

Structures of the Insecticidal Toxin Complex Subunit XptA2 Highlight Roles for Flexible Domains

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Structure of XptA2 by XRC
2.2. Structure of XptA2 by Single-Particle Cryo-EM
2.3. Properties of the XptA2 Translocation Channel
2.4. Importance of a Continuous Linker Region for Proper Pre-Pore Folding
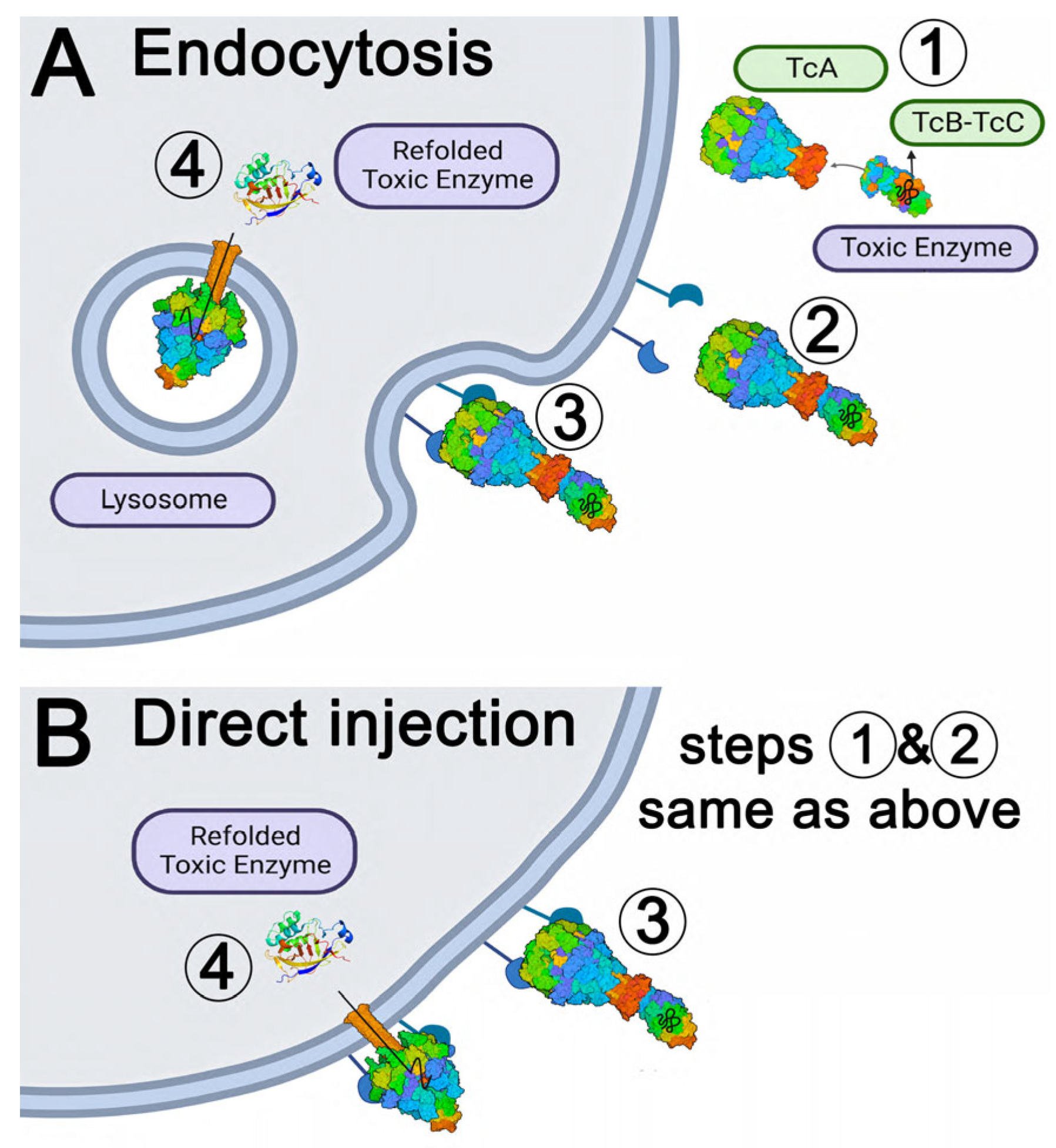
3. Discussion
4. Materials and Methods
4.1. Constructs
4.2. Protein Expression and Purification
4.3. X-ray Crystallography
4.4. Single Particle Cryo-Electron Microscopy
4.4.1. Cryo-EM Grid Preparation
4.4.2. Cryo-EM Imaging
4.4.3. Cryo-EM Data Processing
4.5. Molecular Dynamics Simulations
4.6. Differential Scanning Fluorimetry (DSF)
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheets, J.J.; Hey, T.D.; Fencil, K.J.; Burton, S.L.; Ni, W.; Lang, A.E.; Benz, R.; Aktories, K. Insecticidal toxin complex proteins from Xenorhabdus nematophilus: Structure and pore formation. J. Biol. Chem. 2011, 286, 22742–22749. [Google Scholar] [CrossRef] [PubMed]
- Chaston, J.M.; Suen, G.; Tucker, S.L.; Andersen, A.W.; Bhasin, A.; Bode, E.; Bode, H.B.; Brachmann, A.O.; Cowles, C.E.; Cowles, K.N.; et al. The entomopathogenic bacterial endosymbionts Xenorhabdus and Photorhabdus: Convergent lifestyles from divergent genomes. PLoS ONE 2011, 6, e27909. [Google Scholar] [CrossRef] [PubMed]
- Sicard, M.; Hinsinger, J.; Le Brun, N.; Pages, S.; Boemare, N.; Moulia, C. Interspecific competition between entomopathogenic nematodes (Steinernema) is modified by their bacterial symbionts (Xenorhabdus). BMC Evol Biol. 2006, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Shawer, R.; Donati, I.; Cellini, A.; Spinelli, F.; Mori, N. Insecticidal Activity of Photorhabdus luminescens against Drosophila suzukii. Insects 2018, 9, 148. [Google Scholar] [CrossRef]
- Blackburn, M.B.; Domek, J.M.; Gelman, D.B.; Hu, J.S. The broadly insecticidal Photorhabdus luminescens toxin complex a (Tca): Activity against the Colorado potato beetle, Leptinotarsa decemlineata, and sweet potato whitefly, Bemisia tabaci. J Insect Sci. 2005, 5, 32. [Google Scholar] [CrossRef]
- Castagnola, A.; Stock, S.P. Common Virulence Factors and Tissue Targets of Entomopathogenic Bacteria for Biological Control of Lepidopteran Pests. Insects 2014, 5, 139–166. [Google Scholar] [CrossRef]
- da Silva, W.J.; Pilz-Junior, H.L.; Heermann, R.; da Silva, O.S. The great potential of entomopathogenic bacteria Xenorhabdus and Photorhabdus for mosquito control: A review. Parasit Vectors 2020, 13, 376. [Google Scholar] [CrossRef]
- Fukruksa, C.; Yimthin, T.; Suwannaroj, M.; Muangpat, P.; Tandhavanant, S.; Thanwisai, A.; Vitta, A. Isolation and identification of Xenorhabdus and Photorhabdus bacteria associated with entomopathogenic nematodes and their larvicidal activity against Aedes aegypti. Parasit. Vectors 2017, 10, 440. [Google Scholar] [CrossRef]
- Ng Ang, A.P.N.; Ebner, J.K.; Plessner, M.; Aktories, K.; Schmidt, G. Engineering Photorhabdus luminescens toxin complex (PTC) into a recombinant injection nanomachine. Life Sci. Alliance 2019, 2, e201900485. [Google Scholar] [CrossRef]
- Roderer, D.; Schubert, E.; Sitsel, O.; Raunser, S. Towards the application of Tc toxins as a universal protein translocation system. Nat. Commun. 2019, 10, 5263. [Google Scholar] [CrossRef]
- Domínguez-Arrizabalaga, M.; Villanueva, M.; Escriche, B.; Ancín-Azpilicueta, C.; Caballero, P. Insecticidal Activity of Bacillus thuringiensis Proteins Against Coleopteran Pests. Toxins 2020, 12, 430. [Google Scholar] [CrossRef] [PubMed]
- Dubovskiy, I.M.; Grizanova, E.V.; Tereshchenko, D.; Krytsyna, T.I.; Alikina, T.; Kalmykova, G.; Kabilov, M.; Coates, C.J. Bacillus thuringiensis Spores and Cry3A Toxins Act Synergistically to Expedite Colorado Potato Beetle Mortality. Toxins 2021, 13, 746. [Google Scholar] [CrossRef] [PubMed]
- Bravo, A.; Likitvivatanavong, S.; Gill, S.S.; Soberón, M. Bacillus thuringiensis: A story of a successful bioinsecticide. Insect Biochem. Mol. Biol. 2011, 41, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Valtierra-de-Luis, D.; Villanueva, M.; Berry, C.; Caballero, P. Potential for Bacillus thuringiensis and Other Bacterial Toxins as Biological Control Agents to Combat Dipteran Pests of Medical and Agronomic Importance. Toxins 2020, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Silva-Filha, M.H.N.L.; Romão, T.P.; Rezende, T.M.T.; Carvalho, K.d.S.; Gouveia de Menezes, H.S.; Alexandre do Nascimento, N.; Soberón, M.; Bravo, A. Bacterial Toxins Active against Mosquitoes: Mode of Action and Resistance. Toxins 2021, 13, 523. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Merino, F.; Roderer, D.; Balchin, D.; Schubert, E.; Kuhlee, A.; Hayer-Hartl, M.; Raunser, S. Tc toxin activation requires unfolding and refolding of a beta-propeller. Nature 2018, 563, 209–213. [Google Scholar] [CrossRef]
- Meusch, D.; Gatsogiannis, C.; Efremov, R.G.; Lang, A.E.; Hofnagel, O.; Vetter, I.R.; Aktories, K.; Raunser, S. Mechanism of Tc toxin action revealed in molecular detail. Nature 2014, 508, 61–65. [Google Scholar] [CrossRef]
- Morgan, J.A.; Sergeant, M.; Ellis, D.; Ousley, M.; Jarrett, P. Sequence analysis of insecticidal genes from Xenorhabdus nematophilus PMFI296. Appl. Environ. Microbiol. 2001, 67, 2062–2069. [Google Scholar] [CrossRef]
- Sergeant, M.; Jarrett, P.; Ousley, M.; Morgan, J.A.W. Interactions of insecticidal toxin gene products from Xenorhabdus nematophilus PMFI296. Appl. Environ. Microbiol. 2003, 69, 3344–3349. [Google Scholar] [CrossRef]
- Leidreiter, F.; Roderer, D.; Meusch, D.; Gatsogiannis, C.; Benz, R.; Raunser, S. Common architecture of Tc toxins from human and insect pathogenic bacteria. Sci. Adv. 2019, 5, eaax6497. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Lang, A.E.; Meusch, D.; Pfaumann, V.; Hofnagel, O.; Benz, R.; Aktories, K.; Raunser, S. A syringe-like injection mechanism in Photorhabdus luminescens toxins. Nature 2013, 495, 520–523. [Google Scholar] [CrossRef]
- Landsberg, M.J.; Jones, S.A.; Rothnagel, R.; Busby, J.N.; Marshall, S.D.G.; Simpson, R.M.; Lott, J.S.; Hankamer, B.; Hurst, M.R.H. 3D structure of the Yersinia entomophaga toxin complex and implications for insecticidal activity. Proc. Natl. Acad. Sci. USA 2011, 108, 20544–20549. [Google Scholar] [CrossRef]
- Piper, S.J.; Brillault, L.; Rothnagel, R.; Croll, T.I.; Box, J.K.; Chassagnon, I.; Scherer, S.; Goldie, K.N.; Jones, S.A.; Schepers, F.; et al. Cryo-EM structures of the pore-forming A subunit from the Yersinia entomophaga ABC toxin. Nat. Commun. 2019, 10, 1952. [Google Scholar] [CrossRef]
- Roderer, D.; Raunser, S. Tc Toxin Complexes: Assembly, Membrane Permeation, and Protein Translocation. Annu. Rev. Microbiol. 2019, 73, 247–265. [Google Scholar] [CrossRef] [PubMed]
- Roderer, D.; Bröcker, F.; Sitsel, O.; Kaplonek, P.; Leidreiter, F.; Seeberger, P.H.; Raunser, S. Glycan-dependent cell adhesion mechanism of Tc toxins. Nat. Commun. 2020, 11, 2694. [Google Scholar] [CrossRef] [PubMed]
- Belyy, A.; Lindemann, F.; Roderer, D.; Funk, J.; Bardiaux, B.; Protze, J.; Bieling, P.; Oschkinat, H.; Raunser, S. Mechanism of threonine ADP-ribosylation of F-actin by a Tc toxin. Nat. Commun. 2022, 13, 4202. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.N.; McCoy, A.J.; Terwilliger, T.C.; Read, R.J.; Wiedenheft, B. X-ray structure determination using low-resolution electron microscopy maps for molecular replacement. Nat. Protoc. 2015, 10, 1275–1284. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.W.; Jain, S.; Kapral, G.J.; Grosse Kunstleve, R.W.; et al. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef]
- Gatsogiannis, C.; Merino, F.; Prumbaum, D.; Roderer, D.; Leidreiter, F.; Meusch, D.; Raunser, S. Membrane insertion of a Tc toxin in near-atomic detail. Nat. Struct. Mol. Biol. 2016, 23, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Madduri, K.; Badger, M.; Li, Z.S.; Xu, X.; Thornburgh, S.; Evans, S.; Dhadialla, T.S. Development of stable isotope and selenomethionine labeling methods for proteins expressed in Pseudomonas fluorescens. Protein Expr. Purif. 2009, 65, 57–65. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Rohou, A.; Grigorieff, N. CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015, 192, 216–221. [Google Scholar] [CrossRef]
- Zivanov, J.; Nakane, T.; Forsberg, B.O.; Kimanius, D.; Hagen, W.J.; Lindahl, E.; Scheres, S.H. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife 2018, 7, e42166. [Google Scholar] [CrossRef] [PubMed]
- Rice, L.M.; Earnest, T.N.; Brunger, A.T. Single-wavelength anomalous diffraction phasing revisited. Acta Crystallogr. D Biol. Crystallogr. 2000, 56 Pt 11, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Zivanov, J.; Nakane, T.; Scheres, S.H.W. Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 2020, 7 Pt 2, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Zuckerkandl, E. Evolutionary Divergence and Convergence in Proteins; Academic Press: New York, NY, USA, 1965. [Google Scholar]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, C.L.; Chester, D.W.; Radka, C.D.; Pan, L.; Yang, Z.; Hart, R.C.; Binshtein, E.M.; Wang, Z.; Nagy, L.; DeLucas, L.J.; et al. Structures of the Insecticidal Toxin Complex Subunit XptA2 Highlight Roles for Flexible Domains. Int. J. Mol. Sci. 2023, 24, 13221. https://doi.org/10.3390/ijms241713221
Martin CL, Chester DW, Radka CD, Pan L, Yang Z, Hart RC, Binshtein EM, Wang Z, Nagy L, DeLucas LJ, et al. Structures of the Insecticidal Toxin Complex Subunit XptA2 Highlight Roles for Flexible Domains. International Journal of Molecular Sciences. 2023; 24(17):13221. https://doi.org/10.3390/ijms241713221
Chicago/Turabian StyleMartin, Cole L., David W. Chester, Christopher D. Radka, Lurong Pan, Zhengrong Yang, Rachel C. Hart, Elad M. Binshtein, Zhao Wang, Lisa Nagy, Lawrence J. DeLucas, and et al. 2023. "Structures of the Insecticidal Toxin Complex Subunit XptA2 Highlight Roles for Flexible Domains" International Journal of Molecular Sciences 24, no. 17: 13221. https://doi.org/10.3390/ijms241713221
APA StyleMartin, C. L., Chester, D. W., Radka, C. D., Pan, L., Yang, Z., Hart, R. C., Binshtein, E. M., Wang, Z., Nagy, L., DeLucas, L. J., & Aller, S. G. (2023). Structures of the Insecticidal Toxin Complex Subunit XptA2 Highlight Roles for Flexible Domains. International Journal of Molecular Sciences, 24(17), 13221. https://doi.org/10.3390/ijms241713221