D.; Perrin, N.; Harte, A.H.; Nungesser, R.; Quigley, N.; Potter, N.T.; Weiss, G.; Koenig, T.; deVos, T. Uptake of a colorectal cancer screening blood test is higher than of a fecal test offered in clinic: A randomized trial. Cancer Treat. Res. Commun. 2017, 10, 27–31. [Google Scholar] [CrossRef]
- Song, L.; Jia, J.; Peng, X.; Xiao, W.; Li, Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci. Rep. 2017, 7, 3032. [Google Scholar] [CrossRef]
- Hu, J.; Hu, B.; Gui, Y.C.; Tan, Z.B.; Xu, J.W. Diagnostic Value and Clinical Significance of Methylated SEPT9 for Colorectal Cancer: A Meta-Analysis. Med. Sci. Monit. 2019, 25, 5813–5822. [Google Scholar] [CrossRef]
- Lin, J.S.; Perdue, L.A.; Henrikson, N.B.; Bean, S.I.; Blasi, P.R. Screening for Colorectal Cancer: An Evidence Update for the U.S. Preventive Services Task Force [Internet]. Rockville (MD): Agency for Healthcare Research and Quality (US); 2021 May. (Evidence Synthesis, No. 202). Available online: https://www.ncbi.nlm.nih.gov/books/NBK570913/ (accessed on 15 May 2023).
- Recommendation: Colorectal Cancer: Screening|United States Preventive Services Taskforce. Available online: https://www.uspreventiveservicestaskforce.org/uspstf/recommendation/colorectal-cancer-screening#fullrecommendationstart (accessed on 15 May 2023).
- Longitudinal Performance of Epi proColon—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03218423 (accessed on 15 May 2023).
- Guler, G.D.; Ning, Y.; Ku, C.-J.; Phillips, T.; McCarthy, E.; Ellison, C.K.; Bergamaschi, A.; Collin, F.; Lloyd, P.; Scott, A.; et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat. Commun. 2020, 11, 5270. [Google Scholar] [CrossRef]
- New Onset Diabetes Management for Earlier Detection of Pancreatic Cancer (NODMED)—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05188586 (accessed on 20 May 2023).
- EpiDetect Study: Clinical Validation of a Pancreatic Cancer Detection Test in New-Onset Diabetes Patients—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT05188573 (accessed on 20 May 2023).
- ClearNote Health Clinical Trials for Pancreatic Cancer. ClearNote Health. Available online: https://www.clearnotehealth.com/clinical-trials/ (accessed on 20 May 2023).
- Detecting Cancers Earlier Through Elective Plasma-based CancerSEEK Testing—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04213326 (accessed on 20 May 2023).
- The Circulating Cell-free Genome Atlas Study—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT02889978 (accessed on 20 May 2023).
- The STRIVE Study: Development of a Blood Test for Early Detection of Multiple Cancer Types—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03085888 (accessed on 20 May 2023).
- The SUMMIT Study: A Cancer Screening Study—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03934866 (accessed on 20 May 2023).
- REFLECTION: A Clinical Practice Learning Program for Galleri®—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05205967 (accessed on 20 May 2023).
- Beer, T.M.; McDonnell, C.H.; Nadauld, L.; Liu, M.C.; Klein, E.A.; Reid, R.L.; Marinac, C.; Chung, K.; Lopatin, M.; Fung, E.T.; et al. Interim results of PATHFINDER, a clinical use study using a methylation-based multi-cancer early detection test. J. Clin. Oncol. 2021, 39, 3010. [Google Scholar] [CrossRef]
- Nadauld, L.D.; McDonnell, C.H.I.I.I.; Beer, T.M.; Liu, M.C.; Klein, E.A.; Hudnut, A.; Whittington, R.A.; Taylor, B.; Oxnard, G.R.; Lipson, J.; et al. The PATHFINDER Study: Assessment of the Implementation of an Investigational Multi-Cancer Early Detection Test into Clinical Practice. Cancers 2021, 13, 3501. [Google Scholar] [CrossRef]
- Assessment of the Implementation of an Investigational Multi-Cancer Early Detection Test Into Clinical Practice—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04241796 (accessed on 20 May 2023).
- GRAIL, LLC. The PATHFINDER 2 Study: Evaluating the Safety and Performance of the GRAIL Multi-Cancer Early Detection Test in an Eligible Screening Population. Available online: https://clinicaltrials.gov/ct2/show/NCT05155605 (accessed on 20 May 2023).
- A Proof of Concept Study of Pan-cancer Early Detection by Liquid Biopsy—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04972201 (accessed on 20 May 2023).
- Pan-canceR Early DetectIon project—Full Text View—ClinicalTrials.gov. Available online: https://www.clinicaltrials.gov/ct2/show/NCT04383353 (accessed on 21 May 2023).
- Pan-canceR Early-Stage deteCtion by lIquid Biopsy tEchNique project—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04822792 (accessed on 21 May 2023).
- Multi-canceR Early-detection Test in Asymptomatic Individuals (PREVENT)—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT05227534 (accessed on 21 May 2023).
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef]
- Rolfo, C.; Mack, P.; Scagliotti, G.V.; Aggarwal, C.; Arcila, M.E.; Barlesi, F.; Bivona, T.; Diehn, M.; Dive, C.; Dziadziuszko, R.; et al. Liquid Biopsy for Advanced NSCLC: A Consensus Statement From the International Association for the Study of Lung Cancer. J. Thorac. Oncol. 2021, 16, 1647–1662. [Google Scholar] [CrossRef]
- Fiala, C.; Diamandis, E.P. Utility of circulating tumor DNA in cancer diagnostics with emphasis on early detection. BMC Med. 2018, 16, 166. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.D.; Javed, A.A.; Thoburn, C.; Wong, F.; Tie, J.; Gibbs, P.; Schmidt, C.M.; Yip-Schneider, M.T.; Allen, P.J.; Schattner, M.; et al. Combined circulating tumor DNA and protein biomarker-based liquid biopsy for the earlier detection of pancreatic cancers. Proc. Natl. Acad. Sci. USA 2017, 114, 10202–10207. [Google Scholar] [CrossRef]
- Tangvarasittichai, O.; Jaiwang, W.; Tangvarasittichai, S. The Plasma DNA Concentration as a Potential Breast Cancer Screening Marker. Indian J. Clin. Biochem. 2013, 30, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Lambros, M.B.; Seed, G.; Sumanasuriya, S.; Gil, V.; Crespo, M.; Fontes, M.; Chandler, R.; Mehra, N.; Fowler, G.; Ebbs, B.; et al. Single-Cell Analyses of Prostate Cancer Liquid Biopsies Acquired by Apheresis. Clin. Cancer Res. 2018, 24, 5635–5644. [Google Scholar] [CrossRef] [PubMed]
- Fehm, T.N.; Meier-Stiegen, F.; Driemel, C.; Jäger, B.; Reinhardt, F.; Naskou, J.; Franken, A.; Neubauer, H.; Neves, R.P.; Dalum, G.; et al. Diagnostic leukapheresis for CTC analysis in breast cancer patients: CTC frequency, clinical experiences and recommendations for standardized reporting. Cytom. Part A 2018, 93, 1213–1219. [Google Scholar] [CrossRef]
- Downs, B.M.; Sukumar, S. Capturing ctDNA from Unaltered Stationary and Flowing Plasma with dCas9. ACS Appl. Mater. Interfaces 2022, 14, 24113–24121. [Google Scholar] [CrossRef]
- Lin, J.S.; Perdue, L.A.; Henrikson, N.B.; Bean, S.I.; Blasi, P.R. Screening for Colorectal Cancer: Updated Evidence Report and Systematic Review for the US Preventive Services Task Force. JAMA 2021, 325, 1978–1998. [Google Scholar] [CrossRef]
- In Vitro Companion Diagnostic Devices Guidance for Industry and Food and Drug Administration Staff. 2014. Available online: https://www.fda.gov/media/81309/download (accessed on 25 May 2023).
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf14/P140020B.pdf (accessed on 25 May 2023).
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Golan, T.; Hammel, P.; Reni, M.; van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.v.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P140020S016 (accessed on 25 May 2023).
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P140020S020 (accessed on 25 May 2023).
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200006B.pdf (accessed on 25 May 2023).
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032B.pdf (accessed on 25 May 2023).
- Wu, Y.-L.; Lee, V.; Liam, C.-K.; Lu, S.; Park, K.; Srimuninnimit, V.; Wang, J.; Zhou, C.; Appius, A.; Button, P.; et al. Clinical utility of a blood-based EGFR mutation test in patients receiving first-line erlotinib therapy in the ENSURE, FASTACT-2, and ASPIRATION studies. Lung Cancer 2018, 126, 1–8. [Google Scholar] [CrossRef]
- Goss, G.; Tsai, C.-M.; Shepherd, F.A.; Bazhenova, L.; Lee, J.S.; Chang, G.-C.; Crino, L.; Satouchi, M.; Chu, Q.; Hida, T.; et al. Osimertinib for pretreated EGFR Thr790Met-positive advanced non-small-cell lung cancer (AURA2): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2016, 17, 1643–1652. [Google Scholar] [CrossRef] [PubMed]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P120019S019 (accessed on 1 June 2023).
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef] [PubMed]
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010B.pdf (accessed on 1 June 2023).
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010S002B.pdf (accessed on 1 June 2023).
- Bauml, J.M.; Li, B.T.; Velcheti, V.; Govindan, R.; Curioni-Fontecedro, A.; Dooms, C.; Takahashi, T.; Duda, A.W.; Odegaard, J.I.; Cruz-Guilloty, F.; et al. Clinical validation of Guardant360 CDx as a blood-based companion diagnostic for sotorasib. Lung Cancer 2022, 166, 270–278. [Google Scholar] [CrossRef]
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf21/P210040B.pdf (accessed on 15 June 2023).
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Matulonis, U.A.; Penson, R.T.; Domchek, S.M.; Kaufman, B.; Shapira-Frommer, R.; Audeh, M.W.; Kaye, S.; Molife, L.R.; Gelmon, K.A.; Robertson, J.D.; et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: A multistudy analysis of response rates and safety. Ann. Oncol. 2016, 27, 1013–1019. [Google Scholar] [CrossRef]
- Poveda, A.; Floquet, A.; Ledermann, J.A.; Asher, R.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Pignata, S.; Friedlander, M.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A final analysis of a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2021, 22, 620–631. [Google Scholar] [CrossRef]
- Penson, R.T.; Valencia, R.V.; Cibula, D.; Colombo, N.; Leath, C.A., III; Bidziński, M.; Kim, J.-W.; Nam, J.H.; Madry, R.; Hernández, C.; et al. Olaparib Versus Nonplatinum Chemotherapy in Patients with Platinum-Sensitive Relapsed Ovarian Cancer and a Germline BRCA1/2 Mutation (SOLO3): A Randomized Phase III Trial. J. Clin. Oncol. 2020, 38, 1164–1174. [Google Scholar] [CrossRef]
- BRACAnalysis CDx® Technical Information. Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf14/P140020S020C.pdf (accessed on 25 May 2023).
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- Kristeleit, R.S.; Oaknin, A.; Ray-Coquard, I.; Leary, A.; Balmaña, J.; Drew, Y.; Oza, A.M.; Shapira-Frommer, R.; Domchek, S.M.; Cameron, T.; et al. Antitumor activity of the poly(ADP-ribose) polymerase inhibitor rucaparib as monotherapy in patients with platinum-sensitive, relapsed, BRCA-mutated, high-grade ovarian cancer, and an update on safety. Int. J. Gynecol. Cancer 2019, 29, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Corcoran, C.; Sibilla, C.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Mateo, J.; Olmos, D.; Mehra, N.; et al. Tumor Genomic Testing for >4,000 Men with Metastatic Castration-resistant Prostate Cancer in the Phase III Trial PROfound (Olaparib). Clin. Cancer Res. 2022, 28, 1518–1530. [Google Scholar] [CrossRef] [PubMed]
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf12/P120019B.pdf (accessed on 1 June 2023).
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Benlloch, S.; Botero, M.L.; Beltran-Alamillo, J.; Mayo, C.; Gimenez-Capitán, A.; de Aguirre, I.; Queralt, C.; Ramirez, J.L.; Cajal, S.R.Y.; Klughammer, B.; et al. Clinical Validation of a PCR Assay for the Detection of EGFR Mutations in Non–Small-Cell Lung Cancer: Retrospective Testing of Specimens from the EURTAC Trial. PLoS ONE 2014, 9, e89518. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Zhou, C.; Liam, C.-K.; Wu, G.; Liu, X.; Zhong, Z.; Lu, S.; Cheng, Y.; Han, B.; Chen, L.; et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: Analyses from the phase III, randomized, open-label, ENSURE study. Ann. Oncol. 2015, 26, 1883–1889. [Google Scholar] [CrossRef]
- Park, K.; Yu, C.-J.; Kim, S.-W.; Lin, M.-C.; Sriuranpong, V.; Tsai, C.-M.; Lee, J.-S.; Kang, J.-H.; Chan, K.C.A.; Perez-Moreno, P.; et al. First-Line Erlotinib Therapy Until and Beyond Response Evaluation Criteria in Solid Tumors Progression in Asian Patients With Epidermal Growth Factor Receptor Mutation–Positive Non–Small-Cell Lung Cancer. JAMA Oncol. 2016, 2, 305–308. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Lee, J.S.; Thongprasert, S.; Yu, C.-J.; Zhang, L.; Ladrera, G.; Srimuninnimit, V.; Sriuranpong, V.; Sandoval-Tan, J.; Zhu, Y.; et al. Intercalated combination of chemotherapy and erlotinib for patients with advanced stage non-small-cell lung cancer (FASTACT-2): A randomised, double-blind trial. Lancet Oncol. 2013, 14, 777–786. [Google Scholar] [CrossRef]
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf15/P150047B.pdf (accessed on 1 June 2023).
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf15/P150044B.pdf (accessed on 1 June 2023).
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P120019S018 (accessed on 1 June 2023).
- Cheng, Y.; He, Y.; Li, W.; Zhang, H.; Zhou, Q.; Wang, B.; Liu, C.; Walding, A.; Saggese, M.; Huang, X.; et al. Osimertinib Versus Comparator EGFR TKI as First-Line Treatment for EGFR-Mutated Advanced NSCLC: FLAURA China, A Randomized Study. Target. Oncol. 2021, 16, 165–176. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P120019S031 (accessed on 1 June 2023).
- SUMMARY of SAFETY and EFFECTIVENESS DATA (SSED). Available online: https://www.accessdata.fda.gov/cdrh_docs/pdf17/P170019B.pdf (accessed on 1 June 2023).
- Milbury, C.A.; Creeden, J.; Yip, W.-K.; Smith, D.L.; Pattani, V.; Maxwell, K.; Sawchyn, B.; Gjoerup, O.; Meng, W.; Skoletsky, J.; et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PLoS ONE 2022, 17, e0264138. [Google Scholar] [CrossRef] [PubMed]
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P170019S033 (accessed on 1 June 2023).
- Premarket Approval (PMA). Available online: https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=P190032S008 (accessed on 1 June 2023).
- A Phase 1/2, Study Evaluating the Safety, Tolerability, PK, and Efficacy of Sotorasib (AMG 510) in Subjects with Solid Tumors With a Specific KRAS Mutation (CodeBreaK 100)—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03600883 (accessed on 20 June 2023).
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Phase 1/2 Study of MRTX849 in Patients With Cancer Having a KRAS G12C Mutation KRYSTAL-1—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03785249 (accessed on 20 June 2023).
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.-H.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Engl. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Pekker, I.; Pollak, J.; Potts, K.; Chan, P.; Tsai, C.-H.; Liao, A.; Garrison, C.; Brown, T.; Stull, P.; Bianchi-Frias, D.; et al. Resolution ctDx FIRST plasma assay as a companion diagnostic for adagrasib and its application to longitudinal monitoring. J. Clin. Oncol. 2022, 40, 3057. [Google Scholar] [CrossRef]
- Prognosis and Targeted Therapy Related Molecular Screening Program for Patients of Breast Cancer in China—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03792529 (accessed on 20 June 2023).
- Liao, H.; Zhang, J.; Zheng, T.; Liu, X.; Zhong, J.; Shao, B.; Dong, X.; Wang, X.; Du, P.; King, B.L.; et al. Identification of mutation patterns and circulating tumour DNA-derived prognostic markers in advanced breast cancer patients. J. Transl. Med. 2022, 20, 211. [Google Scholar] [CrossRef]
- Gerratana, L.; Zhang, Q.; Shah, A.N.; Davis, A.A.; Zhang, Y.; Wehbe, F.; Qiang, W.; Flaum, L.; Finkelman, B.S.; Gradishar, W.J.; et al. Performance of a novel Next Generation Sequencing circulating tumor DNA (ctDNA) platform for the evaluation of samples from patients with metastatic breast cancer (MBC). Crit. Rev. Oncol./Hematol. 2020, 145, 102856. [Google Scholar] [CrossRef]
- Developing and Labeling in Vitro Companion Diagnostic Devices for a Specific Group or Class of Oncology Therapeutic Products Guidance for Industry DRAFT GUIDANCE. 2018. Available online: https://www.fda.gov/media/120340/download (accessed on 20 June 2023).
- Chao, M.; Gibbs, P. Caution Is Required Before Recommending Routine Carcinoembryonic Antigen and Imaging Follow-Up for Patients With Early-Stage Colon Cancer. J. Clin. Oncol. 2009, 27, e279–e280. [Google Scholar] [CrossRef]
- Ettinger, D.S.; Wood, D.E.; Aisner, D.L.; Akerley, W.; Bauman, J.; Chirieac, L.R.; D’Amico, T.A.; DeCamp, M.M.; Dilling, T.J.; Dobelbower, M.; et al. Non–Small Cell Lung Cancer, Version 5.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 504–535. [Google Scholar] [CrossRef]
- Erdi, Y.E. Limits of Tumor Detectability in Nuclear Medicine and PET. Mol. Imaging Radionucl. Ther. 2012, 21, 23–28. [Google Scholar] [CrossRef]
- Huang, K.; Dahele, M.; Senan, S.; Guckenberger, M.; Rodrigues, G.B.; Ward, A.; Boldt, R.G.; Palma, D.A. Radiographic changes after lung stereotactic ablative radiotherapy (SABR)—Can we distinguish recurrence from fibrosis? A systematic review of the literature. Radiother. Oncol. 2012, 102, 335–342. [Google Scholar] [CrossRef]
- Garcia-Murillas, I.; Schiavon, G.; Weigelt, B.; Ng, C.; Hrebien, S.; Cutts, R.J.; Cheang, M.; Osin, P.; Nerurkar, A.; Kozarewa, I.; et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Sci. Transl. Med. 2015, 7, 302ra133. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.; Wang, Y.; Tomasetti, C.; Li, L.; Springer, S.; Kinde, I.; Silliman, N.; Tacey, M.; Wong, H.-L.; Christie, M.; et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci. Transl. Med. 2016, 8, 346ra92. [Google Scholar] [CrossRef]
- Chaudhuri, A.A.; Chabon, J.J.; Lovejoy, A.F.; Newman, A.M.; Stehr, H.; Azad, T.D.; Khodadoust, M.S.; Esfahani, M.S.; Liu, C.L.; Zhou, L.; et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017, 7, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Gale, D.; Heider, K.; Ruiz-Valdepenas, A.; Hackinger, S.; Perry, M.; Marsico, G.; Rundell, V.; Wulff, J.; Sharma, G.; Knock, H.; et al. Residual ctDNA after treatment predicts early relapse in patients with early-stage non-small cell lung cancer. Ann. Oncol. 2022, 33, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Flach, S.; Howarth, K.; Hackinger, S.; Pipinikas, C.; Ellis, P.; McLay, K.; Marsico, G.; Forshew, T.; Walz, C.; Reichel, C.A.; et al. Liquid BIOpsy for MiNimal RESidual DiSease Detection in Head and Neck Squamous Cell Carcinoma (LIONESS)—A personalised circulating tumour DNA analysis in head and neck squamous cell carcinoma. Br. J. Cancer 2022, 126, 1186–1195. [Google Scholar] [CrossRef]
- Cutts, R.J.; Coakley, M.; Garcia-Murillas, I.; Ulrich, L.; Howarth, K.; Emmett, W.; Perry, M.; Ellis, P.; Knape, C.; Johnston, S.R.; et al. Abstract 536: Molecular residual disease detection in early stage breast cancer with a personalized sequencing approach. Cancer Res. 2021, 81, 536. [Google Scholar] [CrossRef]
- Janni, W.; Huober, J.; Huesmann, S.; Pipinikas, C.; Braun, T.; Müller, V.; Marsico, G.; Fink, A.; Freire-Pritchett, P.; Koretz, K.; et al. Abstract P2-01-07: Detection of early-stage breast cancer recurrence using a personalised liquid biopsy-based sequencing approach. Cancer Res. 2022, 82, P2-01-07. [Google Scholar] [CrossRef]
- Reinert, T.; Henriksen, T.V.; Christensen, E.; Sharma, S.; Salari, R.; Sethi, H.; Knudsen, M.; Nordentoft, I.K.; Wu, H.-T.; Tin, A.S.; et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients with Stages I to III Colorectal Cancer. JAMA Oncol. 2019, 5, 1124–1131. [Google Scholar] [CrossRef]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.-T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients With Urothelial Bladder Carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef]
- Loupakis, F.; Sharma, S.; Derouazi, M.; Murgioni, S.; Biason, P.; Rizzato, M.D.; Rasola, C.; Renner, D.; Shchegrova, S.; Malashevich, A.K.; et al. Detection of Molecular Residual Disease Using Personalized Circulating Tumor DNA Assay in Patients With Colorectal Cancer Undergoing Resection of Metastases. JCO Precis. Oncol. 2021, 5, 1166–1177. [Google Scholar] [CrossRef]
- Plagnol, V.; Woodhouse, S.; Howarth, K.; Lensing, S.; Smith, M.; Epstein, M.; Madi, M.; Smalley, S.; Leroy, C.; Hinton, J.; et al. Analytical validation of a next generation sequencing liquid biopsy assay for high sensitivity broad molecular profiling. PLoS ONE 2018, 13, e0193802. [Google Scholar] [CrossRef] [PubMed]
- LUCID—LUng Cancer CIrculating Tumour Dna Study—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT04153526 (accessed on 20 June 2023).
- Multi-Omic Assessment of Squamous Cell Cancers Receiving Systemic Therapy—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03712566 (accessed on 20 June 2023).
- Taylor, K.; Zou, J.; Fonseca Magalhaes Filho, M.A.; Howarth, K.; Marsico, G.; Terrell, S.; Jones, G.; Knape, C.; Forshew, T.; Oliva, M.; et al. Personalized circulating tumor DNA (ctDNA) analysis in patients with recurrent/metastatic head and neck squamous cell cancer (R/M HNSCC). J. Clin. Oncol. 2022, 40, 6052. [Google Scholar] [CrossRef]
- Liquid Biopsy Evaluation and Repository Development at Princess Margaret—Full Text View—ClinicalTrials.gov. Available online: https://classic.clinicaltrials.gov/ct2/show/NCT03702309 (accessed on 20 June 2023).
- Elliott, M.J.; Veitch, Z.; Bedard, P.; Amir, E.; Dou, A.; Antras, J.F.; Nadler, M.; Meti, N.; Gregorio, N.; Shah, E.; et al. Abstract P6-01-16: Circulating Tumour DNA (ctDNA) Detection and Dynamics in Patients with Early Breast Cancer (EBC): Results of the Neoadjuvant TRACER cohort. Cancer Res. 2023, 83, P6-01-16. [Google Scholar] [CrossRef]
- Coakley, M.; Sritharan, P.; Villacampa, G.; Swift, C.; Dunne, K.; Kilburn, L.; Goddard, K.; Rojas, P.; Joad, A.; Emmett, W.; et al. Abstract PD5-03: PD5-03 Comparison of a personalized sequencing assay and digital PCR for circulating tumor DNA based Molecular Residual Disease detection in early-stage triple negative breast cancer in the cTRAK-TN trial. Cancer Res. 2023, 83, PD5-03. [Google Scholar] [CrossRef]
- Genta, S.; Araujo, D.V.; Keshavarzi, S.; Pimentel Muniz, T.; Saeed Kamil, Z.; Howarth, K.; Terrell, S.; Joad, A.; Ventura, R.; Covelli, A.; et al. Leveraging personalized circulating tumor DNA (ctDNA) for detection and monitoring of molecular residual disease in high-risk melanoma. J. Clin. Oncol. 2022, 40, 9579. [Google Scholar] [CrossRef]
- Stockem, C.; Gil Jimenez, A.; van Dorp, J.; van Dijk, N.; Alkemade, M.; Seignette, I.; Pipinikas, C.; Jones, G.; Rosenfeld, N.; van Montfoort, M.; et al. 1770P Updated follow-up data and biomarker analysis of pre-operative ipilimumab and nivolumab in locoregional advanced urothelial cancer (NABUCCO). Ann. Oncol. 2022, 33, S1347. [Google Scholar] [CrossRef]
- Taniguchi, H.; Nakamura, Y.; Kotani, D.; Yukami, H.; Mishima, S.; Sawada, K.; Shirasu, H.; Ebi, H.; Yamanaka, T.; Aleshin, A.; et al. CIRCULATE-Japan: Circulating tumor DNA–guided adaptive platform trials to refine adjuvant therapy for colorectal cancer. Cancer Sci. 2021, 112, 2915–2920. [Google Scholar] [CrossRef]
- NIPH Clinical Trials Search. Available online: https://rctportal.niph.go.jp/en/detail?trial_id=jRCT1031200006 (accessed on 20 June 2023).
- Kotani, D.; Oki, E.; Nakamura, Y.; Yukami, H.; Mishima, S.; Bando, H.; Shirasu, H.; Yamazaki, K.; Watanabe, J.; Kotaka, M.; et al. Molecular residual disease and efficacy of adjuvant chemotherapy in patients with colorectal cancer. Nat. Med. 2023, 29, 127–134. [Google Scholar] [CrossRef]
- BESPOKE Study of ctDNA Guided Therapy in Colorectal Cancer—Full Text View—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04264702 (accessed on 20 June 2023).
- Kasi, P.M.; Sawyer, S.; Guilford, J.; Munro, M.; Ellers, S.; Wulff, J.; Hook, N.; Krinshpun, S.; Malashevich, A.K.; Malhotra, M.; et al. BESPOKE study protocol: A multicentre, prospective observational study to evaluate the impact of circulating tumour DNA guided therapy on patients with colorectal cancer. BMJ Open 2021, 11, e047831. [Google Scholar] [CrossRef]
- Powles, T.; Assaf, Z.J.; Davarpanah, N.; Banchereau, R.; Szabados, B.E.; Yuen, K.C.; Grivas, P.; Hussain, M.; Oudard, S.; Gschwend, J.E.; et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature 2021, 595, 432–437. [Google Scholar] [CrossRef]

{kind=link}
| Cancer Type | # of Patients | cfDNA concentration | PFS HR | OS HR | Additional Info | Citation |
|---|---|---|---|---|---|---|
| PDAC | 74 | >9.71 ng/mL | 6.85 | 4.16 | Average of cfDNA concentration before and after chemotherapy treatment. | [59] |
| PDAC | - | - | 1.96 | 3.39 | Subgroup analysis of cfDNA during treatment. | [60] |
| PCa | - | >cut-off | log(HR) = 0.84 | log(HR) = 0.60 | Meta-analysis of studies using different cut-off points. | [61] |
| CRPC | - | >cut-off | log(HR) = 0.65 | log(HR) = 0.59 | Meta-analysis of studies using different cut-off points. | [61] |
| MBC | 194 | >0.306 ng/uL | 1.193 | 1.199 | Majority of samples were collected during treatment. | [62] |
| MBC | 117 | high cfDNA | 1.64 | 2.73 | High cfDNA was determined by comparing to previous samples. HR based on comparing low and high cfDNA levels. | [63] |
| mCRC | 1076 | >cut-off | - | 2.39 | Meta-analysis of studies using different cut-off points. | [64] |
| mCRC | 43 | >26 ng/mL | 1.51 | 2.02 | PFS and OS determined from samples before treatment. | [65] |
| NSCLC | - | >cut-off | 1.32 | 1.64 | meta-analysis of studies using different cut-off points. | [66] |
| NSCLC | 177 | >70 ng/mL | 2.6 | 2.63 | PFS and OS determined from samples before treatment. | [67] |
| Technology Used | Analytical Sensitivity | Cancer Type | Test | Citation |
|---|---|---|---|---|
| RT-PCR | 0.1–1% MAF | NSCLC | cobas EGFR | [101] |
| ddPCR | 0.001% MAF | BRAF V600E | [80] | |
| BEAMing | 0.01% MAF | CRC, MBC | [82,83] | |
| ARMS-qPCR | 0.1% MAF | mCRC | [85] | |
| SNPase-ARMS qPCR | 0.001% MAF | Melanoma | [86] | |
| TEC-Seq | 0.1% MAF | BC, LC, OVC, CRC | [40] | |
| TAm-Seq | 0.002% MAF | Various | RaDaR | [102] |
| TAm-Seq | 0.01% MAF | BC | Signatera | [103] |
| SafeSEQ | 0.05% MAF | Various | CancerSEEK | [88,89,104] |
| CAPP-Seq | 0.02% MAF | NSCLC | [90] | |
| PARE | 0.001% MAF | BC, CRC | [91] | |
| WGBS | 0.023% MAF | Various | Galleri | [95,96] |
| ELSA-seq | 0.02% MAF | Various | OverC | [97,98] |
| cfMeDIP-seq | 0.1% MAF | Various | PanSeer | [99] |
| Test | # of Patients * | Cancer Type (Stage) | Sensitivity | Specificity | Citation |
|---|---|---|---|---|---|
| Epi proColon | 1544 | CRC | 68% | 80% | [118] |
| Epi proColon | 290 | CRC | 73.3% | 81.5% | [119] |
| Bluestar | 748 | PDAC | 51.9% | 100% | [120] |
| CancerSEEK ** | 9911 | Various | 27.1% | 98.9% | [121] |
| CancerSEEK | 1817 | Various (1–3) | 70% | >99% | [104] |
| Galleri | 944 | Various | 36–74% | 98% | [122] |
| Galleri | 1264 | Various | 54.9% | >99% | [95] |
| Galleri | 4077 | Various | 51.5% | >99% | [123] |
| OverC | 492 | Various | 72.4% | 99.2% | [124] |
| OverC | 639 | Various (1–3) | 80.6% | >99% | [125] |
| OverC | 360 | Various | 74.8% | 98.1% | [126] |
| OverC | 1010 | Various (1–3) | 68.5% | 96.3% | [127] |
| Diagnostic Test | Biomarker/ Details | Cancer Type | Drug Name | Clinical Trial # | Compared Test | Concordance | Citations |
|---|---|---|---|---|---|---|---|
| BRACAnalysis | gBRCA1/2 | OC | Olaparib | NCT00753545 NCT01874353 | local testing | 99.2% (259/261) | [161] |
| BRACAnalysis | gBRCA1/2 | BC | Olaparib | NCT02000622 | BRACAnalysis | n/a | [162] |
| BRACAnalysis | gBRCA1/2 | BC | Talazoparib | NCT01945775 | BRACAnalysis | n/a | [163] |
| BRACAnalysis | gBRCA1/2 | PDAC | Olaparib | NCT02184195 | BRACAnalysis | n/a | [164] |
| BRACAnalysis | gBRCA1/2 | OC | Rucaparib | NCT01891344 | Foundation Medicine T5 | n/a | [165,166] |
| BRACAnalysis | gBRCA1/2 | PC | Olaparib | NCT02987543 | FoundationOne CDx | n/a | [167,168] |
| FoundationOne Liquid CDx | BRCA1/2 | OC | Rucaparib | NCT01891344 | Foundation Medicine T5 | 96.3% (209/217) | [169] |
| FoundationOne Liquid CDx | BRCA1/2 | mCRPC | Rucaparib | NCT02952534 | FoundationOne LDT FoundationOne Liquid LDT local testing | 89.4% (144/161) | [170] |
| FoundationOne Liquid CDx | BRCA1/2 | mCRPC | Olaparib | NCT02987543 | FoundationOne CDx | n/a | [170] |
| cobas EGFR Plasma Test v2 | EGFR * | NSCLC | Erlotinib | NCT01310036 | cobas EGFR Mutation Test v1 | 84.4% (757/897) | [171] |
| cobas EGFR Plasma Test v2 | EGFR ** | NSCLC | Osimertinib | NCT02094261 | cobas EGFR Mutation Test v1 | 65.0% (206/317) | [172] |
| cobas EGFR Plasma Test v2 | EGFR * | NSCLC | Gefitinib | - | - | - | [173] |
| FoundationOne Liquid CDx | EGFR * EGFR ** | NSCLC | Erlotinib Osimertinib Gefitinib | - | cobas EGFR Mutation Test v2 | 94.4% (167/177) | [174] |
| Guardant360 CDx | EGFR * EGFR ** | NSCLC | Osimertinib | NCT02296125 | cobas EGFR Mutation Test v2 | 79.5% (372/468) | [175] |
| Guardant360 CDx | KRAS G12C | NSCLC | Sotorasib | NCT03600883 | therascreen KRAS RGQ PCR Kit | 82.0% (155/189) | [176,177] |
| Resolution ctDx | KRAS G12C | NSCLC | Adagrasib | NCT03785249 | local testing | 95.1% (212/223) | [178] |
| MRD Test | Cancer Type | # of Patients | Specificity | Sensitivity | Lead Time | Additional Info | Citation |
|---|---|---|---|---|---|---|---|
| RaDaR™ | NSCLC | 77 | 95.9% (47/49) | 64.8% (18/28) | 212.5 days | ctDNA detected >2 weeks after surgery | [219] |
| RaDaR™ | HNSCC | 17 | 100% (12/12) | 100% (5/5) | 122 days | ctDNA detected from samples taken during routine follow-up visits | [220] |
| RaDaR™ | BC | 22 | 100% (5/5) | 100% (17/17) | 386.7 days | ctDNA detected from samples taken during routine follow-up visits | [221] |
| RaDaR™ | BC | 38 | 94.1% (16/17) | 71.4% (15/21) | 92 days | ctDNA detected from samples taken at detection of recurrence or 3 years later | [222] |
| Signatera | BC | 49 | 100% (31/31) | 89% (16/18) | 267 days | ctDNA detected from samples taken every 6 months | [103] |
| Signatera | CRC | 75 | 98.3% (58/59) | 87.5% (14/16) | 261 days * | ctDNA detected from samples taken at 1 month and every 3 months. | [223] |
| Signatera | UC | 68 | 98% (48/49) | 100% (13/13) | 96 days | ctDNA detected from samples taken during routine follow-up visits | [224] |
| Signatera | mCRC | 112 | 93.3% (14/15) | 91.4% (32/35) | 95 days | ctDNA detection with samples from two-time points | [225] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dao, J.; Conway, P.J.; Subramani, B.; Meyyappan, D.; Russell, S.; Mahadevan, D. Using cfDNA and ctDNA as Oncologic Markers: A Path to Clinical Validation. Int. J. Mol. Sci. 2023, 24, 13219. https://doi.org/10.3390/ijms241713219
Dao J, Conway PJ, Subramani B, Meyyappan D, Russell S, Mahadevan D. Using cfDNA and ctDNA as Oncologic Markers: A Path to Clinical Validation. International Journal of Molecular Sciences. 2023; 24(17):13219. https://doi.org/10.3390/ijms241713219
Chicago/Turabian StyleDao, Jonathan, Patrick J. Conway, Baskaran Subramani, Devi Meyyappan, Sammy Russell, and Daruka Mahadevan. 2023. "Using cfDNA and ctDNA as Oncologic Markers: A Path to Clinical Validation" International Journal of Molecular Sciences 24, no. 17: 13219. https://doi.org/10.3390/ijms241713219
APA StyleDao, J., Conway, P. J., Subramani, B., Meyyappan, D., Russell, S., & Mahadevan, D. (2023). Using cfDNA and ctDNA as Oncologic Markers: A Path to Clinical Validation. International Journal of Molecular Sciences, 24(17), 13219. https://doi.org/10.3390/ijms241713219