

Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents


Abstract
:1. Introduction
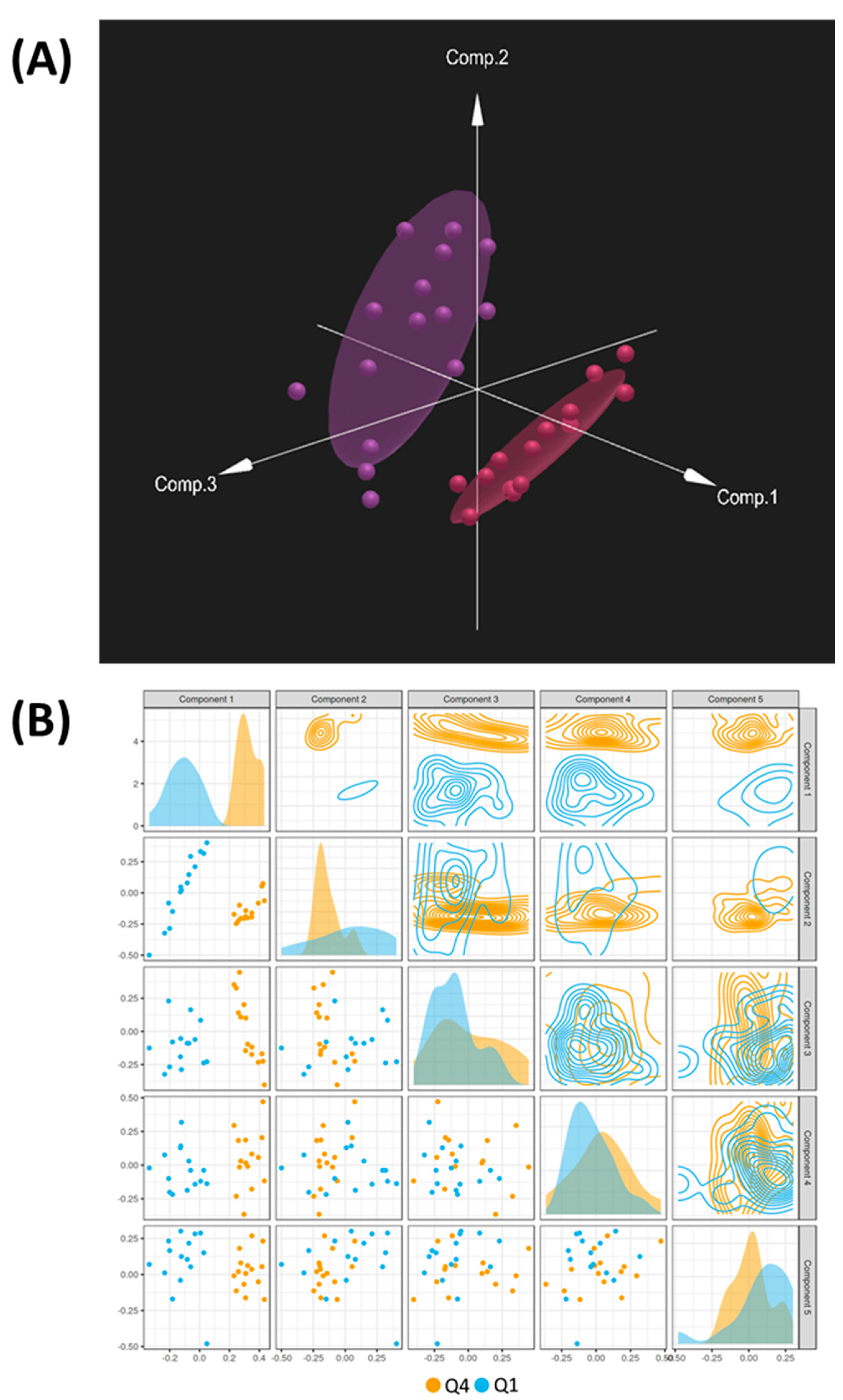
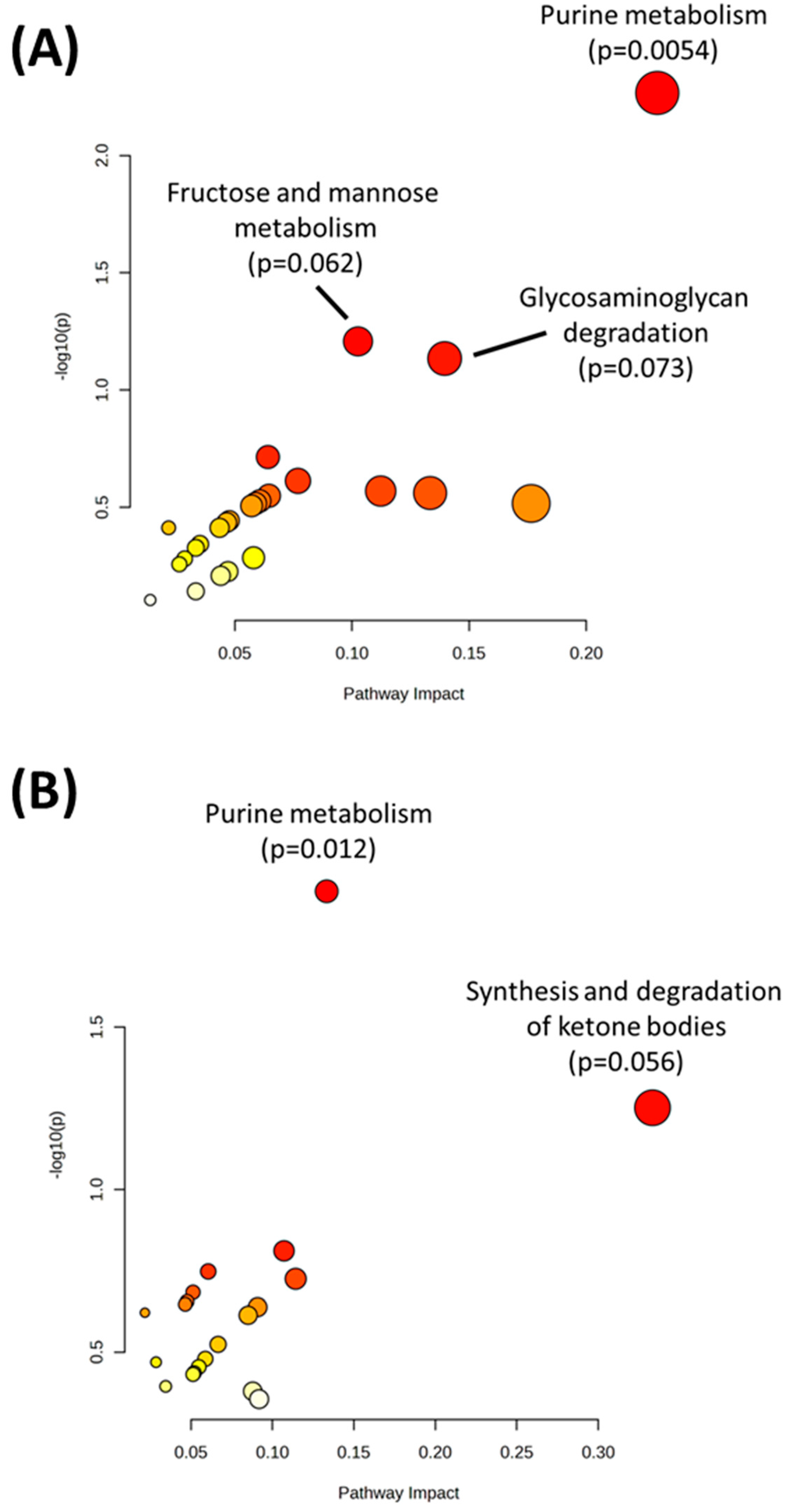
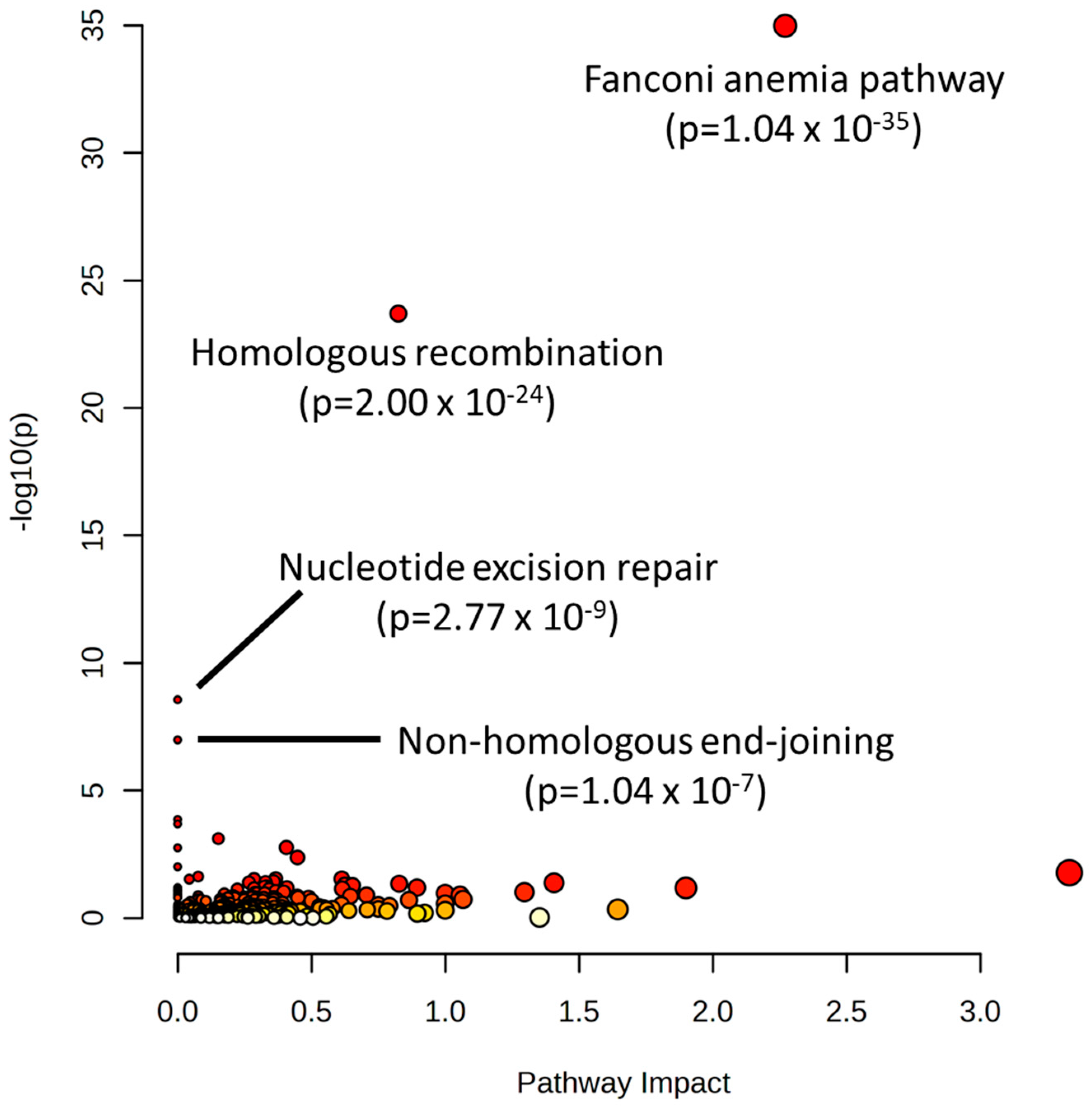
2. Results
3. Discussion
4. Materials and Methods
4.1. Acquisition of Public NCI-60 Cell Line Molecular Data
4.2. Compilation of Drug Response Data for Alkylating Agents in the NCI-60 Cell Line Panel
4.3. Univariate Statistics, Pathway Analysis, and Network Analysis of Selected Features
4.4. Multi-Omic Multivariate Analysis and Joint Pathway Analysis
4.5. CNV Analysis
4.6. Integrative Pathway Visualization
4.7. Data Mining and Pathway Analysis of CRISPR Screen Hits of Alkylating Agents
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ralhan, R.; Kaur, J. Alkylating agents and cancer therapy. Expert Opin. Ther. Pat. 2007, 17, 1061–1075. [Google Scholar] [CrossRef]
- Kobayashi, H.; Man, S.; Graham, C.H.; Kapitain, S.J.; Teicher, B.A.; Kerbel, R.S. Acquired Multicellular-Mediated Resistance to Alkyalting Agents in Cancer. PNAS 1993, 90, 3294–3298. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Kitange, G.J.; James, C.D.; Plummer, R.; Calvert, H.; Weller, M.; Wick, W. Mechanisms of chemoresistance to alkylating agents in malignant glioma. Clin. Cancer Res. 2008, 14, 2900–2908. [Google Scholar] [CrossRef]
- Zhang, J.; Tian, Q.; Chan, S.Y.; Li, S.C.; Zhou, S.; Duan, W.; Zhu, Y.-Z. Metabolism and Transport of Oxazaphosphorines and the Clinical Implications. Drug Metab. Rev. 2005, 37, 611–703. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O6-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2020, 9, 1547. [Google Scholar] [CrossRef] [PubMed]
- Li, L.Y.; Guan, Y.D.; Chen, X.S.; Yang, J.M.; Cheng, Y. DNA Repair Pathways in Cancer Therapy and Resistance. Front. Pharmacol. 2021, 11, 629266. [Google Scholar] [CrossRef]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef]
- Delou, J.M.A.; Souza, A.S.O.; Souza, L.C.M.; Borges, H.L. Highlights in Resistance Mechanism Pathways for Combination Therapy. Cells 2019, 8, 1013. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.H.; Wong, E.W.; Lehmann, B.; Terrian, D.M.; Basecke, J.; Stivala, F.; et al. Targeting the RAF/MEK/ERK, PI3K/AKT and P38 pathways in hematoietic drug resistance. Adv Enzym. Regul 2007, 47, 64–103. [Google Scholar] [CrossRef]
- Mollaei, M.; Hassan, Z.M.; Khorshidi, F.; Langroudi, L. Chemotherapeutic drugs: Cell death- and resistance-related signaling pathways. Are they really as smart as the tumor cells? Transl. Oncol. 2021, 14, 101056. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Woolley, J.F.; Moustafa, A.A.; Malki, M.I. DNA Damage / Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Wang, M.; Chen, S.; Ao, D. Targeting DNA repair pathway in cancer: Mechanisms and clinical application. MedComm 2021, 2, 654–691. [Google Scholar] [CrossRef]
- Kutuk, O.; Arisan, E.D.; Tezil, T.; Shoshan, M.C.; Basaga, H. Cisplatin overcomes Bcl-2-mediated resistance to apoptosis via preferential engagement of Bak: Critical role of Noxa-mediated lipid peroxidation. Carcinogenesis 2009, 30, 1517–1527. [Google Scholar] [CrossRef]
- García-Aranda, M.; Pérez-Ruiz, E.; Redondo, M. Bcl-2 inhibition to overcome resistance to chemo-and immunotherapy. Int. J. Mol. Sci. 2018, 19, 3950. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Quinn, B.A.; Das, S.K.; Dash, R.; Emdad, L.; Dasgupta, S.; Wang, X.Y.; Dent, P.; Reed, J.C.; Pellecchia, M.; et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin. Ther. Targets 2013, 17, 61–75. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Yoshida, G.J. Metabolic reprogramming: The emerging concept and associated therapeutic strategies. J. Exp. Clin. Cancer Res. 2015, 34, 111. [Google Scholar] [CrossRef]
- Zaal, E.A.; Berkers, C.R. The influence of metabolism on drug response in cancer. Front. Oncol. 2018, 8, 500. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013, 4, 500. [Google Scholar] [CrossRef] [PubMed]
- Chabner, B.A. NCI-60 Cell Line Screening: A Radical Departure in Its Time. J. Natl. Cancer Inst. 2016, 108, djv388. [Google Scholar] [CrossRef]
- Yingtaweesittikul, H.; Wu, J.; Mongia, A.; Peres, R.; Ko, K.; Nagarajan, N.; Suphavilai, C. CREAMMIST: An integrative probabilistic database for cancer drug response prediction. Nucleic Acids Res. 2023, 51, D1242–D1248. [Google Scholar] [CrossRef]
- Casiraghi, A.; Bensimon, A.; Superti-Furga, G. Recent developments in ligands and chemical probes targeting solute carrier transporters. Curr. Opin. Chem. Biol. 2021, 62, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Liu, T.; Shang, Y.; Dai, P.; Zhang, W.; Lee, B.J.; Huang, M.; Yang, D.; Wu, Q.; Liu, L.D.; et al. ERCC6L2 promotes DNA orientation-specific recombination in mammalian cells. Cell Res. 2020, 30, 732–744. [Google Scholar] [CrossRef]
- Olivieri, M.; Cho, T.; Álvarez-Quilón, A.; Li, K.; Schellenberg, M.J.; Zimmermann, M.; Hustedt, N.; Rossi, S.E.; Adam, S.; Melo, H.; et al. A Genetic Map of the Response to DNA Damage in Human Cells. Cell 2020, 182, 481–496.e21. [Google Scholar] [CrossRef]
- Ramaker, R.C.; Hardigan, A.A.; Gordon, E.R.; Wright, C.A.; Myers, R.M.; Cooper, S.J. Pooled CRISPR screening in pancreatic cancer cells implicates co-repressor complexes as a cause of multiple drug resistance via regulation of epithelial-to-mesenchymal transition. BMC Cancer 2021, 21, 632. [Google Scholar] [CrossRef]
- Lin, J.F.; Hu, P.S.; Wang, Y.Y.; Tan, Y.T.; Yu, K.; Liao, K.; Wu, Q.N.; Li, T.; Meng, Q.; Lin, J.Z.; et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct. Target. Ther. 2022, 7, 54. [Google Scholar] [CrossRef]
- Yin, J.; Ren, W.; Huang, X.; Deng, J.; Li, T.; Yin, Y. Potential Mechanisms Connecting Purine Metabolism and Cancer Therapy. Front. Immunol. 2018, 9, 1697. [Google Scholar] [CrossRef]
- Furuhashi, M. New insights into purine metabolism in metabolic diseases: Role of xanthine oxidoreductase activity. Am. J. Physiol.-Endocrinol. Metab. 2020, 319, E827–E834. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Torres, A.G.; Ribas de Pouplana, L. Inosine in biology and disease. Genes 2021, 12, 600. [Google Scholar] [CrossRef]
- Pang, B.; McFaline, J.L.; Burgis, N.E.; Dong, M.; Taghizadeh, K.; Sullivan, M.R.; Elmquist, C.E.; Cunningham, R.P.; Dedon, P.C. Defects in purine nucleotide metabolism lead to substantial incorporation of xanthine and hypoxanthine into DNA and RNA. Proc. Natl. Acad. Sci. USA 2012, 109, 2319–2324. [Google Scholar] [CrossRef]
- Lee, A.J.X.; Endesfelder, D.; Rowan, A.J.; Walther, A.; Nicolai, J.; Kschischo, M.; Swanton, C. Chromosomal Instability Confers Intrinsic Multi-Drug Resistance. Cancer Res 2011, 71, 1858–1870. [Google Scholar] [CrossRef] [PubMed]
- Lukow, D.A.; Sausville, E.L.; Suri, P.; Chunduri, N.K.; Wieland, A.; Leu, J.; Smith, J.C.; Girish, V.; Kumar, A.A.; Kendall, J.; et al. Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. Cell 2021, 56, 2427–2439.e4. [Google Scholar] [CrossRef] [PubMed]
- Khongkow, P.; Middleton, A.K.; Wong, J.P.M.; Kandola, N.K.; Kongsema, M.; De Moraes, G.N.; Gomes, A.R.; Lam, E.W.F. In vitro methods for studying the mechanisms of resistance to DNA-damaging therapeutic drugs. In Cancer Drug Resistance; Springer: Berlin/Heidelberg, Germany, 2016; Volume 1395, ISBN 9781493933457. [Google Scholar] [CrossRef]
- Battelli, M.G.; Bortolotti, M.; Polito, L.; Bolognesi, A. Metabolic syndrome and cancer risk: The role of xanthine oxidoreductase. Redox Biol. 2019, 21, 101070. [Google Scholar] [CrossRef]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef]
- Li, H.; Li, B.; Yang, F.; Duan, C.; Bai, Y.; Yang, J.J.; Chen, J.; von Stackelberg, A.; Chen, H.; Tang, J.; et al. De Novo Purine Biosynthesis in Drug Resistance and Tumor Relapse of Childhood ALL. Blood 2015, 126, 2627. [Google Scholar] [CrossRef]
- Shireman, J.M.; Atashi, F.; Lee, G.; Ali, E.S.; Saathoff, M.R.; Park, C.H.; Savchuk, S.; Baisiwala, S.; Miska, J.; Lesniak, M.S.; et al. De novo purine biosynthesis is a major driver of chemoresistance in glioblastoma. Brain 2021, 144, 1230–1246. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Wang, H.; Zuo, X.; Hong, L. Comprehensive Analysis of Purine-Metabolism-Related Gene Signature for Predicting Ovarian Cancer Prognosis, Immune Landscape, and Potential Treatment Options. J. Pers. Med. 2023, 13, 776. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, Y.; Sun, W.; Song, T.; Jiang, X.; Zeng, K.; Zeng, S.; Chen, L.; Yu, L. Blockade LAT1 Mediates Methionine Metabolism to Overcome Oxaliplatin Resistance under Hypoxia in Renal Cell Carcinoma. Cancers 2022, 14, 2551. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Spinelli, J.B.; Asara, J.M.; Toker, A. Adaptive reprogramming of De novo pyrimidine synthesis is a metabolic vulnerability in triple-negative breast cancer. Cancer Discov. 2017, 7, 391–399. [Google Scholar] [CrossRef]
- Chong, Y.C.; Toh, T.B.; Chan, Z.; Lin, Q.X.X.; Thng, D.K.H.; Hooi, L.; Ding, Z.; Shuen, T.; Toh, H.C.; Dan, Y.Y.; et al. Targeted Inhibition of Purine Metabolism Is Effective in Suppressing Hepatocellular Carcinoma Progression. Hepatol. Commun. 2020, 4, 1362–1381. [Google Scholar] [CrossRef]
- Mullen, N.J.; Singh, P.K. Nucleotide metabolism: A pan-cancer metabolic dependency. Nat. Rev. Cancer 2023, 23, 275–294. [Google Scholar] [CrossRef]
- Jiang, X.; Ma, Y.; Wang, T.; Zhou, H.; Wang, K.; Shi, W.; Qin, L.; Guan, J.; Li, L.; Long, B.; et al. Targeting UBE2T Potentiates Gemcitabine Efficacy in Pancreatic Cancer by Regulating Pyrimidine Metabolism and Replication Stress. Gastroenterology 2023, 164, 1232–1247. [Google Scholar] [CrossRef]
- Abu-Remaileh, M.; Aqeilan, R.I. Tumor suppressor WWOX regulates glucose metabolism via HIF1α modulation. Cell Death Differ. 2014, 21, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Pospiech, K.; Pluciennik, E.; Bednarek, A.K. WWOX tumor suppressor gene in breast cancer, a historical perspective and future directions. Front. Oncol. 2018, 8, 345. [Google Scholar] [CrossRef] [PubMed]
- Richards, R.I.; Choo, A.; Lee, C.S.; Dayan, S.; O’Keefe, L. WWOX, the chromosomal fragile site FRA16D spanning gene: Its role in metabolism and contribution to cancer. Exp. Biol. Med. 2015, 240, 338–344. [Google Scholar] [CrossRef]
- Baryła, I.; Kośla, K.; Bednarek, A.K. WWOX and metabolic regulation in normal and pathological conditions. J. Mol. Med. 2022, 100, 1691–1702. [Google Scholar] [CrossRef]
- Yin, F.; Liu, X.; Li, D.; Wang, Q.; Zhang, W.; Li, L. Tumor suppressor genes associated with drug resistance in ovarian cancer (Review). Oncol. Rep. 2013, 30, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Si, W.; Shen, J.; Zheng, H.; Fan, W. The role and mechanisms of action of microRNAs in cancer drug resistance. Clin. Epigenetics 2019, 11, 25. [Google Scholar] [CrossRef]
- Biersack, B. Alkylating anticancer agents and their relations to microRNAs. Cancer Drug Resist. 2019, 2, 1–17. [Google Scholar] [CrossRef]
- Jia, M.; Wei, Z.; Liu, P.; Zhao, X. Silencing of ABCG2 by microRNA-3163 inhibits multidrug resistance in retinoblastoma cancer stem cells. J. Korean Med. Sci. 2016, 31, 836–842. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Wang, C.; Xie, H.; Wang, Y.; Huang, J.; Rong, Y.; Zhang, H.; Kong, H.; Yang, Y.; Lu, Y. MicroRNA-3163 targets ADAM-17 and enhances the sensitivity of hepatocellular carcinoma cells to molecular targeted agents. Cell Death Dis. 2019, 10, 784. [Google Scholar] [CrossRef]
- Li, W.; Wang, W.; Ding, M.; Zheng, X.; Ma, S.; Wang, X. MiR-1244 sensitizes the resistance of non-small cell lung cancer A549 cell to cisplatin. Cancer Cell Int. 2016, 16, 30. [Google Scholar] [CrossRef]
- Stranger, B.E.; Forrest, M.S.; Dunning, M.; Ingle, C.E.; Beazley, C.; Thorne, N.; Redon, R.; Bird, C.P.; Grassi, A.D.; Lee, C.; et al. Relative Impact of Nucleotide and Copy Number Variation on Gene Expression Phenotypes. Science 2007, 315, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, V.; Wang, F.; Mills, G.B.; Chen, K. Uncoupling of gene expression from copy number presents therapeutic opportunities in aneuploid cancers. Cell Reports Med. 2021, 2, 100349. [Google Scholar] [CrossRef] [PubMed]
- Lauer, S.; Gresham, D. An evolving view of copy number variants. Curr. Genet. 2019, 65, 1287–1295. [Google Scholar] [CrossRef]
- Bock, C.; Datlinger, P.; Chardon, F.; Coelho, M.A.; Dong, M.B.; Lawson, K.A.; Lu, T.; Maroc, L.; Norman, T.M.; Song, B.; et al. High-content CRISPR screening. Nat. Rev. Methods Prim. 2022, 2, 8. [Google Scholar] [CrossRef]
- Turgeon, M.O.; Perry, N.J.S.; Poulogiannis, G. DNA damage, repair, and cancer metabolism. Front. Oncol. 2018, 8, 15. [Google Scholar] [CrossRef]
- Frejno, M.; Meng, C.; Ruprecht, B.; Oellerich, T.; Scheich, S.; Kleigrewe, K.; Drecoll, E.; Samaras, P.; Hogrebe, A.; Helm, D.; et al. Proteome activity landscapes of tumor cell lines determine drug responses. Nat. Commun. 2020, 11, 3639. [Google Scholar] [CrossRef]
- Kohn, K.W.; Zeeberg, B.M.; Reinhold, W.C.; Pommier, Y. Gene expression correlations in human cancer cell lines define molecular interaction networks for epithelial phenotype. PLoS ONE 2014, 9, e99269. [Google Scholar] [CrossRef]
- Pfister, T.D.; Reinhold, W.C.; Agama, K.; Gupta, S.; Khin, S.A.; Kinders, R.J.; Parchment, R.E.; Tomaszewski, J.E.; Doroshow, J.H.; Pommier, Y. Topoisomerase I levels in the NCI-60 cancer cell line panel determined by validated ELISA and microarray analysis and correlation with indenoisoquinoline sensitivity. Mol. Cancer Ther. 2009, 8, 1878–1884. [Google Scholar] [CrossRef]
- Shankavaram, U.T.; Varma, S.; Kane, D.; Sunshine, M.; Chary, K.K.; Reinhold, W.C.; Pommier, Y.; Weinstein, J.N. CellMiner: A relational database and query tool for the NCI-60 cancer cell lines. BMC Genomics 2009, 10, 277. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; De Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, 388–396. [Google Scholar] [CrossRef]
- Zhou, G.; Pang, Z.; Lu, Y.; Ewald, J.; Xia, J. OmicsNet 2.0: A web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 2022, 50, W527–W533. [Google Scholar] [CrossRef]
- Zhou, G.; Ewald, J.; Xia, J. OmicsAnalyst: A comprehensive web-based platform for visual analytics of multi-omics data. Nucleic Acids Res. 2021, 49, W476–W482. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Shannon, C.P.; Gautier, B.; Rohart, F.; Vacher, M.; Tebbutt, S.J.; Cao, K.A.L. DIABLO: An integrative approach for identifying key molecular drivers from multi-omics assays. Bioinformatics 2019, 35, 3055–3062. [Google Scholar] [CrossRef]
- Luo, W.; Pant, G.; Bhavnasi, Y.K.; Blanchard, S.G.; Brouwer, C. Pathview Web: User friendly pathway visualization and data integration. Nucleic Acids Res. 2017, 45, W501–W508. [Google Scholar] [CrossRef]
- Luo, W.; Brouwer, C. Pathview: An R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 2013, 29, 1830–1831. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. Molecular network analysis of diseases and drugs in KEGG. Methods Mol Biol 2013, 939, 263–275. [Google Scholar] [CrossRef]
- Wiggs, A.; Molina, S.; Sumner, S.J.; Rushing, B.R. A Review of Metabolic Targets of Anticancer Nutrients and Nutraceuticals in Pre-Clinical Models of Triple-Negative Breast Cancer. Nutrients 2022, 14, 1990. [Google Scholar] [CrossRef] [PubMed]
- Rushing, B.R.; Wiggs, A.; Molina, S.; Schroder, M.; Sumner, S. Metabolomics Analysis Reveals Novel Targets of Chemosensitizing Polyphenols and Omega-3 Polyunsaturated Fatty Acids in Triple Negative Breast Cancer Cells. Int. J. Mol. Med. 2023, 24, 4406. [Google Scholar] [CrossRef] [PubMed]
- D’Eliseo, D.; Velotti, F. Omega-3 Fatty Acids and Cancer Cell Cytotoxicity: Implications for Multi-Targeted Cancer Therapy. J. Clin. Med. 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Lewandowska, U.; Gorlach, S.; Owczarek, K.; Hrabec, E.; Szewczyk, K. Synergistic interactions between anticancer chemotherapeutics and phenolic compounds and anticancer synergy between polyphenols. Postepy Hig. Med. Dosw. 2014, 68, 528–540. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Cancer Type | Median z-Score Rank Order | Quartile Number |
|---|---|---|---|
| SR | Leukemia | 3.5 | 1 |
| HL-60(TB) | Leukemia | 4 | 1 |
| CCRF-CEM | Leukemia | 6 | 1 |
| MOLT-4 | Leukemia | 6 | 1 |
| NCI-H460 | Non-Small Cell Lung | 6 | 1 |
| UACC-62 | Melanoma | 8 | 1 |
| ACHN | Renal | 10 | 1 |
| CAKI-1 | Renal | 11 | 1 |
| SF-295 | Central nervous system | 12 | 1 |
| SF-539 | Central nervous system | 14 | 1 |
| LOX IMVI | Melanoma | 15 | 1 |
| 786-0 | Renal | 15 | 1 |
| MCF7 | Breast | 17 | 1 |
| HOP-62 | Non-Small Cell Lung | 18 | 1 |
| SN12C | Renal | 19 | 1 |
| SF-268 | Central nervous system | 20 | 2 |
| U251 | Central nervous system | 20 | 2 |
| A549/ATCC | Non-Small Cell Lung | 20 | 2 |
| NCI-H23 | Non-Small Cell Lung | 21 | 2 |
| NCI-H522 | Non-Small Cell Lung | 22.5 | 2 |
| SNB-75 | Central nervous system | 23 | 2 |
| M14 | Melanoma | 23 | 2 |
| SK-MEL-5 | Melanoma | 25 | 2 |
| SW-620 | Colon | 26 | 2 |
| HOP-92 | Non-Small Cell Lung | 26 | 2 |
| DU-145 | Genitourinary | 26 | 2 |
| HCT-116 | Colon | 29 | 2 |
| RPMI-8226 | Leukemia | 30 | 2 |
| RXF-393 | Renal | 30 | 2 |
| MALME-3M | Melanoma | 32 | 3 |
| OVCAR-8 | Genitourinary | 32 | 3 |
| T-47D | Breast | 33.5 | 3 |
| HCC-2998 | Colon | 34 | 3 |
| BT-549 | Breast | 34.5 | 3 |
| SNB-19 | Central nervous system | 35 | 3 |
| K-562 | Leukemia | 35 | 3 |
| NCI-H226 | Non-Small Cell Lung | 35.5 | 3 |
| SK-OV-3 | Genitourinary | 35.5 | 3 |
| IGROV1 | Genitourinary | 36 | 3 |
| NCI/ADR-RES | Genitourinary | 36 | 3 |
| HCT-15 | Colon | 37 | 3 |
| COLO 205 | Colon | 38 | 3 |
| UACC-257 | Melanoma | 38 | 3 |
| OVCAR-3 | Genitourinary | 39 | 4 |
| UO-31 | Renal | 39 | 4 |
| HT29 | Colon | 40 | 4 |
| MDA-MB-435 | Melanoma | 40 | 4 |
| A498 | Renal | 42 | 4 |
| PC-3 | Genitourinary | 44 | 4 |
| OVCAR-4 | Genitourinary | 45 | 4 |
| OVCAR-5 | Genitourinary | 45 | 4 |
| KM12 | Colon | 47 | 4 |
| SK-MEL-28 | Melanoma | 47 | 4 |
| EKVX | Non-Small Cell Lung | 48 | 4 |
| MDA-MB-231/ATCC | Breast | 51 | 4 |
| HS 578T | Breast | 51 | 4 |
| NCI-H322M | Non-Small Cell Lung | 53 | 4 |
| TK-10 | Renal | 53 | 4 |
| Pathway | p-Value | FDR |
|---|---|---|
| EGFR tyrosine kinase inhibitor resistance | 4.24 × 10−73 | 1.42 × 10−70 |
| Cysteine and methionine metabolism | 4.36 × 10−14 | 5.62 × 10−12 |
| Pyrimidine metabolism | 5.02 × 10−14 | 5.62 × 10−12 |
| Starch and sucrose metabolism | 1.3 × 10−12 | 9.03 × 10−11 |
| Purine metabolism | 1.34 × 10−12 | 9.03 × 10−11 |
| ABC transporters | 7.98 × 10−09 | 4.47 × 10−07 |
| Nicotinate and nicotinamide metabolism | 9.81 × 10−09 | 4.71 × 10−07 |
| Alanine, aspartate, and glutamate metabolism | 9.69 × 10−08 | 4.07 × 10−06 |
| Valine, leucine, and isoleucine degradation | 1.91 × 10−07 | 7.12 × 10−06 |
| Platinum drug resistance | 1.21 × 10−06 | 4.07 × 10−05 |
| Pathway | p-Value |
|---|---|
| Purine metabolism | 0.005412 |
| Fructose and mannose metabolism | 0.062016 |
| Glycosaminoglycan degradation | 0.073378 |
| Amino sugar and nucleotide sugar metabolism | 0.193000 |
| Arginine biosynthesis | 0.243890 |
| Pyrimidine metabolism | 0.269520 |
| One carbon pool by folate | 0.274760 |
| Pentose and glucuronate interconversions | 0.282290 |
| Pantothenate and CoA biosynthesis | 0.297110 |
| Selenocompound metabolism | 0.304420 |
| Pathway | p-Value |
|---|---|
| Purine metabolism | 0.012062 |
| Synthesis and degradation of ketone bodies | 0.056019 |
| Butanoate metabolism | 0.154510 |
| Pantothenate and CoA biosynthesis | 0.178800 |
| Terpenoid backbone biosynthesis | 0.188330 |
| Fructose and mannose metabolism | 0.207080 |
| Starch and sucrose metabolism | 0.220880 |
| Beta-Alanine metabolism | 0.225430 |
| Pyruvate metabolism | 0.229960 |
| Biosynthesis of unsaturated fatty acids | 0.238940 |
| Gene Symbol | Gene Name | Number of CNV Markers | Direction of Change in Q4 |
|---|---|---|---|
| MIR1302-2 | microRNA 1302-2 | 149 | Increased |
| MIR3163 | microRNA 3163 | 87 | Increased |
| MIR1244-3 | microRNA 1244-3 | 27 | Decreased |
| WWOX | WW domain containing oxidoreductase | 22 | Decreased |
| CNTN5 | contactin 5 | 17 | Increased |
| ARL17B | ADP ribosylation factor like GTPase 17B | 12 | Decreased |
| VAT1L | vesicle amine transport 1 like | 10 | Decreased |
| OR4F3 | olfactory receptor family 4 subfamily F member 3 | 9 | Decreased |
| MIR1302-9 | microRNA 1302-9 | 8 | Increased |
| DDAH1 | dimethylarginine dimethylaminohydrolase 1 | 7 | Increased |
| DISC1FP1 | DISC1 fusion partner 1 | 7 | Increased |
| PGR | PGR | 7 | Increased |
| RNU6-2 | RNA, U6 small nuclear 2 | 7 | Increased |
| FRG2 | FSHD region gene 2 | 6 | Decreased |
| TRPC6 | transient receptor potential cation channel subfamily C member 6 | 6 | Increased |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rushing, B.R. Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents. Int. J. Mol. Sci. 2023, 24, 13242. https://doi.org/10.3390/ijms241713242
Rushing BR. Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents. International Journal of Molecular Sciences. 2023; 24(17):13242. https://doi.org/10.3390/ijms241713242
Chicago/Turabian StyleRushing, Blake R. 2023. "Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents" International Journal of Molecular Sciences 24, no. 17: 13242. https://doi.org/10.3390/ijms241713242
APA StyleRushing, B. R. (2023). Multi-Omics Analysis of NCI-60 Cell Line Data Reveals Novel Metabolic Processes Linked with Resistance to Alkylating Anti-Cancer Agents. International Journal of Molecular Sciences, 24(17), 13242. https://doi.org/10.3390/ijms241713242