
Calcium Dyshomeostasis Drives Pathophysiology and Neuronal Demise in Age-Related Neurodegenerative Diseases

Abstract
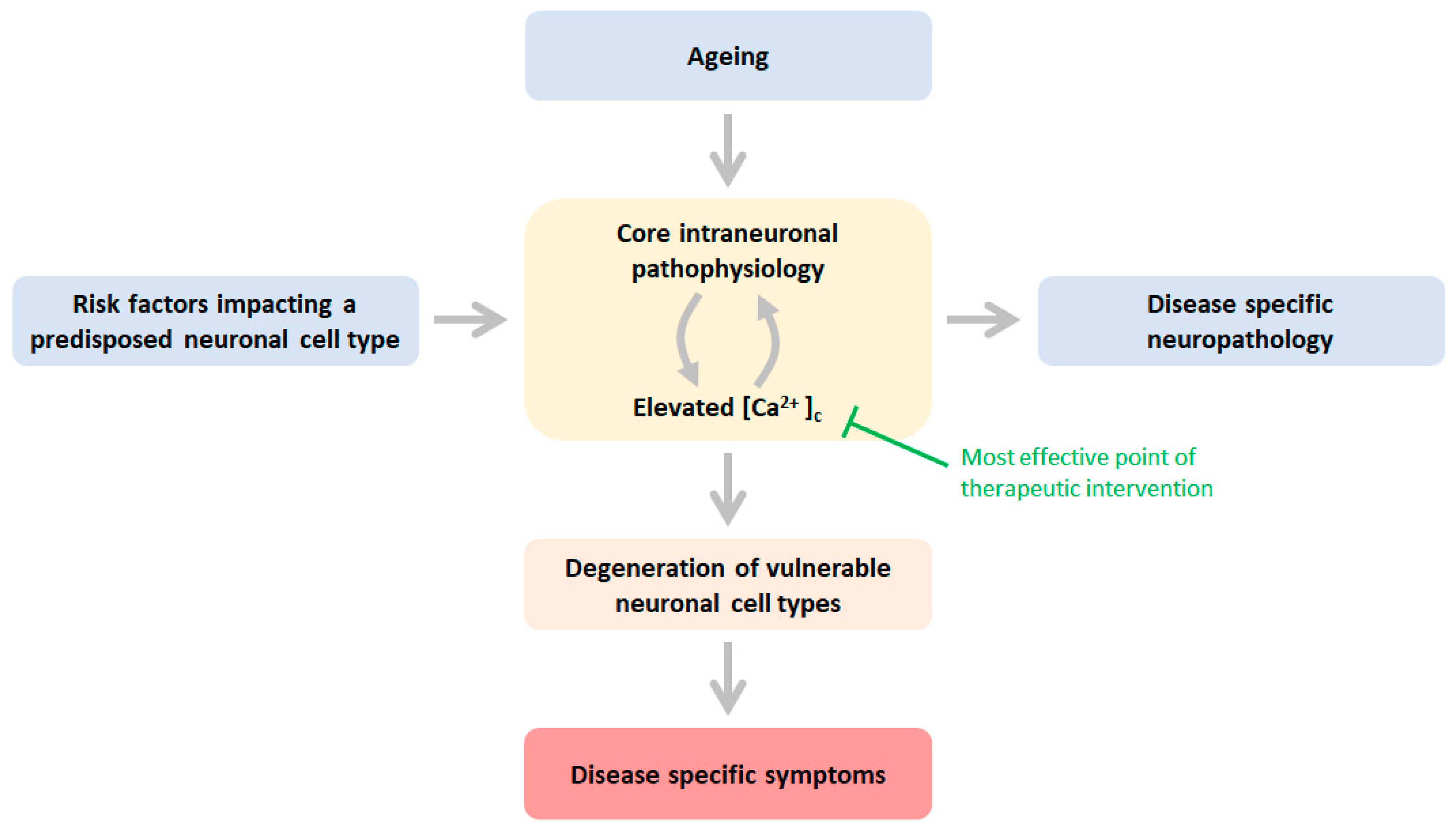
:1. Introduction
2. Making and Breaking Neuronal Connections: An Intrinsic Property of the Brain
3. Intracellular Calcium: The Concentration Gradient Specifies Its Function for Better and for Worse
4. Risk Factors of Neurodegeneration Compromise the Ca2+ Concentration Gradient
5. Selective Neuronal Vulnerability to Risk Factors (Except Ageing)
6. Different Risk Factors Impacting Different Neuronal Cell Types but Yet a Common Outcome
7. Therapeutic Approaches: Restoring the Distorted Calcium Gradient
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Webber, E.K.; Fivaz, M.; Stutzmann, G.E.; Griffioen, G. Cytosolic calcium: Judge, jury and executioner of neurodegeneration in Alzheimer’s disease and beyond. Alzheimer’s Dement. 2023, 19, 3701–3717. [Google Scholar] [CrossRef] [PubMed]
- Kolb, B.; Whishaw, I.Q. Brain Plasticity and Behavior. Annu. Rev. Psychol. 1998, 49, 43–64. [Google Scholar] [CrossRef]
- Schaefer, N.; Rotermund, C.; Blumrich, E.-M.; Lourenco, M.V.; Joshi, P.; Hegemann, R.U.; Jamwal, S.; Ali, N.; Romero, E.M.G.; Sharma, S.; et al. The malleable brain: Plasticity of neural circuits and behavior—A review from students to students. J. Neurochem. 2017, 142, 790–811. [Google Scholar] [CrossRef]
- Burek, M.J.; Oppenheim, R.W. Programmed Cell Death in the Developing Nervous System. Brain Pathol. 1996, 6, 427–446. [Google Scholar] [CrossRef]
- Reese, L.C.; Taglialatela, G. A Role for Calcineurin in Alzheimers Disease. Curr. Neuropharmacol. 2011, 9, 685–692. [Google Scholar] [CrossRef]
- Wang, H.-G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-Induced Apoptosis Through Calcineurin Dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Han, X.-J.; Lu, Y.-F.; Li, S.-A.; Tomizawa, K.; Takei, K.; Matsushita, M.; Matsui, H. Involvement of calcineurin in glutamate-induced mitochondrial dynamics in neurons. Neurosci. Res. 2008, 60, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Roman, R.J.; Fan, F. Hippocampus is more susceptible to hypoxic injury: Has the Rosetta Stone of regional variation in neurovascular coupling been deciphered? Geroscience 2021, 44, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Dineley, K.T.; Hogan, D.; Zhang, W.-R.; Taglialatela, G. Acute inhibition of calcineurin restores associative learning and memory in Tg2576 APP transgenic mice. Neurobiol. Learn. Mem. 2007, 88, 217–224. [Google Scholar] [CrossRef]
- Taglialatela, G.; Rastellini, C.; Cicalese, L. Reduced Incidence of Dementia in Solid Organ Transplant Patients Treated with Calcineurin Inhibitors. J. Alzheimer’s Dis. 2015, 47, 329–333. [Google Scholar] [CrossRef]
- Abdul, H.M.; Sama, M.A.; Furman, J.L.; Mathis, D.M.; Beckett, T.L.; Weidner, A.M.; Patel, E.S.; Baig, I.; Murphy, M.P.; LeVine, H.; et al. Cognitive Decline in Alzheimer’s Disease Is Associated with Selective Changes in Calcineurin/NFAT Signaling. J. Neurosci. 2009, 29, 12957–12969. [Google Scholar] [CrossRef]
- Alhaider, A. Sub-chronic treatment of calcineurin inhibitor averts impairment of cognitive function in animal model of amnesia. Afr. J. Pharm. Pharmacol. 2013, 7, 2998–3003. [Google Scholar] [CrossRef]
- Reese, L.C.; Taglialatela, G. Neuroimmunomodulation by calcineurin in aging and Alzheimer’s disease. Aging Dis. 2010, 1, 245–253. [Google Scholar]
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.-H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E3773–E3781. [Google Scholar] [CrossRef] [PubMed]
- Baumgärtel, K.; Mansuy, I.M. Neural functions of calcineurin in synaptic plasticity and memory. Learn. Mem. 2012, 19, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Norris, C.M. Calcineurin: Directing the damage in Alzheimer disease. J. Neurochem. 2018, 147, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Lautermilch, N.J.; Spitzer, N.C. Regulation of Calcineurin by Growth Cone Calcium Waves Controls Neurite Extension. J. Neurosci. 2000, 20, 315–325. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, J.-L.; Xu, X.; Zhou, X.-P.; Du, J.; Wang, X.; Zhou, Y.; Zhu, Q.; Yao, L.-L.; Wang, Y.-G.; et al. NMDA receptors inhibit axonal outgrowth by inactivating Akt and activating GSK-3β via calcineurin in cultured immature hippocampal neurons. Exp. Cell Res. 2018, 371, 389–398. [Google Scholar] [CrossRef]
- Zhuo, M.; Zhang, W.; Son, H.; Mansuy, I.; Sobel, R.A.; Seidman, J.; Kandel, E.R. A selective role of calcineurin Aα in synaptic depotentiation in hippocampus. Proc. Natl. Acad. Sci. USA 1999, 96, 4650–4655. [Google Scholar] [CrossRef]
- Descazeaud, V.; Mestre, E.; Marquet, P.; Essig, M. Calcineurin regulation of cytoskeleton organisation: A new paradigm to analyse the effects of calcineurin inhibitors on the kidney. J. Cell. Mol. Med. 2012, 16, 218–227. [Google Scholar] [CrossRef]
- Coleman, M.P.; Höke, A. Programmed axon degeneration: From mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020, 21, 183–196. [Google Scholar] [CrossRef]
- Hernández, D.E.; Salvadores, N.A.; Moya-Alvarado, G.; Catalán, R.J.; Bronfman, F.C.; Court, F.A. Axonal degeneration induced by glutamate-excitotoxicity is mediated by necroptosis. J. Cell Sci. 2018, 131, jcs214684. [Google Scholar] [CrossRef]
- Hopkins, E.L.; Gu, W.; Kobe, B.; Coleman, M.P. A Novel NAD Signaling Mechanism in Axon Degeneration and its Relationship to Innate Immunity. Front. Mol. Biosci. 2021, 8, 703532. [Google Scholar] [CrossRef] [PubMed]
- Metwally, E.; Zhao, G.; Zhang, Y.Q. The calcium-dependent protease calpain in neuronal remodeling and neurodegeneration. Trends Neurosci. 2021, 44, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Mahaman, Y.A.R.; Huang, F.; Afewerky, H.K.; Maibouge, T.M.S.; Ghose, B.; Wang, X. Involvement of calpain in the neuropathogenesis of Alzheimer’s disease. Med. Res. Rev. 2018, 39, 608–630. [Google Scholar] [CrossRef] [PubMed]
- Roufayel, R.; Murshid, N. CDK5: Key Regulator of Apoptosis and Cell Survival. Biomedicines 2019, 7, 88. [Google Scholar] [CrossRef]
- Pao, P.-C.; Tsai, L.-H. Three decades of Cdk5. J. Biomed. Sci. 2021, 28, 79. [Google Scholar] [CrossRef] [PubMed]
- Goñi-Oliver, P.; Lucas, J.J.; Avila, J.; Hernández, F. N-terminal Cleavage of GSK-3 by Calpain. J. Biol. Chem. 2007, 282, 22406–22413. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Bigio, E.H. Calpain-Mediated Tau Cleavage: A Mechanism Leading to Neurodegeneration Shared by Multiple Tauopathies. Mol. Med. 2011, 17, 676–685. [Google Scholar] [CrossRef]
- Zhang, X.; Connelly, J.; Levitan, E.S.; Sun, D.; Wang, J.Q. Calcium/Calmodulin–Dependent Protein Kinase II in Cerebrovascular Diseases. Transl. Stroke Res. 2021, 12, 513–529. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Giese, K.P. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Mol. Brain 2015, 8, 78. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-Reticulum Calcium Depletion and Disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef]
- Chami, M. Calcium Signalling in Alzheimer’s Disease: From Pathophysiological Regulation to Therapeutic Approaches. Cells 2021, 10, 140. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; LaFerla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed]
- Chami, M.; Checler, F. Alterations of the Endoplasmic Reticulum (ER) Calcium Signaling Molecular Components in Alzheimer’s Disease. Cells 2020, 9, 2577. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, D.; Checler, F.; Chami, M. Ryanodine receptors: Physiological function and deregulation in Alzheimer disease. Mol. Neurodegener. 2014, 9, 21. [Google Scholar] [CrossRef]
- Marshall, C. Specificity of receptor tyrosine kinase signaling: Transient versus sustained extracellular signal-regulated kinase activation. Cell 1995, 80, 179–185. [Google Scholar] [CrossRef]
- Stanciu, M.; DeFranco, D.B. Prolonged Nuclear Retention of Activated Extracellular Signal-regulated Protein Kinase Promotes Cell Death Generated by Oxidative Toxicity or Proteasome Inhibition in a Neuronal Cell Line. J. Biol. Chem. 2002, 277, 4010–4017. [Google Scholar] [CrossRef]
- Stanciu, M.; Wang, Y.; Kentor, R.; Burke, N.; Watkins, S.; Kress, G.; Reynolds, I.; Klann, E.; Angiolieri, M.R.; Johnson, J.W.; et al. Persistent Activation of ERK Contributes to Glutamate-induced Oxidative Toxicity in a Neuronal Cell Line and Primary Cortical Neuron Cultures. J. Biol. Chem. 2000, 275, 12200–12206. [Google Scholar] [CrossRef]
- Grewal, S.S.; York, R.D.; Stork, P.J. Extracellular-signal-regulated kinase signalling in neurons. Curr. Opin. Neurobiol. 1999, 9, 544–553. [Google Scholar] [CrossRef]
- Cai, Q.; Sheng, Z.-H. Moving or Stopping Mitochondria: Miro as a Traffic Cop by Sensing Calcium. Neuron 2009, 61, 493–496. [Google Scholar] [CrossRef]
- Tadic, V.; Prell, T.; Lautenschlaeger, J.; Grosskreutz, J. The ER mitochondria calcium cycle and ER stress response as therapeutic targets in amyotrophic lateral sclerosis. Front. Cell. Neurosci. 2014, 8, 147. [Google Scholar] [CrossRef] [PubMed]
- Baev, A.Y.; Vinokurov, A.Y.; Novikova, I.N.; Dremin, V.V.; Potapova, E.V.; Abramov, A.Y. Interaction of Mitochondrial Calcium and ROS in Neurodegeneration. Cells 2022, 11, 706. [Google Scholar] [CrossRef]
- Müller, M.; Ahumada-Castro, U.; Sanhueza, M.; Gonzalez-Billault, C.; Felipe, A.; Court, F.A.; Cárdenas, C. Mitochondria and Calcium Regulation as Basis of Neurodegeneration Associated with Aging. Front. Neurosci. 2018, 12, 470. [Google Scholar] [CrossRef]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Thakkar, A. Interactions of Mitochondria/Metabolism and Calcium Regulation in Alzheimer’s Disease: A Calcinist Point of View. Neurochem. Res. 2017, 42, 1636–1648. [Google Scholar] [CrossRef]
- Park, J.Y.; Jang, S.Y.; Shin, Y.K.; Suh, D.J.; Park, H.T. Calcium-dependent proteasome activation is required for axonal neurofilament degradation. Neural Regen. Res. 2013, 8, 3401–3409. [Google Scholar] [CrossRef] [PubMed]
- Djakovic, S.N.; Schwarz, L.A.; Barylko, B.; DeMartino, G.N.; Patrick, G.N. Regulation of the Proteasome by Neuronal Activity and Calcium/Calmodulin-dependent Protein Kinase II. J. Biol. Chem. 2009, 284, 26655–26665. [Google Scholar] [CrossRef] [PubMed]
- Court, F.; Arrazola, M.S. Compartmentalised necroptosis activation in excitotoxicity-induced axonal degeneration: A novel mechanism implicated in neurodegenerative disease pathology. Neural Regen. Res. 2019, 14, 1385–1386. [Google Scholar] [CrossRef]
- Mustaly-Kalimi, S.; Gallegos, W.; Marr, R.A.; Gilman-Sachs, A.; Peterson, D.A.; Sekler, I.; Stutzmann, G.E. Protein mishandling and impaired lysosomal proteolysis generated through calcium dysregulation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2022, 119, e2211999119. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-S.; McNeil, B.D.; Xu, J.; Fan, J.; Xue, L.; Melicoff, E.; Adachi, R.; Bai, L.; Wu, L.-G. Ca2+ and calmodulin initiate all forms of endocytosis during depolarisation at a nerve terminal. Nat. Neurosci. 2009, 12, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.-G.; Hamid, E.; Shin, W.; Chiang, H.-C. Exocytosis and Endocytosis: Modes, Functions, and Coupling Mechanisms. Annu. Rev. Physiol. 2014, 76, 301–331. [Google Scholar] [CrossRef]
- Burrinha, T.; Martinsson, I.; Gomes, R.; Terrasso, A.P.; Gouras, G.K.; Almeida, C.G. Upregulation of APP endocytosis by neuronal aging drives amyloid-dependent synapse loss. J. Cell Sci. 2021, 134, jcs255752. [Google Scholar] [CrossRef] [PubMed]
- Choy, R.W.-Y.; Cheng, Z.; Schekman, R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. USA 2012, 109, E2077–E2082. [Google Scholar] [CrossRef]
- Cirrito, J.R.; Kang, J.-E.; Lee, J.; Stewart, F.R.; Verges, D.K.; Silverio, L.M.; Bu, G.; Mennerick, S.; Holtzman, D.M. Endocytosis Is Required for Synaptic Activity-Dependent Release of Amyloid-β In Vivo. Neuron 2008, 58, 42–51. [Google Scholar] [CrossRef]
- Pooler, A.M.; Usardi, A.; Evans, C.J.; Philpott, K.L.; Noble, W.; Hanger, D.P. Dynamic association of tau with neuronal membranes is regulated by phosphorylation. Neurobiol. Aging 2012, 33, 431.e27–431.e38. [Google Scholar] [CrossRef] [PubMed]
- Hefti, M.M.; Kim, S.; Bell, A.J.; Betters, R.K.; Fiock, K.L.; Iida, M.A.; Smalley, M.E.; Farrell, K.; Fowkes, M.E.; Crary, J.F. Tau Phosphorylation and Aggregation in the Developing Human Brain. J. Neuropathol. Exp. Neurol. 2019, 78, 930–938. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Drew, K.L.; Perry, G.; Smith, M.A.; Zhu, X. Physiological regulation of tau phosphorylation during hibernation. J. Neurochem. 2008, 105, 2098–2108. [Google Scholar] [CrossRef]
- Arendt, T.; Bullmann, T. Neuronal plasticity in hibernation and the proposed role of the microtubule-associated protein tau as a “master switch” regulating synaptic gain in neuronal networks. Am. J. Physiol. Integr. Comp. Physiol. 2013, 305, R478–R489. [Google Scholar] [CrossRef] [PubMed]
- Goulay, R.; Romo, L.M.; Hol, E.M.; Dijkhuizen, R.M. From Stroke to Dementia: A Comprehensive Review Exposing Tight Interactions Between Stroke and Amyloid-β Formation. Transl. Stroke Res. 2020, 11, 601–614. [Google Scholar] [CrossRef]
- Liu, W.; Wong, A.; Law, A.C.; Mok, V.C. Cerebrovascular Disease, Amyloid Plaques, and Dementia. Stroke 2015, 46, 1402–1407. [Google Scholar] [CrossRef] [PubMed]
- Bootman, M.D.; Bultynck, G. Fundamentals of Cellular Calcium Signaling: A Primer. Cold Spring Harb. Perspect. Biol. 2020, 12, a038802. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Calcium regulation of neural rhythms, memory and Alzheimer’s disease. J. Physiol. 2014, 592 Pt 2, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Elementary and global aspects of calcium signalling. J. Physiol. 1997, 499 Pt 2, 291–306. [Google Scholar] [CrossRef]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Stutzmann, G.E. The Pathogenesis of Alzheimers Disease—Is It a Lifelong “Calciumopathy”? Neurosci. 2007, 13, 546–559. [Google Scholar] [CrossRef]
- Sun, N.; Youle, R.J.; Finkel, T. The Mitochondrial Basis of Aging. Mol. Cell 2016, 61, 654–666. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Simoes, I.C.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef]
- Adam-Vizi, V.; Starkov, A.A. Calcium and Mitochondrial Reactive Oxygen Species Generation: How to Read the Facts. J. Alzheimer’s Dis. 2010, 20 (Suppl. S2), S413–S426. [Google Scholar] [CrossRef]
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Bell, J.D.; Baker, A.J. Traumatic brain injury: Can the consequences be stopped? Can. Med. Assoc. J. 2008, 178, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.-H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef]
- Khachaturian, Z.S. Calcium Hypothesis of Alzheimer’s Disease and Brain Aginga. Ann. N. Y. Acad. Sci. 2006, 747, 1–11. [Google Scholar] [CrossRef]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed]
- Khachaturian, Z.S. Introduction and Overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium Dysregulation and Membrane Disruption as a Ubiquitous Neurotoxic Mechanism of Soluble Amyloid Oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef]
- Ferreira, I.; Bajouco, L.; Mota, S.; Auberson, Y.; Oliveira, C.; Rego, A. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106. [Google Scholar] [CrossRef]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar] [CrossRef]
- Pierrot, N.; Ghisdal, P.; Caumont, A.-S.; Octave, J.-N. Intraneuronal amyloid-β1-42 production triggered by sustained increase of cytosolic calcium concentration induces neuronal death. J. Neurochem. 2004, 88, 1140–1150. [Google Scholar] [CrossRef]
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897. [Google Scholar] [CrossRef]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The Many Faces of Tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef]
- Buxbaum, J.D.; Ruefli, A.A.; Parker, C.A.; Cypess, A.M.; Greengard, P. Calcium regulates processing of the Alzheimer amyloid protein precursor in a protein kinase C-independent manner. Proc. Natl. Acad. Sci. USA 1994, 91, 4489–4493. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Dreses-Werringloer, U.; Vingtdeux, V. Calcium signaling in neurodegeneration. Mol. Neurodegener. 2009, 4, 20. [Google Scholar] [CrossRef] [PubMed]
- Cortes, L.; Malva, J.; Rego, A.C.; Pereira, C.F. Calcium Signaling in Aging and Neurodegenerative Diseases 2019. Int. J. Mol. Sci. 2020, 21, 1125. [Google Scholar] [CrossRef] [PubMed]
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100. [Google Scholar] [CrossRef]
- Kasumu, A.; Bezprozvanny, I. Deranged Calcium Signaling in Purkinje Cells and Pathogenesis in Spinocerebellar Ataxia 2 (SCA2) and Other Ataxias. Cerebellum 2010, 11, 630–639. [Google Scholar] [CrossRef]
- Imamura, K.; Sahara, N.; Kanaan, N.M.; Tsukita, K.; Kondo, T.; Kutoku, Y.; Ohsawa, Y.; Sunada, Y.; Kawakami, K.; Hotta, A.; et al. Calcium dysregulation contributes to neurodegeneration in FTLD patient iPSC-derived neurons. Sci. Rep. 2016, 6, 34904. [Google Scholar] [CrossRef]
- Llorens, F.; Thüne, K.; Sikorska, B.; Schmitz, M.; Tahir, W.; Fernández-Borges, N.; Cramm, M.; Gotzmann, N.; Carmona, M.; Streichenberger, N.; et al. Altered Ca2+ homeostasis induces Calpain-Cathepsin axis activation in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. Commun. 2017, 5, 35. [Google Scholar] [CrossRef] [PubMed]
- Canet, G.; Chevallier, N.; Zussy, C.; Desrumaux, C.; Givalois, L. Central Role of Glucocorticoid Receptors in Alzheimer’s Disease and Depression. Front. Neurosci. 2018, 12, 739. [Google Scholar] [CrossRef]
- Storch, A.; Ludolph, A.C.; Schwarz, J. Dopamine transporter: Involvement in selective dopaminergic neurotoxicity and degeneration. J. Neural Transm. 2004, 111, 1267–1286. [Google Scholar] [CrossRef]
- Praschberger, R.; Kuenen, S.; Schoovaerts, N.; Kaempf, N.; Singh, J.; Janssens, J.; Swerts, J.; Nachman, E.; Calatayud, C.; Aerts, S.; et al. Neuronal identity defines α-synuclein and tau toxicity. Neuron 2023, 111, 1577–1590.e11. [Google Scholar] [CrossRef] [PubMed]
- Lotharius, J.; Brundin, P. Pathogenesis of parkinson’s disease: Dopamine, vesicles and α-synuclein. Nat. Rev. Neurosci. 2002, 3, 932–942. [Google Scholar] [CrossRef] [PubMed]
- Redox-Active Metals in Dopaminergic Neuronal Death. Available online: https://encyclopedia.pub/entry/40373 (accessed on 28 June 2023).
- Ma, L.; Azad, M.G.; Dharmasivam, M.; Richardson, V.; Quinn, R.J.; Feng, Y.; Pountney, D.L.; Tonissen, K.F.; Mellick, G.D.; Yanatori, I.; et al. Parkinson’s disease: Alterations in iron and redox biology as a key to unlock therapeutic strategies. Redox Biol. 2021, 41, 101896. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.-P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- De Vos, K.J.; Hafezparast, M. Neurobiology of axonal transport defects in motor neuron diseases: Opportunities for translational research? Neurobiol. Dis. 2017, 105, 283–299. [Google Scholar] [CrossRef]
- Perosa, V.; Priester, A.; Ziegler, G.; Cardenas-Blanco, A.; Dobisch, L.; Spallazzi, M.; Assmann, A.; Maass, A.; Speck, O.; Oltmer, J.; et al. Hippocampal vascular reserve associated with cognitive performance and hippocampal volume. Brain 2020, 143, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Kurtishi, A.; Rosen, B.; Patil, K.S.; Alves, G.W.; Møller, S.G. Cellular Proteostasis in Neurodegeneration. Mol. Neurobiol. 2018, 56, 3676–3689. [Google Scholar] [CrossRef]
- Morfini, G.A.; Burns, M.; Binder, L.I.; Kanaan, N.M.; Lapointe, N.; Bosco, D.A.; Brown, R.H., Jr.; Brown, H.; Tiwari, A.; Hayward, L.; et al. Axonal Transport Defects in Neurodegenerative Diseases. J. Neurosci. 2009, 29, 12776–12786. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Zhang, B.; Lee, V.M.-Y.; Trojanowski, J.Q. Axonal transport defects: A common theme in neurodegenerative diseases. Acta Neuropathol. 2005, 109, 5–13. [Google Scholar] [CrossRef]
- Peng, J.; Liang, G.; Inan, S.; Wu, Z.; Joseph, D.J.; Meng, Q.; Peng, Y.; Eckenhoff, M.F.; Wei, H. Dantrolene ameliorates cognitive decline and neuropathology in Alzheimer triple transgenic mice. Neurosci. Lett. 2012, 516, 274–279. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, B.; Liu, C.; Liang, G.; Liu, W.; Pickup, S.; Meng, Q.; Tian, Y.; Li, S.; Eckenhoff, M.F.; et al. Long-term Dantrolene Treatment Reduced Intraneuronal Amyloid in Aged Alzheimer Triple Transgenic Mice. Alzheimer Dis. Assoc. Disord. 2015, 29, 184–191. [Google Scholar] [CrossRef]
- Oulès, B.; Del Prete, D.; Greco, B.; Zhang, X.; Lauritzen, I.; Sevalle, J.; Moreno, S.; Paterlini-Bréchot, P.; Trebak, M.; Checler, F.; et al. Ryanodine Receptor Blockade Reduces Amyloid-β Load and Memory Impairments in Tg2576 Mouse Model of Alzheimer Disease. J. Neurosci. 2012, 32, 11820–11834. [Google Scholar] [CrossRef] [PubMed]
- Anekonda, T.S.; Quinn, J.F. Calcium channel blocking as a therapeutic strategy for Alzheimer’s disease: The case for isradipine. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Anekonda, T.S.; Quinn, J.F.; Harris, C.; Frahler, K.; Wadsworth, T.L.; Woltjer, R.L. L-type voltage-gated calcium channel blockade with isradipine as a therapeutic strategy for Alzheimer’s disease. Neurobiol. Dis. 2011, 41, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Gholamipour-Badie, H.; Naderi, N.; Khodagholi, F.; Shaerzadeh, F.; Motamedi, F. L-type calcium channel blockade alleviates molecular and reversal spatial learning and memory alterations induced by entorhinal amyloid pathology in rats. Behav. Brain Res. 2013, 237, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Birks, J.; López-Arrieta, J. Nimodipine for primary degenerative, mixed and vascular dementia. Cochrane Database Syst. Rev. 2002, 2002, CD000147. [Google Scholar] [CrossRef]
- Novotny, M.; Klimova, B.; Valis, M. Nitrendipine and Dementia: Forgotten Positive Facts? Front. Aging Neurosci. 2018, 10, 418. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.E.; Zhu, L.; Verret, L.; Vossel, K.A.; Orr, A.G.; Cirrito, J.R.; Devidze, N.; Ho, K.; Yu, G.-Q.; Palop, J.J.; et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 2012, 109, E2895–E2903. [Google Scholar] [CrossRef]
- Winblad, B.; Jones, R.W.; Wirth, Y.; Stöffler, A.; Möbius, H.J. Memantine in Moderate to Severe Alzheimer’s Disease: A Meta-Analysis of Randomised Clinical Trials. Dement. Geriatr. Cogn. Disord. 2007, 24, 20–27. [Google Scholar] [CrossRef]
- Li, P.; Xu, J.; Gu, H.; Peng, H.; Yin, Y.; Zhuang, J. Memantine ameliorates cognitive deficit in AD mice via enhancement of entorhinal–CA1 projection. BMC Neurosci. 2021, 22, 41. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Coria, H.; Green, K.N.; Billings, L.M.; Kitazawa, M.; Albrecht, M.; Rammes, G.; Parsons, C.G.; Gupta, S.; Banerjee, P.; LaFerla, F.M. Memantine Improves Cognition and Reduces Alzheimer’s-like Neuropathology in Transgenic Mice. Am. J. Pathol. 2010, 176, 870–880. [Google Scholar] [CrossRef]
- Stazi, M.; Wirths, O. Chronic Memantine Treatment Ameliorates Behavioral Deficits, Neuron Loss, and Impaired Neurogenesis in a Model of Alzheimer’s Disease. Mol. Neurobiol. 2020, 58, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Dahl, R.; Moore, A.C.; Knight, C.; Mauger, C.; Zhang, H.; Schiltz, G.E.; Koss, W.A.; Bezprozvanny, I. Positive Allosteric Modulator of SERCA Pump NDC-1173 Exerts Beneficial Effects in Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 11057. [Google Scholar] [CrossRef]
- Krajnak, K.; Dahl, R. A new target for Alzheimer’s disease: A small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorganic Med. Chem. Lett. 2018, 28, 1591–1594. [Google Scholar] [CrossRef]
- Bussiere, R.; Lacampagne, A.; Reiken, S.; Liu, X.; Scheuerman, V.; Zalk, R.; Martin, C.; Checler, F.; Marks, A.R.; Chami, M. Amyloid β production is regulated by β2-adrenergic signaling-mediated post-translational modifications of the ryanodine receptor. J. Biol. Chem. 2017, 292, 10153–10168. [Google Scholar] [CrossRef]
- Lacampagne, A.; Liu, X.; Reiken, S.; Bussiere, R.; Meli, A.C.; Lauritzen, I.; Teich, A.F.; Zalk, R.; Saint, N.; Arancio, O.; et al. Post-translational remodeling of ryanodine receptor induces calcium leak leading to Alzheimer’s disease-like pathologies and cognitive deficits. Acta Neuropathol. 2017, 134, 749–767. [Google Scholar] [CrossRef]
- Angelova, P.R.; Vinogradova, D.; Neganova, M.E.; Serkova, T.P.; Sokolov, V.V.; Bachurin, S.O.; Shevtsova, E.F.; Abramov, A.Y. Pharmacological Sequestration of Mitochondrial Calcium Uptake Protects Neurons Against Glutamate Excitotoxicity. Mol. Neurobiol. 2018, 56, 2244–2255. [Google Scholar] [CrossRef] [PubMed]
- Cavallucci, V.; Berretta, N.; Nobili, A.; Nisticò, R.; Mercuri, N.B.; D’amelio, M. Calcineurin Inhibition Rescues Early Synaptic Plasticity Deficits in a Mouse Model of Alzheimer’s Disease. Neuromol. Med. 2013, 15, 541–548. [Google Scholar] [CrossRef]

{kind=link}
| Ca2+ Sensitive Effectors (Not Exhaustive) | Anticipated Intraneuronal Pathophysiology/Neuronal Destruction Pathways under Conditions of Ca2+ Dyshomeostasis | References |
|---|---|---|
| Calcineurin | Synaptic depression, dendritic spine loss, apoptosis, altered mitochondrial dynamics, inhibition of axonal outgrowth | [5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20] |
| Calpains | Dendritic pruning, axonal degeneration | [21,22,23,24,25] |
| Calpain-p25-CDK5 Calpain-GSK3 Calpain-tau | Apoptosis, phosphorylation tau, neurotoxic Aβ generation, synaptic dysfunction, mitochondrial dysfunction, excessive ROS production, neurotoxic tau fragments | [26,27,28,29] |
| CamKII | Phosphorylation tau, apoptosis, necrosis, synaptic degeneration | [30,31] |
| ER function | Impaired proteostasis, ER-stress-induced apoptosis (because of depleted Ca2+ levels in the ER) | [32,33] |
| IP3 and ryanodine receptors | Ca2+ dyshomeostasis (by elevated CICR) | [34,35,36,37] |
| MAPK (ERK) | Apoptosis | [38,39,40,41] |
| Miro | Stalled axonal trafficking mitochondria | [42] |
| Mitochondrial function | Ca2+ dyshomeostasis, apoptosis, necroptosis, excessive ROS production, ATP production | [43,44,44,45,46,47] |
| Proteasome | Axonal degeneration | [48,49] |
| RIPK | Necroptosis | [22,50] |
| vATPase function | Impaired lysosomal-autophagosome function | [51] |
| Vesicular trafficking | APP processing, tau-exocytosis | [52,53,54,55,56,57] |
| Compound | Target | Effects in Non-Clinical Models and (Where Indicated) in Patients | References |
|---|---|---|---|
| Dantrolene | RyR | Reduces amyloid pathology, normalises synaptic plasticity, and improves behavioural performance. | [102,103,104] |
| Isradipine, nimodipine, nitrendipine | Cav1.2 channel | Reduces amyloid and tau pathology, improves autophagy, and mitigates cognitive impairment. Possibly some benefit in patients. | [105,106,107,108,109] |
| Levetiracetam | SV2a | Mitigates network hyperactivity and improves learning and memory. | [110] |
| Memantine | NMDA receptor | Dendritic spine regeneration, rescue of synaptic plasticity, reduced hippocampal CA1 neuron loss reduction Aβ/tau pathology, and improved learning and memory performance. Benefits cognitive, functional, global, and behavioural endpoints in patients. | [111,112,113,114] |
| NDC-1173, CDN1163 | SERCA pump activator | Improves memory and other behavioural read-outs. | [115,116] |
| REM0046127 | SOCE modulator | Full rescue of synaptic plasticity, EEG, and cognition. Reduces inflammation and Aβ/tau pathology. | Personal communication GG |
| S107 (Rycal) | RyR2 macromolecular complex | Reduces APP cleavage and Aβ production and restores synaptic plasticity and cognitive deficits. | [117,118] |
| TG-2112x | Lowers mitochondrial Ca2+ uptake | Mitigates glutamate excitotoxicity. | [119] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Griffioen, G. Calcium Dyshomeostasis Drives Pathophysiology and Neuronal Demise in Age-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 13243. https://doi.org/10.3390/ijms241713243
Griffioen G. Calcium Dyshomeostasis Drives Pathophysiology and Neuronal Demise in Age-Related Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(17):13243. https://doi.org/10.3390/ijms241713243
Chicago/Turabian StyleGriffioen, Gerard. 2023. "Calcium Dyshomeostasis Drives Pathophysiology and Neuronal Demise in Age-Related Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 17: 13243. https://doi.org/10.3390/ijms241713243
APA StyleGriffioen, G. (2023). Calcium Dyshomeostasis Drives Pathophysiology and Neuronal Demise in Age-Related Neurodegenerative Diseases. International Journal of Molecular Sciences, 24(17), 13243. https://doi.org/10.3390/ijms241713243