

Brain Dopamine–Clock Interactions Regulate Cardiometabolic Physiology: Mechanisms of the Observed Cardioprotective Effects of Circadian-Timed Bromocriptine-QR Therapy in Type 2 Diabetes Subjects


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Temporal Synergisms of CNS Circadian Neural Oscillations Dictate Physiological Status
3. CNS Circadian Clock Mechanisms Generating Seasonal Physiology
4. CNS Circadian Dopaminergic Neuronal Input Modulation of CNS Clock Output Control of Cardiometabolic Status
5. SCN Output Circuits Controlling Metabolism and Cardiovascular Health
6. Targeting Low CNS Dopaminergic–Clock Activity to Treat Cardiometabolic Disease: Circadian-Timed Bromocriptine Therapy for the Treatment of Cardiometabolic Syndrome—Preclinical Findings
7. Circadian Rhythm Influence on Human Physiology
8. Shift in CNS Circadian Dopamine–Clock Interactions to Initiate the CNS Treacherous Triad Are Coupled to Known Peripheral Neuroendocrine Pathologies That Potentiate Downstream Cardiovascular Disease in Humans
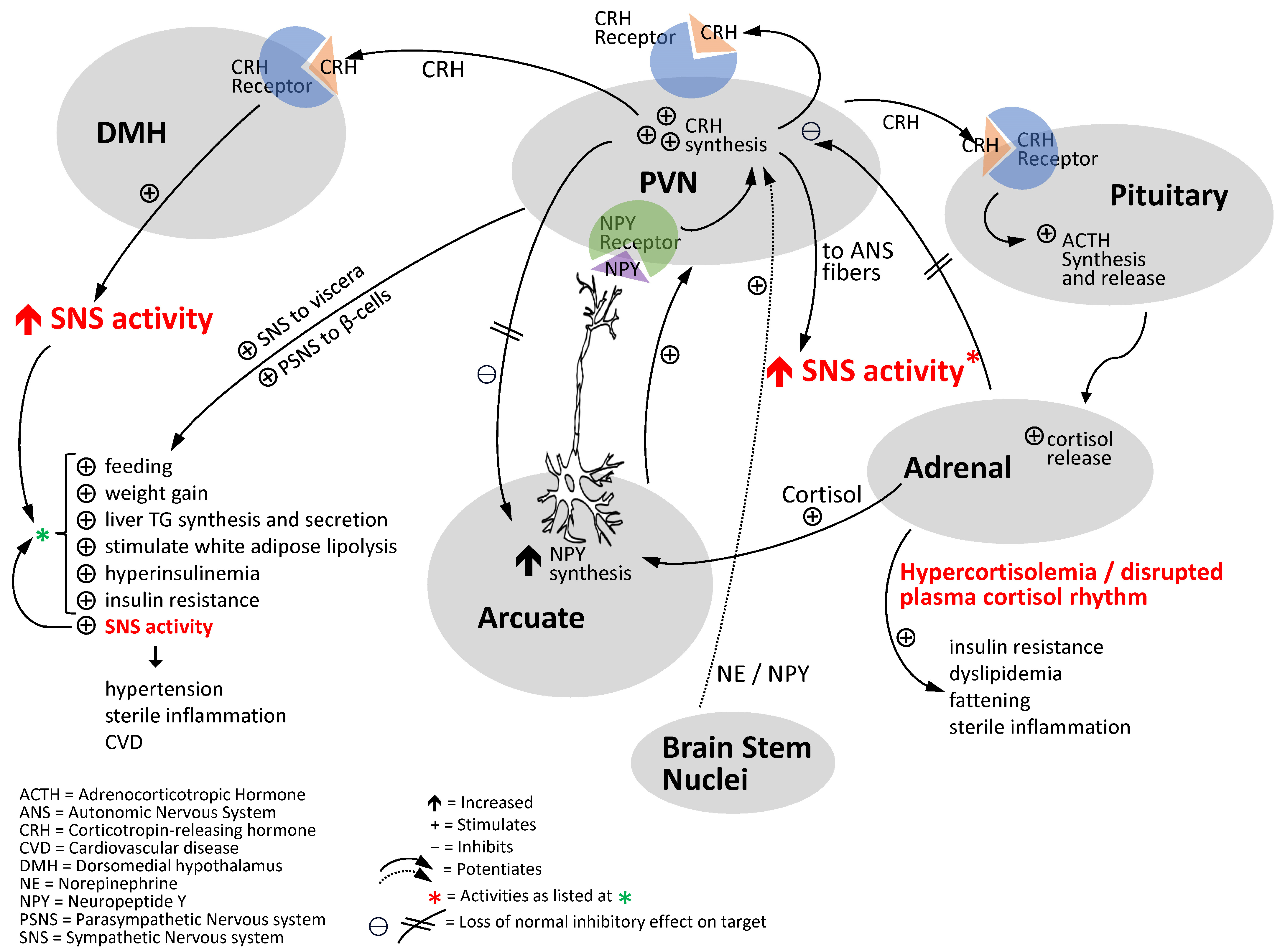
9. Common Chronic Environmental Stress Factors of Modern Man Disrupt the CNS Dopaminergic–Clock Circuit, Reduce CNS Dopaminergic Activity and Increase SNS Tone to Potentiate Cardiometabolic Disease and CVD
10. Circadian-Timed Bromocriptine-QR for the Treatment of Cardiometabolic Disease–Human Studies
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HTP | 5-hydroxy-tryptophan |
| αMSH | α-melanocyte stimulating hormone |
| Bromocriptine-QR | bromocriptine-quick release |
| CNS | central nervous system |
| CRH | corticotropin releasing hormone |
| CVD | cardiovascular disease |
| FFA | free fatty acids |
| HbA1c | hemoglobin A1C |
| HPA | hypothalamic-pituitary-adrenal |
| HyperFFAemia | HyperFreeFattyAcidemia |
| NE | norepinephrine |
| NO | nitric oxide |
| NPY | neuropeptide Y |
| PBMC | peripheral blood mononuclear cells |
| PVN | paraventricular nucleus |
| SCN | suprachiasmatic nucleus |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| S | serotonin |
| SuMN | supramammillary nucleus |
| SNS | sympathetic nervous system |
| T1DM | type 1 Diabetes Mellitus |
| T2D | type 2 diabetes |
| TH | tyrosine hydroxylase |
| VMH | ventromedial hypothalamus |
| BMAL1 | BasicHelix-Loop-HelixARNTLike1 |
| CLOCK | Circadian Locomotor Output Cycles Protein Kaput |
| CRY1/2 | Cryptochrome Circadian Regulator 1/2 |
| DBP | D-Box BindingPAR BZIP Transcription Factor |
| eNOS | Nitric Oxide Synthase3, Endothelial |
| IKKαβ | Nuclear Factor NFkappaB Inhibitor Kinase Alpha/Beta |
| IL-6 | Interleukin 6 |
| JAK2 | Janus Kinase 2 |
| JNK | Mitogen-Activated Protein Kinase 8 |
| NFIL3 | Nuclear Factor, Interleukin 3 Regulated |
| NFkB | Nuclear Factor Kappa B |
| PER1/2/3 | Circadian Clock Protein PERIOD 1/2/3 |
| PPAR-γ | Peroxisome Proliferator Activated Receptor Gamma |
| REV-ERBα/β | Nuclear Receptor Subfamily 1 Group D Member 1/2 |
| RORα/β/γ | RAR Related Orphan Receptor A/B/C |
| SOCS3 | Suppressor Of Cytokine Signaling 3 |
| STAT3 | Signal Transducer And Activator Of Transcription 3 |
| TLR | Toll-Like Receptor |
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global Burden of Cardiovascular Diseases and Risk Factors, 1990–2019: Update From the GBD 2019 Study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef]
- Wang, W.; Hu, M.; Liu, H.; Zhang, X.; Li, H.; Zhou, F.; Liu, Y.-M.; Lei, F.; Qin, J.-J.; Zhao, Y.-C.; et al. Global Burden of Disease Study 2019 suggests that metabolic risk factors are the leading drivers of the burden of ischemic heart disease. Cell Metab. 2021, 33, 1943–1956.e2. [Google Scholar] [CrossRef]
- Dwivedi, A.K.; Dubey, P.; Reddy, S.Y.; Clegg, D.J. Associations of Glycemic Index and Glycemic Load with Cardiovascular Disease: Updated Evidence from Meta-analysis and Cohort Studies. Curr. Cardiol. Rep. 2022, 24, 141–161. [Google Scholar] [PubMed]
- Jenkins, D.J.A.; Dehghan, M.; Mente, A.; Bangdiwala, S.I.; Rangarajan, S.; Srichaikul, K.; Mohan, V.; Avezum, A.; Diaz, R.; Rosengren, A.; et al. Glycemic Index, Glycemic Load, and Cardiovascular Disease and Mortality. N. Engl. J. Med. 2021, 384, 1312–1322. [Google Scholar] [CrossRef]
- Wang, D.D.; Hu, F.B. Dietary Fat and Risk of Cardiovascular Disease: Recent Controversies and Advances. Annu. Rev. Nutr. 2017, 37, 423–446. [Google Scholar] [CrossRef] [PubMed]
- Wali, J.A.; Jarzebska, N.; Raubenheimer, D.; Simpson, S.J.; Rodionov, R.N.; O’Sullivan, J.F. Cardio-Metabolic Effects of High-Fat Diets and Their Underlying Mechanisms—A Narrative Review. Nutrients 2020, 12, 1505. [Google Scholar] [CrossRef]
- Hooper, L.; Martin, N.; Jimoh, O.F.; Kirk, C.; Foster, E.; Abdelhamid, A.S. Reduction in saturated fat intake for cardiovascular disease. Cochrane Database Syst. Rev. 2020, 8, CD011737. [Google Scholar]
- KKorostovtseva, L.; Bochkarev, M.; Sviryaev, Y. Sleep and Cardiovascular Risk. Sleep Med. Clin. 2021, 16, 485–497. [Google Scholar]
- Thosar, S.S.; Butler, M.P.; Shea, S.A. Role of the circadian system in cardiovascular disease. J. Clin. Investig. 2018, 128, 2157–2167. [Google Scholar] [CrossRef]
- Cappuccio, F.P.; Miller, M.A. Sleep and Cardio-Metabolic Disease. Curr. Cardiol. Rep. 2017, 19, 110. [Google Scholar] [CrossRef]
- Gottlieb, D.J. Sleep Apnea and Cardiovascular Disease. Curr. Diab. Rep. 2021, 21, 64. [Google Scholar] [CrossRef]
- Jike, M.; Itani, O.; Watanabe, N.; Buysse, D.J.; Kaneita, Y. Long sleep duration and health outcomes: A systematic review, meta-analysis and meta-regression. Sleep. Med. Rev. 2018, 39, 25–36. [Google Scholar]
- Covassin, N.; Singh, P. Sleep Duration and Cardiovascular Disease Risk: Epidemiologic and Experimental Evidence. Sleep Med. Clin. 2016, 11, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Powell-Wiley, T.M.; Baumer, Y.; Baah, F.O.; Baez, A.S.; Farmer, N.; Mahlobo, C.T.; Pita, M.A.; Potharaju, K.A.; Tamura, K.; Wallen, G.R. Social Determinants of Cardiovascular Disease. Circ. Res. 2022, 130, 782–799. [Google Scholar] [PubMed]
- Kivimäki, M.; Kawachi, I. Work Stress as a Risk Factor for Cardiovascular Disease. Curr. Cardiol. Rep. 2015, 17, 630. [Google Scholar] [PubMed]
- Wirtz, P.H.; von Kanel, R. Psychological Stress, Inflammation, and Coronary Heart Disease. Curr. Cardiol. Rep. 2017, 19, 111. [Google Scholar] [PubMed]
- Halaris, A. Co-morbidity between cardiovascular pathology and depression: Role of inflammation. Mod Trends Pharmacopsychiatry 2013, 28, 144–161. [Google Scholar] [PubMed]
- Hare, D.L. Depression and cardiovascular disease. Curr. Opin. Lipidol. 2021, 32, 167–174. [Google Scholar] [CrossRef]
- Halaris, A. Inflammation-Associated Co-morbidity Between Depression and Cardiovascular Disease. Curr. Top Behav. Neurosci. 2017, 31, 45–70. [Google Scholar]
- Jakobsen, M.U.; O’Reilly, E.J.; Heitmann, B.L.; Pereira, M.A.; Balter, K.; Fraser, G.E.; Goldbourt, U.; Hallmans, G.; Knekt, P.; Liu, S.; et al. Major types of dietary fat and risk of coronary heart disease: A pooled analysis of 11 cohort studies. Am. J. Clin. Nutr. 2009, 89, 1425–1432. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Karin, M. “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. 2021, 33, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Taskinen, M.-R.; Söderlund, S.; Bogl, L.H.; Hakkarainen, A.; Matikainen, N.; Pietiläinen, K.H.; Räsänen, S.; Lundbom, N.; Björnson, E.; Eliasson, B.; et al. Adverse effects of fructose on cardiometabolic risk factors and hepatic lipid metabolism in subjects with abdominal obesity. J. Intern. Med. 2017, 282, 187–201. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, K.L.; Bremer, A.A.; Medici, V.; Nakajima, K.; Ito, Y.; Nakano, T.; Chen, G.; Fong, T.H.; Lee, V.; Menorca, R.I.; et al. Consumption of fructose and high fructose corn syrup increase postprandial triglycerides, LDL-cholesterol, and apolipoprotein-B in young men and women. J. Clin. Endocrinol. Metab. 2011, 96, E1596–E1605. [Google Scholar] [CrossRef]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar]
- Meier, A.H.; Cincotta, A.H. Circadian rhythms regulate the expression of the thrifty genotype/phenotype. Diabetes Rev. 1996, 4, 464–487. Available online: https://veroscience.com/publications/ (accessed on 16 August 2023).
- Cincotta, A.H. Hypothalamic role in the insulin resistance syndrome. In Insulin Resistance and Insulin Resistance Syndrome, Frontiers in Animal Diabetes Research Series; Hansen, B., Shafrir, E., Eds.; Taylor and Francis: London, UK, 2002; pp. 271–312. [Google Scholar]
- Guh, Y.-J.; Tamai, T.K.; Yoshimura, T. The underlying mechanisms of vertebrate seasonal reproduction. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 343–357. [Google Scholar]
- Martin, L.B.; Weil, Z.M.; Nelson, R.J. Seasonal changes in vertebrate immune activity: Mediation by physiological trade-offs. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 321–339. [Google Scholar]
- Renthlei, Z.; Yatung, S.; Lalpekhlui, R.; Trivedi, A.K. Seasonality in tropical birds. J. Exp. Zool. A Ecol. Integr. Physiol. 2022, 337, 952–966. [Google Scholar] [PubMed]
- Turbek, S.P.; Scordato, E.S.; Safran, R.J. The Role of Seasonal Migration in Population Divergence and Reproductive Isolation. Trends Ecol. Evol. 2018, 33, 164–175. [Google Scholar] [CrossRef]
- Dardente, H.; Wood, S.; Ebling, F.; De Miera, C.S. An integrative view of mammalian seasonal neuroendocrinology. J. Neuroendocr. 2019, 31, e12729. [Google Scholar]
- Hazlerigg, D.G.; Wagner, G.C. Seasonal photoperiodism in vertebrates: From coincidence to amplitude. Trends Endocrinol. Metab. 2006, 17, 83–91. [Google Scholar]
- Bacon, B.; Khatiri, A.; Palmer, J.; Freeth, T.; Pettitt, P.; Kentridge, R. An Upper Palaeolithic Proto-writing System and Phenological Calendar. Camb. Archaeol. J. 2023, 33, 1–19. [Google Scholar]
- Meier, A.H.; Davis, K.B. Diurnal variations of the fattening response to prolactin in the White-throated Sparrow, Zonotrichia albicollis. Gen. Comp. Endocrinol. 1967, 8, 110–114. [Google Scholar] [CrossRef]
- Meier, A.H. Circadian basis for neuroendocrine regulation. In Proceedings International Symposium on Rhythms in Fishes; Ali, M.A., Ed.; Plenum: New York, NY, USA, 1993; pp. 109–126. [Google Scholar]
- Meier, A.H.; Russo, A.C. Circadian organization of the avian annual cycle. In Current Ornithology; Johnston, R.E., Ed.; Plenum: New York, NY, USA, 1984; Volume 2, pp. 303–343. [Google Scholar]
- Meier, A.H. Temporal Synergism of Circadian Neuroendocrine Oscillations Regulates Seasonal Conditions in the Gulf Killifish. Trans. Am. Fish. Soc. 1984, 113, 422–431. [Google Scholar] [CrossRef]
- Meier, A.H.; Trobec, T.N.; Joseph, M.M.; John, T.M. Temporal Synergism of Prolactin and Adrenal Steroids in the Regulation of Fat Stores. Proc. Soc. Exp. Biol. Med. 1971, 137, 408–415. [Google Scholar] [CrossRef]
- Meier, A.H. Temporal synergism of prolactin and adrenal steroids. Gen. Comp. Endocrinol. 1972, 3, 499–508. [Google Scholar] [CrossRef]
- Meier, A.H.; Burns, J.T. Circadian Hormone Rhythms in Lipid Regulation. Am. Zool. 1976, 16, 649–659. [Google Scholar] [CrossRef][Green Version]
- Cincotta, A.H.; Wilson, J.M.; Desouza, C.J.; Meier, A.H. Properly timed injections of cortisol and prolactin produce long-term reductions in obesity, hyperinsulinaemia and insulin resistance in the Syrian hamster (Mesocricetus auratus). J. Endocrinol. 1989, 120, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Schiller, B.C.; Meier, A.H. Bromocriptine inhibits the seasonally occurring obesity, hyperinsulinemia, insulin resistance, and impaired glucose tolerance in the Syrian hamster, Mesocricetus auratus. Metabolism 1991, 40, 639–644. [Google Scholar] [CrossRef]
- Revel, F.G.; Saboureau, M.; Masson-Pevet, M.; Pevet, P.; Mikkelsen, J.D.; Simonneaux, V. Kisspeptin mediates the photoperiodic control of reproduction in hamsters. Curr. Biol. 2006, 16, 1730–1735. [Google Scholar] [CrossRef] [PubMed]
- Goldman, B.D. Mammalian Photoperiodic System: Formal Properties and Neuroendocrine Mechanisms of Photoperiodic Time Measurement. J. Biol. Rhythm. 2001, 16, 283–301. [Google Scholar]
- Mendoza, J.; Challet, E. Brain Clocks: From the Suprachiasmatic Nuclei to a Cerebral Network. Neuroscientist 2009, 15, 477–488. [Google Scholar] [CrossRef]
- Okamura, H. Suprachiasmatic Nucleus Clock Time in the Mammalian Circadian System. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 551–556. [Google Scholar] [CrossRef]
- Buijs, R.M.; Scheer, F.A.; Kreier, F.; Yi, C.; Bos, N.; Goncharuk, V.D.; Kalsbeek, A. Organization of circadian functions: Interaction with the body. Prog. Brain Res. 2006, 153, 341–360. [Google Scholar] [PubMed]
- Verwey, M.; Dhir, S.; Amir, S. Circadian influences on dopamine circuits of the brain: Regulation of striatal rhythms of clock gene expression and implications for psychopathology and disease. F1000Research 2016, 5, 2062. [Google Scholar]
- Baño-Otálora, B.; Piggins, H.D. Contributions of the lateral habenula to circadian timekeeping. Pharmacol. Biochem. Behav. 2017, 162, 46–54. [Google Scholar]
- Mendoza, J. Circadian neurons in the lateral habenula: Clocking motivated behaviors. Pharmacol. Biochem. Behav. 2017, 162, 55–61. [Google Scholar]
- Cox, K.H.; Takahashi, J.S. Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 2019, 63, R93–R102. [Google Scholar] [CrossRef]
- Healy, K.L.; Morris, A.R.; Liu, A.C. Circadian Synchrony: Sleep, Nutrition, and Physical Activity. Front. Netw. Physiol. 2021, 1, 732243. [Google Scholar]
- Koronowski, K.B.; Sassone-Corsi, P. Communicating clocks shape circadian homeostasis. Science 2021, 371, eabd0951. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, U. Timing to Perfection: The Biology of Central and Peripheral Circadian Clocks. Neuron 2012, 74, 246–260. [Google Scholar] [PubMed]
- Paydar-Ravandi, F.; Meier, A.H. Melatonin Mediates Alternation of Seasonality in Syrian Hamsters1. Biol. Reprod. 1989, 40, 475–480. [Google Scholar] [CrossRef] [PubMed]
- Morin, L.P. Neuroanatomy of the extended circadian rhythm system. Exp. Neurol. 2012, 243, 4–20. [Google Scholar]
- Buijs, R.M.; Ruiz, M.A.G.; Hernández, R.M.; Cortés, B.R. The suprachiasmatic nucleus; a responsive clock regulating homeostasis by daily changing the setpoints of physiological parameters. Auton. Neurosci. 2019, 218, 43–50. [Google Scholar]
- Buijs, R.M.; Soto Tinoco, E.C.; Hurtado Alvarado, G.; Escobar, C. The circadian system: From clocks to physiology. Handb. Clin. Neurol. 2021, 179, 233–247. [Google Scholar]
- Oosterman, J.E.; Kalsbeek, A.; la Fleur, S.E.; Belsham, D.D. Impact of nutrients on circadian rhythmicity. Am. J. Physiol. Integr. Comp. Physiol. 2015, 308, R337–R350. [Google Scholar] [CrossRef]
- Buijs, F.N.; León-Mercado, L.; Guzmán-Ruiz, M.; Guerrero-Vargas, N.N.; Romo-Nava, F.; Buijs, R.M. The Circadian System: A Regulatory Feedback Network of Periphery and Brain. Physiology 2016, 31, 170–181. [Google Scholar] [CrossRef]
- Flanagan, A.; Bechtold, D.A.; Pot, G.K.; Johnston, J.D. Chrono-nutrition: From molecular and neuronal mechanisms to human epidemiology and timed feeding patterns. J. Neurochem. 2021, 157, 53–72. [Google Scholar] [CrossRef]
- Helfrich-Förster, C. Interactions between psychosocial stress and the circadian endogenous clock. PsyCh J. 2017, 6, 277–289. [Google Scholar] [CrossRef]
- Knight, E.L.; Jiang, Y.; Rodriguez-Stanley, J.; Almeida, D.M.; Engeland, C.G.; Zilioli, S. Perceived stress is linked to heightened biomarkers of inflammation via diurnal cortisol in a national sample of adults. Brain Behav. Immun. 2021, 93, 206–213. [Google Scholar] [CrossRef]
- Kuang, X.D.; Yu, X.B.; Cao, Y.; Li, D.S.; Zhu, H.Y. Interaction between the Circadian Clock and Chronic Stress. Biomed. Environ. Sci. 2018, 31, 686–699. [Google Scholar] [PubMed]
- Kramer, A.C.; Neubauer, A.B.; Stoffel, M.; Voss, A.; Ditzen, B. Tomorrow’s gonna suck: Today’s stress anticipation predicts tomorrow’s post-awakening cortisol increase. Psychoneuroendocrinology 2019, 106, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Pitsillou, E.; Liang, J.; Hung, A.; Karagiannis, T.C. The circadian machinery links metabolic disorders and depression: A review of pathways, proteins and potential pharmacological interventions. Life Sci. 2021, 265, 118809. [Google Scholar] [CrossRef]
- Dallman, M.F.; Akana, S.F.; Bhatnagar, S.; Bell, M.E.; Strack, A.M. Bottomed out: Metabolic significance of the circadian trough in glucocorticoid concentrations. Int. J. Obes. Relat. Metab. Disord. 2000, 24 (Suppl. 2), S40–S46. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Vu, T.-H.; Maas, M.B.; I Braun, R.; Wolf, M.S.; Roenneberg, T.; Daviglus, M.L.; Reid, K.J.; Zee, P.C. Light at night in older age is associated with obesity, diabetes, and hypertension. Sleep 2022, 46, zsac130. [Google Scholar] [CrossRef]
- Cho, Y.; Ryu, S.-H.; Lee, B.R.; Kim, K.H.; Lee, E.; Choi, J. Effects of artificial light at night on human health: A literature review of observational and experimental studies applied to exposure assessment. Chrono-Int. 2015, 32, 1294–1310. [Google Scholar] [CrossRef]
- Wyse, C.A.; Selman, C.; Page, M.M.; Coogan, A.N.; Hazlerigg, D.G. Circadian desynchrony and metabolic dysfunction; did light pollution make us fat? Med. Hypotheses 2011, 77, 1139–1144. [Google Scholar] [CrossRef]
- Punjabi, N.M.; Beamer, B.A. Alterations in Glucose Disposal in Sleep-disordered Breathing. Am. J. Respir. Crit. Care Med. 2009, 179, 235–240. [Google Scholar] [CrossRef]
- Albrecht, U. Circadian rhythms and sleep--the metabolic connection. Pflug. Arch 2012, 463, 23–30. [Google Scholar] [CrossRef]
- Azmi, N.A.S.M.; Juliana, N.; Teng, N.I.M.F.; Azmani, S.; Das, S.; Effendy, N. Consequences of Circadian Disruption in Shift Workers on Chrononutrition and their Psychosocial Well-Being. Int. J. Environ. Res. Public Health 2020, 17, 2043. [Google Scholar] [CrossRef]
- Ishihara, A.; Park, I.; Suzuki, Y.; Yajima, K.; Cui, H.; Yanagisawa, M.; Sano, T.; Kido, J.; Tokuyama, K. Metabolic responses to polychromatic LED and OLED light at night. Sci. Rep. 2021, 11, 12402. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Meier, A.H. Circadian rhythms of lipogenic and hypoglycaemic responses to insulin in the golden hamster (Mesocricetus auratus). J. Endocrinol. 1984, 103, 141–146. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Meier, A.H. Prolactin permits the expression of a circadian variation in lipogenic responsiveness to insulin in hepatocytes of the golden hamster (Mesocricetus auratus). J. Endocrinol. 1985, 106, 173–176. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Meier, A.H. Circadian Rhythm of Hepatic Lipogenic Enzyme Activity; IEEE Engineering in Medicine and Biology Society: Piscataway, NJ, USA, 1988; pp. 1818–1819. [Google Scholar]
- de Souza, C.J.; Meier, A.H. Circadian and seasonal variations of plasma insulin and cortisol concentrations in the Syrian hamster, Mesocricetus auratus. Chronobiol. Int. 1987, 4, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Meier, A.H. Prolactin permits the expression of a circadian variation in insulin receptor profile in hepatocytes of the golden hamster (Mesocricetus auratus). J. Endocrinol. 1985, 106, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Meier, A.H. Prolactin Influences the Circadian Rhythm of Lipogenesis in Primary Cultured Hepatocytes. Horm. Metab. Res. 1989, 21, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.E.; Demarest, K.T.; Johnston, C.A. Influence of prolactin on dopaminergic neuronal systems in the hypothalamus. Fed. Proc. 1980, 39, 2912–2916. [Google Scholar] [PubMed]
- Telegdy, G.; Vermes, I. Effect of adrenocorical hormones on activity of the serotonergic system in limbic structures. Neuroendocrinology 1975, 18, 16–26. [Google Scholar] [CrossRef]
- Meier, A.H.; Ferrell, B.R.; Miller, L.J. Circadian components of the circannual mechanism in the white-throated sparrow. In Acta XVII Congressus Internationalis Ornithologia; Nohring, R., Ed.; Verlag Der Deutschen Ornithologen-Gesellschaft: Berlin, Germany, 1981; Volume 1, pp. 453–462. [Google Scholar]
- Emata, A.C.; Meier, A.H.; Spieler, R.E. Temporal variations in gonadal and body fat responses to daily injections of 5-hydroxytryptophan (5-HTP) and dihydroxyphenylalanine (DOPA) in the gulf killifish, Fundulus grandis. J. Exp. Zool. 1985, 233, 29–34. [Google Scholar] [CrossRef]
- Yadav, S.; Chaturvedi, C.M. Internal coincidence of serotonergic and dopaminergic oscillations modulates photo sexual responses of Japanese quail, Coturnix coturnix japonica. Indian J. Exp. Biol. 2014, 52, 489–495. [Google Scholar] [PubMed]
- Wilson, J.M.; Meier, A.H. Resetting the Annual Cycle with Timed Daily Injections of 5-Hydroxytryptophan and L-Dihydroxyphenylalanine in Syrian Hamsters. Chrono-Int. 1989, 6, 113–121. [Google Scholar] [CrossRef]
- Glass, J.D.; Grossman, G.H.; Farnbauch, L.; DiNardo, L. Midbrain Raphe Modulation of Nonphotic Circadian Clock Resetting and 5-HT Release in the Mammalian Suprachiasmatic Nucleus. J. Neurosci. 2003, 23, 7451–7460. [Google Scholar] [CrossRef]
- Pickard, G.E.; Rea, M.A. Serotonergic innervation of the hypothalamic suprachiasmatic nucleus and photic regulation of circadian rhythms. Biol. Cell 1997, 89, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Rea, M.; Glass, J.; Colwell, C. Serotonin modulates photic responses in the hamster suprachiasmatic nuclei. J. Neurosci. 1994, 14, 3635–3642. [Google Scholar] [CrossRef]
- Moore, R.Y.; Speh, J.C. Serotonin innervation of the primate suprachiasmatic nucleus. Brain Res. 2004, 1010, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Luo, J.; Meier, A.H.; Cincotta, A.H. Dopaminergic neurotoxin administration to the area of the suprachiasmatic nuclei induces insulin resistance. Neuroreport 1997, 8, 3495–3499. [Google Scholar] [CrossRef]
- Luo, S.; Luo, J.; Cincotta, A.H. Suprachiasmatic nuclei monoamine metabolism of glucose tolerant versus intolerant hamsters. Neuroreport 1999, 10, 2073–2077. [Google Scholar] [CrossRef]
- Luo, S.; Zhang, Y.; Ezrokhi, M.; Li, Y.; Tsai, T.-H.; Cincotta, A.H. Circadian peak dopaminergic activity response at the biological clock pacemaker (suprachiasmatic nucleus) area mediates the metabolic responsiveness to a high-fat diet. J. Neuroendocr. 2018, 30, e12563. [Google Scholar] [CrossRef]
- Luo, S.; Ezrokhi, M.; Cominos, N.; Tsai, T.-H.; Stoelzel, C.R.; Trubitsyna, Y.; Cincotta, A.H. Experimental dopaminergic neuron lesion at the area of the biological clock pacemaker, suprachiasmatic nuclei (SCN) induces metabolic syndrome in rats. Diabetol. Metab. Syndr. 2021, 13, 11. [Google Scholar] [CrossRef]
- Zhang, Y.; Stoelzel, C.; Ezrokhi, M.; Tsai, T.-H.; Cincotta, A.H. Activation State of the Supramammillary Nucleus Regulates Body Composition and Peripheral Fuel Metabolism. Neuroscience 2021, 466, 125–147. [Google Scholar] [CrossRef]
- Bartness, T.J.; Song, C.K.; Demas, G.E. SCN efferents to peripheral tissues: Implications for biological rhythms. J. Biol. Rhythms 2001, 16, 196–204. [Google Scholar] [CrossRef]
- Buijs, R.M.; la Fleur, S.E.; Wortel, J.; van Heyningen, C.; Zuiddam, L.; Mettenleiter, T.C.; Kalsbeek, A.; Nagai, K.; Niijima, A. The suprachiasmatic nucleus balances sympathetic and parasympathetic output to peripheral organs through separate preautonomic neurons. J. Comp. Neurol. 2003, 464, 36–48. [Google Scholar] [CrossRef]
- Kalsbeek, A.; Palm, I.F.; La Fleur, S.E.; Scheer, F.A.J.L.; Perreau-Lenz, S.; Ruiter, M.; Kreier, F.; Cailotto, C.; Buijs, R.M. SCN Outputs and the Hypothalamic Balance of Life. J. Biol. Rhythm. 2006, 21, 458–469. [Google Scholar] [CrossRef]
- Hastings, M.; O’neill, J.S.; Maywood, E.S. Circadian clocks: Regulators of endocrine and metabolic rhythms. J. Endocrinol. 2007, 195, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Pazienza, V.; Vinciguerra, M. Clock Genes and Clock-Controlled Genes in the Regulation of Metabolic Rhythms. Chrono-Int. 2012, 29, 227–251. [Google Scholar] [CrossRef] [PubMed]
- Li, J.D.; Hu, W.P.; Zhou, Q.Y. The circadian output signals from the suprachiasmatic nuclei. Prog. Brain Res. 2012, 199, 119–127. [Google Scholar]
- La Fleur, S.E. Daily rhythms in glucose metabolism: Suprachiasmatic nucleus output to peripheral tissue. J. Neuroendocrinol. 2003, 15, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Ruiter, M.; Buijs, R.M.; Kalsbeek, A. Hormones and the autonomic nervous system are involved in suprachiasmatic nucleus modulation of glucose homeostasis. Curr. Diabetes Rev. 2006, 2, 213–226. [Google Scholar] [CrossRef]
- Radziuk, J.M. The suprachiasmatic nucleus, circadian clocks, and the liver. Diabetes 2013, 62, 1017–1019. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coomans, C.P.; Berg, S.A.v.D.; Lucassen, E.A.; Houben, T.; Pronk, A.C.; van der Spek, R.D.; Kalsbeek, A.; Biermasz, N.R.; van Dijk, K.W.; Romijn, J.A.; et al. The Suprachiasmatic Nucleus Controls Circadian Energy Metabolism and Hepatic Insulin Sensitivity. Diabetes 2013, 62, 1102–1108. [Google Scholar] [CrossRef]
- Foppen, E.; Tan, A.A.T.; Ackermans, M.T.; Fliers, E.; Kalsbeek, A. Suprachiasmatic Nucleus Neuropeptides and Their Control of Endogenous Glucose Production. J. Neuroendocr. 2016, 28. [Google Scholar] [CrossRef]
- Kudo, T.; Horikawa, K.; Shibata, S. Circadian Rhythms in the CNS and Peripheral Clock Disorders: The Circadian Clock and Hyperlipidemia. J. Pharmacol. Sci. 2007, 103, 139–143. [Google Scholar] [CrossRef]
- Kolbe, I.; Husse, J.; Salinas, G.; Lingner, T.; Astiz, M.; Oster, H. The SCN Clock Governs Circadian Transcription Rhythms in Murine Epididymal White Adipose Tissue. J. Biol. Rhythm. 2016, 31, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Buijs, R.M.; Chun, S.J.; Niijima, A.; Romijn, H.J.; Nagai, K. Parasympathetic and sympathetic control of the pancreas: A role for the suprachiasmatic nucleus and other hypothalamic centers that are involved in the regulation of food intake. J. Comp. Neurol. 2001, 431, 405–423. [Google Scholar] [CrossRef]
- Kalsbeek, A.; Yi, C.-X.; La Fleur, S.E.; Fliers, E. The hypothalamic clock and its control of glucose homeostasis. Trends Endocrinol. Metab. 2010, 21, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Warren, W.S.; Champney, T.H.; Cassone, V.M. The suprachiasmatic nucleus controls the circadian rhythm of heart rate via the sympathetic nervous system. Physiol. Behav. 1994, 55, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Scheer, F.A.J.L.; Kalsbeek, A.; Buijs, R.M. Cardiovascular Control by the Suprachiasmatic Nucleus: Neural and Neuroendocrine Mechanisms in Human and Rat. Biol. Chem. 2003, 384, 697–709. [Google Scholar] [CrossRef]
- Maemura, K.; Takeda, N.; Nagai, R. Circadian Rhythms in the CNS and Peripheral Clock Disorders: Role of the Biological Clock in Cardiovascular Diseases. J. Pharmacol. Sci. 2007, 103, 134–138. [Google Scholar] [CrossRef]
- Hu, K.; Scheer, F.A.; Buijs, R.M.; Shea, S.A. The circadian pacemaker generates similar circadian rhythms in the fractal structure of heart rate in humans and rats. Cardiovasc. Res. 2008, 80, 62–68. [Google Scholar] [CrossRef]
- Rüger, M.; Scheer, F.A.J.L. Effects of circadian disruption on the cardiometabolic system. Rev. Endocr. Metab. Disord. 2009, 10, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Buijs, F.; Cazarez, F.; Basualdo, M.; Scheer, F.; Perusquía, M.; Centurion, D.; Buijs, R. The suprachiasmatic nucleus is part of a neural feedback circuit adapting blood pressure response. Neuroscience 2014, 266, 197–207. [Google Scholar] [CrossRef]
- Palomino-Segura, M.; Hidalgo, A. Circadian immune circuits. J. Exp. Med. 2020, 218, e20200798. [Google Scholar] [CrossRef]
- Logan, R.W.; Sarkar, D.K. Circadian nature of immune function. Mol. Cell. Endocrinol. 2012, 349, 82–90. [Google Scholar] [CrossRef]
- Prendergast, B.J.; Cable, E.J.; Patel, P.N.; Pyter, L.M.; Onishi, K.G.; Stevenson, T.J.; Ruby, N.F.; Bradley, S.P. Impaired leukocyte trafficking and skin inflammatory responses in hamsters lacking a functional circadian system. Brain Behav. Immun. 2013, 32, 94–104. [Google Scholar] [CrossRef]
- Zhang, Z.; Zeng, P.; Gao, W.; Zhou, Q.; Feng, T.; Tian, X. Circadian clock: A regulator of the immunity in cancer. Cell Commun. Signal. 2021, 19, 37. [Google Scholar] [CrossRef]
- Collis, S.J.; Boulton, S.J. Emerging links between the biological clock and the DNA damage response. Chromosoma 2007, 116, 331–339. [Google Scholar] [CrossRef]
- Antle, M.C.; Silver, R. Orchestrating time: Arrangements of the brain circadian clock. Trends Neurosci. 2005, 28, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic Nucleus: Cell Autonomy and Network Properties. Annu. Rev. Physiol. 2010, 72, 551–577. [Google Scholar] [CrossRef] [PubMed]
- Ramkisoensing, A.; Meijer, J.H. Synchronization of Biological Clock Neurons by Light and Peripheral Feedback Systems Promotes Circadian Rhythms and Health. Front. Neurol. 2015, 6, 128. [Google Scholar] [CrossRef]
- Brancaccio, M.; Enoki, R.; Mazuski, C.N.; Jones, J.; Evans, J.A.; Azzi, A. Network-Mediated Encoding of Circadian Time: The Suprachiasmatic Nucleus (SCN) from Genes to Neurons to Circuits, and Back. J. Neurosci. 2014, 34, 15192–15199. [Google Scholar] [CrossRef] [PubMed]
- Hastings, M.H.; Duffield, G.E.; Ebling, F.J.; Kidd, A.; Maywood, E.S.; Schurov, I. Non-photic signalling in the suprachiasmatic nucleus. Biol. Cell 1997, 89, 495–503. [Google Scholar] [CrossRef]
- Maywood, E.S.; O’neill, J.S.; Chesham, J.E.; Hastings, M.H. Minireview: The Circadian Clockwork of the Suprachiasmatic Nuclei—Analysis of a Cellular Oscillator that Drives Endocrine Rhythms. Endocrinology 2007, 148, 5624–5634. [Google Scholar] [CrossRef]
- Yan, L.; Karatsoreos, I.; LeSauter, J.; Welsh, D.K.; Kay, S.; Foley, D.; Silver, R. Exploring Spatiotemporal Organization of SCN Circuits. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 527–541. [Google Scholar] [CrossRef] [PubMed]
- Gamble, K.L.; Berry, R.; Frank, S.J.; Young, M.E. Circadian clock control of endocrine factors. Nat. Rev. Endocrinol. 2014, 10, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Tousson, E.; Meissl, H. Suprachiasmatic Nuclei Grafts Restore the Circadian Rhythm in the Paraventricular Nucleus of the Hypothalamus. J. Neurosci. 2004, 24, 2983–2988. [Google Scholar] [CrossRef]
- Huang, Y.; Lin, X.; Lin, S. Neuropeptide Y and Metabolism Syndrome: An Update on Perspectives of Clinical Therapeutic Intervention Strategies. Front. Cell Dev. Biol. 2021, 9, 695623. [Google Scholar] [CrossRef] [PubMed]
- Tiesjema, B.; la Fleur, S.E.; Luijendijk, M.C.; Adan, R.A. Sustained NPY Overexpression in the PVN Results in Obesity via Temporarily Increasing Food Intake. Obesity 2009, 17, 1448–1450. [Google Scholar] [CrossRef]
- Baldini, G.; Phelan, K.D. The melanocortin pathway and control of appetite-progress and therapeutic implications. J. Endocrinol. 2019, 241, R1–R33. [Google Scholar] [CrossRef]
- Pyner, S. Neurochemistry of the paraventricular nucleus of the hypothalamus: Implications for cardiovascular regulation. J. Chem. Neuroanat. 2009, 38, 197–208. [Google Scholar] [CrossRef]
- Lu, Q.-B.; Feng, X.-M.; Tong, N.; Sun, H.-J.; Ding, L.; Wang, Y.-J.; Wang, X.; Zhou, Y.-B. Neuronal and Endothelial Nitric Oxide Synthases in the Paraventricular Nucleus Modulate Sympathetic Overdrive in Insulin-Resistant Rats. PLoS ONE 2015, 10, e0140762. [Google Scholar] [CrossRef]
- Wahlestedt, C.; Skagerberg, G.; Ekman, R.; Heilig, M.; Sundler, F.; Håkanson, R. Neuropeptide Y (NPY) in the area of the hypothalamic paraventricular nucleus activates the pituitary-adrenocortical axis in the rat. Brain Res. 1987, 417, 33–38. [Google Scholar] [CrossRef]
- Luo, S.; Meier, A.H.; Cincotta, A.H. Bromocriptine Reduces Obesity, Glucose Intolerance and Extracellular Monoamine Metabolite Levels in the Ventromedial Hypothalamus of Syrian Hamsters. Neuroendocrinology 1998, 68, 1–10. [Google Scholar] [CrossRef]
- Boundy, V.A.; Cincotta, A.H. Hypothalamic adrenergic receptor changes in the metabolic syndrome of genetically obese (ob/ob) mice. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R505–R514. [Google Scholar] [CrossRef]
- Kraszewski, K.Z.; Cincotta, A.H. Increased responsiveness of ventromedial hypothalamic neurons to norepinephrine in obese versus lean mice: Relation to the metabolic syndrome. Int. J. Mol. Med. 2000, 5, 349–355. [Google Scholar] [CrossRef]
- Luo, S.; Luo, J.; Cincotta, A.H. Chronic Ventromedial Hypothalamic Infusion of Norepinephrine and Serotonin Promotes Insulin Resistance and Glucose Intolerance. Neuroendocrinology 1999, 70, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Luo, S.; Zhang, Y.; Liang, Y.; Bina, K.G.; Jetton, T.L.; Scislowski, P.W.D. Chronic infusion of norepinephrine into the VMH of normal rats induces the obese glucose-intolerant state. Am. J. Physiol. Integr. Comp. Physiol. 2000, 278, R435–R444. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Luo, S.; Liang, Y. Hyperinsulinemia increases norepinephrine metabolism in the ventromedial hypothalamus of rats. Neuroreport 2000, 11, 383–387. [Google Scholar] [CrossRef]
- Pocai, A.; Lam, T.K.; Obici, S.; Gutierrez-Juarez, R.; Muse, E.D.; Arduini, A.; Rossetti, L. Restoration of hypothalamic lipid sensing normalizes energy and glucose homeostasis in overfed rats. J. Clin. Invest. 2006, 116, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.K.T. Neuronal regulation of homeostasis by nutrient sensing. Nat. Med. 2010, 16, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T.; Minokoshi, Y. Systemic Glucoregulation by Glucose-Sensing Neurons in the Ventromedial Hypothalamic Nucleus (VMH). J. Endocr. Soc. 2017, 1, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Routh, V.H. Glucose Sensing Neurons in the Ventromedial Hypothalamus. Sensors 2010, 10, 9002–9025. [Google Scholar] [CrossRef] [PubMed]
- Stoelzel, C.R.; Zhang, Y.; Cincotta, A.H. Circadian-timed dopamine agonist treatment reverses high-fat diet-induced diabetogenic shift in ventromedial hypothalamic glucose sensing. Endocrinol. Diabetes Metab. 2020, 3, e00139. [Google Scholar] [CrossRef]
- Luo, S.; Ezrokhi, M.; Trubitsyna, Y.; Li, Y.; Cincotta, A.H. Elevation of norepinephrine (NE) activity at the ventromedial hypothalamus (VMH) of normal rats induces the obese hypertensive insulin restsant state without altered feeding. Diabetes 2015, 64, A540. [Google Scholar]
- Luo, S.; Ezrokhi, M.; Trubitsyna, Y.; Cincotta, A.H. Elevation of Serotonin Activity within the Ventromedial Hypothalamus (VMH) Induces the Hypertensive Insulin Resistant State in Rats. Diabetes 2011, 60, A128. [Google Scholar]
- Luo, S.; Ezrokhi, M.; Trubitsyna, Y.; Cincotta, A.H. Intrahypothalamic circuitry regulating hypothalamic fuel sensing to induce insulin sensitivity or insulin resistance. Diabetologia 2008, 51, S59. [Google Scholar]
- Kalsbeek, A.; Foppen, E.; Schalij, I.; Van Heijningen, C.; van der Vliet, J.; Fliers, E.; Buijs, R.M. Circadian control of the daily plasma glucose rhythm: An interplay of GABA and glutamate. PLoS ONE 2008, 3, e3194. [Google Scholar] [CrossRef]
- Orozco-Cabal, L.; Pollandt, S.; Liu, J.; Shinnick-Gallagher, P.; Gallagher, J.P. Regulation of synaptic transmission by CRF receptors. Rev. Neurosci. 2006, 17, 279–308. [Google Scholar] [CrossRef]
- Herman, J.P.; Tasker, J.G. Paraventricular Hypothalamic Mechanisms of Chronic Stress Adaptation. Front. Endocrinol. 2016, 7, 137. [Google Scholar] [CrossRef] [PubMed]
- Bina, K.G.; Cincotta, A.H. Dopaminergic Agonists Normalize Elevated Hypothalamic Neuropeptide Y and Corticotropin-Releasing Hormone, Body Weight Gain, and Hyperglycemia in ob/ob Mice. Neuroendocrinology 2000, 71, 68–78. [Google Scholar] [CrossRef]
- Husum, H.; Msc, H.H.; Mathé, A.A. Early Life Stress Changes Concentrations of Neuropeptide Y and Corticotropin-releasing Hormone in Adult Rat Brain. Lithium Treatment Modifies These Changes. Neuropsychopharmacology 2002, 27, 756–764. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kalra, S.; Kalra, P. NPY and cohorts in regulating appetite, obesity and metabolic syndrome: Beneficial effects of gene therapy. Neuropeptides 2004, 38, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Denis, R.; Joly-Amado, A.; Cansell, C.; Castel, J.; Martinez, S.; Delbes, A.; Luquet, S. Central orchestration of peripheral nutrient partitioning and substrate utilization: Implications for the metabolic syndrome. Diabetes Metab. 2014, 40, 191–197. [Google Scholar] [CrossRef]
- Stofkova, A.; Skurlova, M.; Kiss, A.; Zelezna, B.; Zorad, S.; Jurcovicova, J. Activation of hypothalamic NPY, AgRP, MC4R, AND IL-6 mRNA levels in young Lewis rats with early-life diet-induced obesity. Endocr. Regul. 2009, 43, 99–106. [Google Scholar]
- Wei, W.; Pham, K.; Gammons, J.W.; Sutherland, D.; Liu, Y.; Smith, A.; Kaczorowski, C.C.; O’Connell, K.M. Diet composition, not calorie intake, rapidly alters intrinsic excitability of hypothalamic AgRP/NPY neurons in mice. Sci. Rep. 2015, 5, 16810. [Google Scholar] [CrossRef]
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.-Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and TNF-α on appetite-regulating NPY neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef]
- Paredes, S.; Ribeiro, L. Cortisol: The villain in metabolic syndrome? Rev. Assoc. Med. Bras. (1992) 2014, 60, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Prpić-Križevac, I.; Canecki-Varžić, S.; Bilić-Ćurčić, I. Hyperactivity of the hypothalamic-pituitary-adrenal axis in patients with type 2 diabetes and relations with insulin resistance and chronic complications. Wien. Klin. Wochenschr. 2012, 124, 403–411. [Google Scholar] [CrossRef]
- Huybrechts, I.; De Vriendt, T.; Breidenassel, C.; Rogiers, J.; Vanaelst, B.; Cuenca-Garcia, M.; Moreno, L.A.; Gonzalez-Gross, M.; Roccaldo, R.; Kafatos, A.; et al. Mechanisms of stress, energy homeostasis and insulin resistance in European adolescents--the HELENA study. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 1082–1089. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Yamada, T.; Mitani, H.; Yamada, S.; Pu, S.; Yamanashi, T.; Matsumura, H.; Nakagome, K.; Kaneko, K. Relationship between hypothalamic–pituitary–adrenal axis dysregulation and insulin resistance in elderly patients with depression. Psychiatry Res. 2015, 226, 494–498. [Google Scholar] [CrossRef]
- Sharma, V.K.; Singh, T.G. Chronic Stress and Diabetes Mellitus: Interwoven Pathologies. Curr. Diabetes Rev. 2020, 16, 546–556. [Google Scholar] [PubMed]
- Plat, L.; Byrne, M.M.; Sturis, J.; Polonsky, K.S.; Mockel, J.; Fery, F.; Van Cauter, E. Effects of morning cortisol elevation on insulin secretion and glucose regulation in humans. Am. J. Physiol. 1996, 270 Pt 1, E36–E42. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.J.; Wang, X.; Roux, A.V.D.; Sanchez, B.N.; Seeman, T.E.; Needham, B.L.; Golden, S.H. Antecedent longitudinal changes in body mass index are associated with diurnal cortisol curve features: The multi-ethnic study of atherosclerosis. Metabolism 2017, 68, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Rajamanickam, S.; Justice, N.J. CRF signaling between neurons in the paraventricular nucleus of the hypothalamus (PVN) coordinates stress responses. Neurobiol. Stress 2019, 11, 100192. [Google Scholar] [CrossRef]
- Nakamura, K.; Nakamura, Y.; Kataoka, N. A hypothalamomedullary network for physiological responses to environmental stresses. Nat. Rev. Neurosci. 2022, 23, 35–52. [Google Scholar] [CrossRef]
- Bernardis, L.L.; Bellinger, L.L. The dorsomedial hypothalamic nucleus revisited: 1998 update. Proc. Soc. Exp. Biol. Med. 1998, 218, 284–306. [Google Scholar] [CrossRef]
- Irwin, M.; Hauger, R.; Brown, M. Central corticotropin-releasing hormone activates the sympathetic nervous system and reduces immune function: Increased responsivity of the aged rat. Endocrinology 1992, 131, 1047–1053. [Google Scholar] [CrossRef]
- Brown, M.R.; Fisher, L.A.; Spiess, J.; Rivier, C.; Rivier, J.; Vale, W. Corticotropin-Releasing Factor: Actions on the Sympathetic Nervous System and Metabolism*. Endocrinology 1982, 111, 928–931. [Google Scholar] [CrossRef]
- Strack, A.; Sawyer, W.; Hughes, J.; Platt, K.; Loewy, A. A general pattern of CNS innervation of the sympathetic outflow demonstrated by transneuronal pseudorabies viral infections. Brain Res. 1989, 491, 156–162. [Google Scholar] [CrossRef]
- Valentino, R.J.; Foote, S.L.; Aston-Jones, G. Corticotropin-releasing factor activates noradrenergic neurons of the locus coeruleus. Brain Res. 1983, 270, 363–367. [Google Scholar] [CrossRef]
- Lowrance, S.A.; Ionadi, A.; McKay, E.; Douglas, X.; Johnson, J.D. Sympathetic nervous system contributes to enhanced corticosterone levels following chronic stress. Psychoneuroendocrinology 2016, 68, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Kenney, M.J.; Weiss, M.L.; Haywood, J.R. The paraventricular nucleus: An important component of the central neurocircuitry regulating sympathetic nerve outflow. Acta Physiol. Scand. 2003, 177, 7–15. [Google Scholar] [CrossRef]
- Grzeda, E.; Ziarniak, K.; Sliwowska, J.H. The paraventricular nucleus of the hypothalamus—The concertmaster of autonomic control. Focus on blood pressure regulation. Acta Neurobiol. Exp. (Wars) 2023, 83, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Ezrokhi, M.; Luo, S.; Trubitsyna, Y.; Cincotta, A.H. Neuroendocrine and metabolic components of dopamine agonist amelioration of metabolic syndrome in SHR rats. Diabetol. Metab. Syndr. 2014, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Ezrokhi, M.; Zhang, Y.; Luo, S.; Cincotta, A.H. Time-of-Day-Dependent Effects of Bromocriptine to Ameliorate Vascular Pathology and Metabolic Syndrome in SHR Rats Held on High Fat Diet. Int. J. Mol. Sci. 2021, 22, 6142. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; MacEachern, T.A.; Meier, A.H. Bromocriptine redirects metabolism and prevents seasonal onset of obese hyperinsulinemic state in Syrian hamsters. Am. J. Physiol. Metab. 1993, 264 Pt 1, E285–E293. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Meier, A.H. Bromocriptine inhibits in vivo free fatty acid oxidation and hepatic glucose output in seasonally obese hamsters (Mesocricetus auratus). Metabolism 1995, 44, 1349–1355. [Google Scholar] [CrossRef]
- Davis, L.M.; Pei, Z.; Trush, M.A.; Cheskin, L.J.; Contoreggi, C.; McCullough, K.; Watkins, P.A.; Moran, T.H. Bromocriptine reduces steatosis in obese rodent models. J. Hepatol. 2006, 45, 439–444. [Google Scholar] [CrossRef]
- Tsai, T.-H.; Cincotta, A. 1868-P: Bromocriptine (BC) Improves Hepatic Steatosis, Inflammation, and ER Stress in a Mouse Obesogenic Dietary Model of Nonalcoholic Steatohepatitis (NASH). Diabetes 2019, 68 (Suppl. 1), 1868-P. [Google Scholar] [CrossRef]
- Tellez, L.A.; Medina, S.; Han, W.; Ferreira, J.G.; Licona-Limón, P.; Ren, X.; Lam, T.T.; Schwartz, G.J.; De Araujo, I.E. A Gut Lipid Messenger Links Excess Dietary Fat to Dopamine Deficiency. Science 2013, 341, 800–802. [Google Scholar] [CrossRef] [PubMed]
- Hryhorczuk, C.; Florea, M.; Rodaros, D.; Poirier, I.; Daneault, C.; Rosiers, C.D.; Arvanitogiannis, A.; Alquier, T.; Fulton, S. Dampened Mesolimbic Dopamine Function and Signaling by Saturated but not Monounsaturated Dietary Lipids. Neuropsychopharmacology 2016, 41, 811–821. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Haburcak, M.; Avena, N.; Moyer, M.; Hoebel, B.; Pothos, E. Deficits of mesolimbic dopamine neurotransmission in rat dietary obesity. Neuroscience 2009, 159, 1193–1199. [Google Scholar] [CrossRef] [PubMed]
- Florian, J.P.; Pawelczyk, J.A. Non-esterified fatty acids increase arterial pressure via central sympathetic activation in humans. Clin. Sci. 2009, 118, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Benthem, L.; Keizer, K.; Wiegman, C.; De Boer, S.F.; Strubbe, J.H.; Steffens, A.B.; Kuipers, F.; Scheurink, A.J.W. Excess portal venous long-chain fatty acids induce syndrome X via HPA axis and sympathetic activation. Am. J. Physiol. Metab. 2000, 279, E1286–E1293. [Google Scholar] [CrossRef]
- Stojiljkovic, M.P.; Zhang, D.; Lopes, H.F.; Lee, C.G.; Goodfriend, T.L.; Egan, B.M. Hemodynamic effects of lipids in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 280, R1674–R1679. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Tarshoby, M.; Monestel, R.; Hook, G.; Cronin, J.; Johnson, A.; Bayazeed, B.; Baron, A.D. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J. Clin. Investig. 1997, 100, 1230–1239. [Google Scholar] [CrossRef]
- Haastrup, A.T.; Stepniakowski, K.T.; Goodfriend, T.L.; Egan, B.M. Intralipid enhances alpha1-adrenergic receptor mediated pressor sensitivity. Hypertension 1998, 32, 693–698. [Google Scholar] [CrossRef]
- Benthem, L.; Kuipers, F.; Steffens, A.B.; Scheurink, A.J.W. Excessive portal venous supply of long-chain free fatty acids to the liver, leading to hypothalamus-pituitary-adrenal-axis and sympathetic activation as a key to the development of syndrome X. A proposed concept for the induction of syndrome X. Ann. N. Y. Acad. Sci. 1999, 892, 308–311. [Google Scholar] [CrossRef][Green Version]
- Reda, E.; Hassaneen, S.; El-Abhar, H.S. Novel Trajectories of Bromocriptine Antidiabetic Action: Leptin-IL-6/JAK2/p-STAT3/SOCS3, p-IR/p-AKT/GLUT4, PPAR-gamma/Adiponectin, Nrf2/PARP-1, and GLP-1. Front. Pharmacol. 2018, 9, 771. [Google Scholar] [CrossRef]
- Moore, M.C.; Smith, M.S.; Swift, L.L.; Cincotta, A.H.; Ezrokhi, M.; Cominos, N.; Zhang, Y.; Farmer, B.; Cherrington, A.D. Bromocriptine mesylate improves glucose tolerance and disposal in a high-fat-fed canine model. Am. J. Physiol. Metab. 2020, 319, E133–E145. [Google Scholar] [CrossRef]
- Ezrokhi, M.; Luo, S.; Trubitsyna, Y.; Cincotta, A.H. Weighted effects of bromocriptine treatment on glucose homeostasis during hyperglycemic versus euglycemic clamp conditions in insulin resistant hamsters: Bromocriptine as a unique post-prandial insulin sensitizer. J. Diabetes Metab. 2012, S2, 1–4. [Google Scholar] [CrossRef]
- Folgueira, C.; Beiroa, D.; Porteiro, B.; Duquenne, M.; Puighermanal, E.; Fondevila, M.F.; Barja-Fernandez, S.; Gallego, R.; Hernandez-Bautista, R.; Castelao, C.; et al. Hypothalamic dopamine signaling regulates brown fat thermogenesis. Nat. Metab. 2019, 1, 811–829. [Google Scholar] [CrossRef]
- Tavares, G.; Marques, D.; Barra, C.; Rosendo-Silva, D.; Costa, A.; Rodrigues, T.; Gasparini, P.; Melo, B.; Sacramento, J.; Seiça, R.; et al. Dopamine D2 receptor agonist, bromocriptine, remodels adipose tissue dopaminergic signalling and upregulates catabolic pathways, improving metabolic profile in type 2 diabetes. Mol. Metab. 2021, 51, 101241. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Meier, A.H. Reductions of body fat stores and total plasma cholesterol and triglyceride concentrations in several species by bromocriptine treatment. Life Sci. 1989, 45, 2247–2254. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Cersosimo, E.; Alatrach, M.; Ezrokhi, M.; Agyin, C.; Adams, J.; Chilton, R.; Triplitt, C.; Chamarthi, B.; Cominos, N.; et al. Bromocriptine-QR Therapy Reduces Sympathetic Tone and Ameliorates a Pro-Oxidative/Pro-Inflammatory Phenotype in Peripheral Blood Mononuclear Cells and Plasma of Type 2 Diabetes Subjects. Int. J. Mol. Sci. 2022, 23, 8851. [Google Scholar] [CrossRef] [PubMed]
- Gaziano, J.M.; Cincotta, A.H.; O’Connor, C.M.; Ezrokhi, M.; Rutty, D.; Ma, Z.J.; Scranton, R.E. Randomized clinical trial of quick-release bromocriptine among patients with type 2 diabetes on overall safety and cardiovascular outcomes. Diabetes Care 2010, 33, 1503–1508. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Cincotta, A.H.; Vinik, A.; Blonde, L.; Bohannon, N.; Scranton, R. Effect of Bromocriptine-QR (a Quick-Release Formulation of Bromocriptine Mesylate) on Major Adverse Cardiovascular Events in Type 2 Diabetes Subjects. J. Am. Hear Assoc. 2012, 1, e002279. [Google Scholar] [CrossRef]
- Chamarthi, B.; Gaziano, J.M.; Blonde, L.; Vinik, A.; Scranton, R.E.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Timed Bromocriptine-QR Therapy Reduces Progression of Cardiovascular Disease and Dysglycemia in Subjects with Well-Controlled Type 2 Diabetes Mellitus. J. Diabetes Res. 2015, 2015, 157698. [Google Scholar] [CrossRef]
- Chamarthi, B.; Ezrokhi, M.; Rutty, D.; Cincotta, A.H. Impact of bromocriptine-QR therapy on cardiovascular outcomes in type 2 diabetes mellitus subjects on metformin. Postgrad. Med. 2016, 128, 761–769. [Google Scholar] [CrossRef]
- Nade, V.S.; Kawale, L.A.; Todmal, U.B.; Tajanpure, A.B. Effect of bromocriptine on cardiovascular complications associated with metabolic syndrome in fructose fed rats. Indian J. Pharmacol. 2012, 44, 688–693. [Google Scholar]
- Gao, J.; Guo, J.; Li, H.; Bai, S.; Li, H.; Wu, B.; Wang, L.; Xi, Y.; Tian, Y.; Yang, G.; et al. Involvement of dopamine D2 receptors activation in ischemic post-conditioning-induced cardioprotection through promoting PKC-epsilon particulate translocation in isolated rat hearts. Mol. Cell. Biochem. 2013, 379, 267–276. [Google Scholar] [CrossRef]
- Li, H.; Wei, C.; Gao, J.; Bai, S.; Li, H.; Zhao, Y.; Li, H.; Han, L.; Tian, Y.; Yang, G.; et al. Mediation of dopamine D2 receptors activation in post-conditioning-attenuated cardiomyocyte apoptosis. Exp. Cell Res. 2014, 323, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Z.; Guo, J.; Gao, J.; Han, L.-P.; Jiang, C.-M.; Bai, S.-Z.; Zhang, W.-H.; Li, G.-W.; Wang, L.-N.; Zhao, Y.-J.; et al. Role of dopamine D2 receptors in ischemia/reperfusion induced apoptosis of cultured neonatal rat cardiomyocytes. J. Biomed. Sci. 2011, 18, 18. [Google Scholar] [CrossRef]
- O’Neill, M.J.; Hicks, C.A.; Ward, M.A.; Cardwell, G.P.; Reymann, J.-M.; Allain, H.; Bentué-Ferrer, D. Dopamine D2 receptor agonists protect against ischaemia-induced hippocampal neurodegeneration in global cerebral ischaemia. Eur. J. Pharmacol. 1998, 352, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Narkar, V.; Kunduzova, O.; Hussain, T.; Cambon, C.; Parini, A.; Lokhandwala, M. Dopamine D2-like receptor agonist bromocriptine protects against ischemia/reperfusion injury in rat kidney. Kidney Int. 2004, 66, 633–640. [Google Scholar] [CrossRef]
- Scislowski, P.W.; Tozzo, E.; Zhang, Y.; Phaneuf, S.; Prevelige, R.; Cincotta, A.H. Biochemical mechanisms responsible for the attenuation of diabetic and obese conditions in ob/ob mice treated with dopaminergic agonists. Int. J. Obes. Relat. Metab. Disord. 1999, 23, 425–431. [Google Scholar] [CrossRef]
- Liang, Y.; Jetton, T.L.; Lubkin, M.; Meier, A.H.; Cincotta, A.H. Bromocriptine/SKF38393 ameliorates islet dysfunction in the diabetic (db/db) mouse. Cell. Mol. Life Sci. 1998, 54, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Scislowski, P.; Phaneuf, S.; Prevelige, R.; Meier, A.H.; Joslin, J. Dopamine agonist treatment ameliorates the obese hyperglycemic condition in lethal yellow (A y/a) mice. Diabetes 1998, 47, A318. [Google Scholar]
- Davis, L.M.; Michaelides, M.; Cheskin, L.J.; Moran, T.H.; Aja, S.; Watkins, P.A.; Pei, Z.; Contoreggi, C.; McCullough, K.; Hope, B.; et al. Bromocriptine Administration Reduces Hyperphagia and Adiposity and Differentially Affects Dopamine D2 Receptor and Transporter Binding in Leptin-Receptor-Deficient Zucker Rats and Rats with Diet-Induced Obesity. Neuroendocrinology 2009, 89, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Southern, L.L.; Cincotta, A.H.; Meier, A.H.; Bidner, T.D.; Watkins, K.L. Bromocriptine-induced reduction of body fat in pigs. J. Anim. Sci. 1990, 68, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Peixoto-Silva, N.; Moura, E.G.; Carvalho, J.C.; Nobre, J.L.; Quitete, F.T.; Pinheiro, C.R.; Santos-Silva, A.P.; de Oliveira, E.; Lisboa, P.C. Bromocriptine treatment at the end of lactation prevents hyperphagia, higher visceral fat and liver triglycerides in early-weaned rats at adulthood. Clin. Exp. Pharmacol. Physiol. 2017, 44, 488–499. [Google Scholar] [CrossRef]
- Cermakian, N.; Boivin, D.B. The regulation of central and peripheral circadian clocks in humans. Obes. Rev. 2009, 10 (Suppl. 2), 25–36. [Google Scholar] [CrossRef]
- Gentry, N.W.; Ashbrook, L.H.; Fu, Y.-H.; Ptáček, L.J. Human circadian variations. J. Clin. Investig. 2021, 131, e148282. [Google Scholar] [CrossRef] [PubMed]
- Lack, L.C.; Wright, H.R. Chronobiology of sleep in humans. Cell. Mol. Life Sci. 2007, 64, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Eckstein, M.L.; Wolf, A.; Zimmer, R.T.; Wachsmuth, N.B.; Moser, O. Eat, Train, Sleep-Retreat? Hormonal Interactions of Intermittent Fasting, Exercise and Circadian Rhythm. Biomolecules 2021, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Joo, Y.; Kim, M.-S.; Choe, H.K.; Tong, Q.; Kwon, O. Effects of Intermittent Fasting on the Circulating Levels and Circadian Rhythms of Hormones. Endocrinol. Metab. 2021, 36, 745–756. [Google Scholar] [CrossRef]
- Tsang, A.H.; Barclay, J.L.; Oster, H. Interactions between endocrine and circadian systems. J. Mol. Endocrinol. 2014, 52, R1–R16. [Google Scholar] [CrossRef]
- Skene, D.J.; Arendt, J. Human circadian rhythms: Physiological and therapeutic relevance of light and melatonin. Ann. Clin. Biochem. 2006, 43 Pt 5, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Vasey, C.; McBride, J.; Penta, K. Circadian Rhythm Dysregulation and Restoration: The Role of Melatonin. Nutrients 2021, 13, 3480. [Google Scholar] [CrossRef]
- Morris, C.J.; Aeschbach, D.; Scheer, F.A. Circadian system, sleep and endocrinology. Mol. Cell Endocrinol. 2012, 349, 91–104. [Google Scholar] [CrossRef]
- Qian, J.; Man, C.D.; Morris, C.J.; Cobelli, C.; Scheer, F.A.J.L. Differential effects of the circadian system and circadian misalignment on insulin sensitivity and insulin secretion in humans. Diabetes Obes. Metab. 2018, 20, 2481–2485. [Google Scholar] [CrossRef] [PubMed]
- Catalano, F.; De Vito, F.; Cassano, V.; Fiorentino, T.V.; Sciacqua, A.; Hribal, M.L. Circadian Clock Desynchronization and Insulin Resistance. Int. J. Environ. Res. Public Health 2022, 20, 29. [Google Scholar] [CrossRef]
- Wefers, J.; van Moorsel, D.; Hansen, J.; Connell, N.J.; Havekes, B.; Hoeks, J.; Lichtenbelt, W.D.v.M.; Duez, H.; Phielix, E.; Kalsbeek, A.; et al. Circadian misalignment induces fatty acid metabolism gene profiles and compromises insulin sensitivity in human skeletal muscle. Proc. Natl. Acad. Sci. USA 2018, 115, 7789–7794. [Google Scholar] [CrossRef]
- Pisu, E.; Diana, A.; Lombardi, A.; Cassader, M.; Pagano, G. Diurnal variations in insulin secretion and insulin sensitivity in aged subjects. Acta Diabetol. Lat. 1980, 17, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Ratzmann, K.P.; Albrecht, G.; Bibergeil, H. Diurnal rhythm of insulin sensitivity in subjects with normal and impaired glucose tolerance. Exp. Clin. Endocrinol. 1983, 81, 263–272. [Google Scholar] [CrossRef]
- Lange, T.; Dimitrov, S.; Born, J. Effects of sleep and circadian rhythm on the human immune system. Ann. N. Y. Acad. Sci. USA 2010, 1193, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.K.; Zitting, K.; Wang, W.; Buxton, O.M.; Williams, J.S.; Duffy, J.F.; Czeisler, C.A. Fasting blood triglycerides vary with circadian phase in both young and older people. Physiol. Rep. 2020, 8, e14453. [Google Scholar] [CrossRef]
- Romon, M.; Le Fur, C.; Lebel, P.; Edmé, J.L.; Fruchart, J.C.; Dallongeville, J. Circadian variation of postprandial lipemia. Am. J. Clin. Nutr. 1997, 65, 934–940. [Google Scholar] [CrossRef]
- Grant, L.K.; Hilaire, M.A.S.; Brainard, G.C.; Czeisler, C.A.; Lockley, S.W.; Rahman, S.A. Endogenous circadian regulation and phase resetting of clinical metabolic biomarkers. J. Pineal Res. 2021, 71, e12752. [Google Scholar] [CrossRef]
- Mazzoccoli, G.; Vinciguerra, M.; Oben, J.; Tarquini, R.; De Cosmo, S. Non-alcoholic fatty liver disease: The role of nuclear receptors and circadian rhythmicity. Liver Int. 2014, 34, 1133–1152. [Google Scholar] [CrossRef]
- Bray, M.S.; Young, M.E. Diurnal variations in myocardial metabolism. Cardiovasc. Res. 2008, 79, 228–237. [Google Scholar] [CrossRef][Green Version]
- Pourcet, B.; Duez, H. Circadian Control of Inflammasome Pathways: Implications for Circadian Medicine. Front. Immunol. 2020, 11, 1630. [Google Scholar] [CrossRef]
- Huang, H.; Li, Z.; Ruan, Y.; Feng, W.; Chen, J.; Li, X.; Ouyang, L.; Huang, H. Circadian rhythm disorder: A potential inducer of vascular calcification? J. Physiol. Biochem. 2020, 76, 513–524. [Google Scholar] [CrossRef]
- Rodrigo, G.C.; Herbert, K.E. Regulation of vascular function and blood pressure by circadian variation in redox signalling. Free. Radic. Biol. Med. 2018, 119, 115–120. [Google Scholar] [CrossRef]
- Stritesky Larssen, K.; Lyberg, T. Oxidative status--age- and circadian variations?--a study in leukocytes/plasma. Neuro. Endocrinol. Lett. 2006, 27, 445–452. [Google Scholar]
- Hardeland, R.; Coto-Montes, A.; Poeggeler, B. Circadian rhythms, oxidative stress, and antioxidative defense mechanisms. Chronobiol. Int. 2003, 20, 921–962. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wu, S.; Li, W.; He, W. A Tissue-Specific Rhythmic Recruitment Pattern of Leukocyte Subsets. Front. Immunol. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jain, M.K. Circadian regulation of cardiac metabolism. J. Clin. Investig. 2021, 131, e148276. [Google Scholar] [CrossRef] [PubMed]
- Martino, T.A.; Young, M.E. Influence of the Cardiomyocyte Circadian Clock on Cardiac Physiology and Pathophysiology. J. Biol. Rhythm. 2015, 30, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Khaper, N.; Bailey, C.D.; Ghugre, N.R.; Reitz, C.; Awosanmi, Z.; Waines, R.; Martino, T.A. Implications of disturbances in circadian rhythms for cardiovascular health: A new frontier in free radical biology. Free. Radic. Biol. Med. 2018, 119, 85–92. [Google Scholar] [CrossRef]
- Peliciari-Garcia, R.A.; Darley-Usmar, V.; Young, M.E. An overview of the emerging interface between cardiac metabolism, redox biology and the circadian clock. Free. Radic. Biol. Med. 2018, 119, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Thosar, S.S.; Shea, S.A. Circadian control of human cardiovascular function. Curr. Opin. Pharmacol. 2021, 57, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Giles, T.D. Circadian rhythm of blood pressure and the relation to cardiovascular events. J. Hypertens. 2006, 24, S11–S16. [Google Scholar] [CrossRef] [PubMed]
- Izzedine, H.; Launay-Vacher, V.; Deray, G. Abnormal blood pressure circadian rhythm: A target organ damage? Int. J. Cardiol. 2006, 107, 343–349. [Google Scholar] [CrossRef]
- Monfredi, O.; Lakatta, E. Complexities in cardiovascular rhythmicity: Perspectives on circadian normality, ageing and disease. Cardiovasc. Res. 2019, 115, 1576–1595. [Google Scholar] [CrossRef] [PubMed]
- Latimer, M.N.; Young, M.E. Circadian Governance of Cardiac Growth. Cells 2022, 11, 1494. [Google Scholar] [CrossRef]
- Crnko, S.; Du Pré, B.C.; Sluijter, J.P.G.; Van Laake, L.W. Circadian rhythms and the molecular clock in cardiovascular biology and disease. Nat. Rev. Cardiol. 2019, 16, 437–447. [Google Scholar] [CrossRef]
- Rabinovich-Nikitin, I.; Lieberman, B.; Martino, T.A.; Kirshenbaum, L.A. Circadian-Regulated Cell Death in Cardiovascular Diseases. Circulation 2019, 139, 965–980. [Google Scholar] [CrossRef]
- Crnko, S.; Cour, M.; Van Laake, L.W.; Lecour, S. Vasculature on the clock: Circadian rhythm and vascular dysfunction. Vasc. Pharmacol. 2018, 108, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Aziz, I.S.; McMahon, A.M.; Friedman, D.; Rabinovich-Nikitin, I.; Kirshenbaum, L.A.; Martino, T.A. Circadian influence on inflammatory response during cardiovascular disease. Curr. Opin. Pharmacol. 2021, 57, 60–70. [Google Scholar] [CrossRef]
- Hyun, M.H.; Kang, J.H.; Kim, S.; Na, J.O.; Choi, C.U.; Kim, J.W.; Kim, E.J.; Rha, S.-W.; Park, C.G.; Lee, E.; et al. Patterns of Circadian Variation in 24-Hour Ambulatory Blood Pressure, Heart Rate, and Sympathetic Tone Correlate with Cardiovascular Disease Risk: A Cluster Analysis. Cardiovasc. Ther. 2020, 2020, 4354759. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Zhong, Q.; Liu, C.; Zhou, D.; Li, X.; Zhang, Y.; Feng, Y.; Zhou, Y. Associations of systolic and diastolic blood pressure night-to-day ratios with atherosclerotic cardiovascular diseases. Hypertens. Res. 2016, 39, 874–878. [Google Scholar] [CrossRef] [PubMed]
- Manfredini, R.; Boari, B.; Salmi, R.; Fabbian, F.; Pala, M.; Tiseo, R.; Portaluppi, F. Twenty-Four-Hour Patterns in Occurrence and Pathophysiology of Acute Cardiovascular Events and Ischemic Heart Disease. Chrono-Int. 2013, 30, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Brotman, D.J.; Golden, S.H.; Wittstein, I.S. The cardiovascular toll of stress. Lancet 2007, 370, 1089–1100. [Google Scholar] [CrossRef]
- Muller, J.E.; Tofler, G.H.; Willich, S.N.; Stone, P.H. Circadian variation of cardiovascular disease and sympathetic activity. J. Cardiovasc. Pharmacol. 1987, 10 (Suppl. 2), S104–S109; discussion S110–S111. [Google Scholar] [PubMed]
- Winklewski, P.J.; Radkowski, M.; Demkow, U. Relevance of Immune-Sympathetic Nervous System Interplay for the Development of Hypertension. Adv. Exp. Med. Biol. 2016, 884, 37–43. [Google Scholar]
- Scheiermann, C.; Kunisaki, Y.; Lucas, D.; Chow, A.; Jang, J.E.; Zhang, D.; Hashimoto, D.; Merad, M.; Frenette, P.S. Adrenergic nerves govern circadian leukocyte recruitment to tissues. Immunity 2012, 37, 290–301. [Google Scholar] [CrossRef] [PubMed]
- de Juan, A.; Ince, L.M.; Pick, R.; Chen, C.S.; Molica, F.; Zuchtriegel, G.; Wang, C.; Zhang, D.; Druzd, D.; Hessenauer, M.E.T.; et al. Artery-Associated Sympathetic Innervation Drives Rhythmic Vascular Inflammation of Arteries and Veins. Circulation 2019, 140, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Detry, J.M.; Vincent, M. Circadian rhythms in cardiovascular disease: The crucial hours. J. Hum. Hypertens 1992, 6 (Suppl. 1), S3–S8. [Google Scholar]
- Tendler, A.; Bar, A.; Mendelsohn-Cohen, N.; Karin, O.; Kohanim, Y.K.; Maimon, L.; Milo, T.; Raz, M.; Mayo, A.; Tanay, A.; et al. Hormone seasonality in medical records suggests circannual endocrine circuits. Proc. Natl. Acad. Sci. USA 2021, 118, e2003926118. [Google Scholar] [CrossRef]
- Smals, A.; Kloppenborg, P.; Benraad, T. Circannual cycle in plasma testosterone levels in man. J. Clin. Endocrinol. Metab. 1976, 42, 979–982. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Vitale, J.A.; Logoluso, S.; Logoluso, G.; Cocco, N.; Cocco, G.; Cocco, A.; Banfi, G. Circannual rhythm of plasmatic vitamin D levels and the association with markers of psychophysical stress in a cohort of Italian professional soccer players. Chrono- Int. 2017, 34, 471–479. [Google Scholar] [CrossRef]
- Pierre, K.; Schlesinger, N.; Androulakis, I.P. The role of the hypothalamic-pituitary-adrenal axis in modulating seasonal changes in immunity. Physiol. Genom. 2016, 48, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, N.; Okuda, T.; Shinohara, H.; Yamasaki, R.S.; Hirano, N.; Kang, J.; Ogawa, M.; Nishi, N.N. Relationship between Seasonal Changes in Food Intake and Energy Metabolism, Physical Activity, and Body Composition in Young Japanese Women. Nutrients 2022, 14, 506. [Google Scholar] [CrossRef] [PubMed]
- Clarkson-Townsend, D.A.; Kennedy, E.; Everson, T.M.; Deyssenroth, M.A.; Burt, A.A.; Hao, K.; Chen, J.; Pardue, M.T.; Marsit, C.J. Seasonally variant gene expression in full-term human placenta. FASEB J. 2020, 34, 10431–10442. [Google Scholar] [CrossRef]
- Garaulet, M.; Gomez-Abellan, P. Chronobiology and obesity. Nutr. Hosp. 2013, 28 (Suppl. 5), 114–120. [Google Scholar]
- Kanikowska, D.; Sato, M.; Witowski, J. Contribution of daily and seasonal biorhythms to obesity in humans. Int. J. Biometeorol. 2015, 59, 377–384. [Google Scholar] [CrossRef]
- Linden, M.; Larsson, M.; Prellner, T.; Brattsand, R. Seasonal variation in the phagocytic activity and arachidonic-acid metabolism of human blood monocytes in healthy non-smokers, smokers and chronic bronchitics. Agents Actions Suppl. 1990, 30, 121–132. [Google Scholar]
- Ciardullo, S.; Muraca, E.; Cannistraci, R.; Manzoni, G.; Perra, S.; Bianconi, E.; Oltolini, A.; Zerbini, F.; Grassi, G.; Mancia, G.; et al. Seasonal variation in estimated cardiovascular risk in patients with type 2 diabetes. Nutr. Metab. Cardiovasc. Dis. 2021, 31, 1494–1500. [Google Scholar] [CrossRef] [PubMed]
- Matheson, G.J.; Schain, M.; Almeida, R.; Lundberg, J.; Cselényi, Z.; Borg, J.; Varrone, A.; Farde, L.; Cervenka, S. Diurnal and seasonal variation of the brain serotonin system in healthy male subjects. Neuroimage 2015, 112, 225–231. [Google Scholar] [CrossRef]
- Stergiou, G.S.; Palatini, P.; Modesti, P.A.; Asayama, K.; Asmar, R.; Bilo, G.; de la Sierra, A.; Dolan, E.; Head, G.; Kario, K.; et al. Seasonal variation in blood pressure: Evidence, consensus and recommendations for clinical practice. Consensus statement by the European Society of Hypertension Working Group on Blood Pressure Monitoring and Cardiovascular Variability. J. Hypertens 2020, 38, 1235–1243. [Google Scholar] [CrossRef]
- Narita, K.; Hoshide, S.; Kario, K. Seasonal variation in blood pressure: Current evidence and recommendations for hypertension management. Hypertens. Res. 2021, 44, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Dentali, F.; Manfredini, R.; Ageno, W. Seasonal variability of venous thromboembolism. Curr. Opin. Pulm. Med. 2009, 15, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.W. Seasonal changes in preprandial glucose, A1C, and blood pressure in diabetic patients. Diabetes Care 2007, 30, 2501–2502. [Google Scholar] [CrossRef]
- Sohmiya, M.; Kanazawa, I.; Kato, Y. Seasonal Changes in Body Composition and Blood HbA1c Levels Without Weight Change in Male Patients With Type 2 Diabetes Treated With Insulin. Diabetes Care 2004, 27, 1238–1239. [Google Scholar] [CrossRef]
- Gikas, A.; Sotiropoulos, A.; Pastromas, V.; Papazafiropoulou, A.; Apostolou, O.; Pappas, S. Seasonal variation in fasting glucose and HbA1c in patients with type 2 diabetes. Prim. Care Diabetes 2009, 3, 111–114. [Google Scholar] [CrossRef]
- Partonen, T. During winter the body resists insulin. Hypertens. Res. 2013, 36, 390–391. [Google Scholar] [CrossRef] [PubMed]
- Kamezaki, F.; Sonoda, S.; Nakata, S.; Muraoka, Y.; Okazaki, M.; Tamura, M.; Abe, H.; Tekeuchi, M.; Otsuji, Y. Association of seasonal variation in the prevalence of metabolic syndrome with insulin resistance. Hypertens. Res. 2013, 36, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Kamezaki, F.; Sonoda, S.; Tomotsune, Y.; Yunaka, H.; Otsuji, Y. Seasonal variation in metabolic syndrome prevalence. Hypertens. Res. 2010, 33, 568–572. [Google Scholar] [CrossRef]
- Rintamäki, R.; Grimaldi, S.; Englund, A.; Haukka, J.; Partonen, T.; Reunanen, A.; Aromaa, A.; Lönnqvist, J. Seasonal Changes in Mood and Behavior Are Linked to Metabolic Syndrome. PLoS ONE 2008, 3, e1482. [Google Scholar] [CrossRef] [PubMed]
- Marti-Soler, H.; Gonseth, S.; Gubelmann, C.; Stringhini, S.; Bovet, P.; Chen, P.-C.; Wojtyniak, B.; Paccaud, F.; Tsai, D.-H.; Zdrojewski, T.; et al. Seasonal Variation of Overall and Cardiovascular Mortality: A Study in 19 Countries from Different Geographic Locations. PLoS ONE 2014, 9, e113500. [Google Scholar] [CrossRef]
- Bhatia, S.; Bhatia, S.; Mears, J.; Dibu, G.; Deshmukh, A. Seasonal Periodicity of Ischemic Heart Disease and Heart Failure. Hear Fail. Clin. 2017, 13, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Keates, A.K.; Redfern, A.; McMurray, J.J.V. Seasonal variations in cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 654–664. [Google Scholar] [CrossRef]
- Kelly, G.S. Seasonal variations of selected cardiovascular risk factors. Altern. Med. Rev. J. Clin. Ther. 2005, 10, 307–320. [Google Scholar]
- Modesti, P.A.; Rapi, S.; Rogolino, A.; Tosi, B.; Galanti, G. Seasonal blood pressure variation: Implications for cardiovascular risk stratification. Hypertens. Res. 2018, 41, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Castañeda, T.R.; Prado, B.M.; Prieto, D.; Mora, F.; de Prado, B.M. Circadian rhythms of dopamine, glutamate and GABA in the striatum and nucleus accumbens of the awake rat: Modulation by light. J. Pineal Res. 2004, 36, 177–185. [Google Scholar] [CrossRef]
- Schade, R.; Vick, K.; Ott, T.; Sohr, R.; Pfister, C.; Bellach, J.; Golor, G.; Lemmer, B. Circadian rhythms of dopamine and cholecystokinin in nucleus accumbens and striatum of rats--influence on dopaminergic stimulation. Chronobiol. Int. 1995, 12, 87–99. [Google Scholar] [CrossRef]
- Dunn, J.P.; Kessler, R.M.; Feurer, I.D.; Volkow, N.D.; Patterson, B.W.; Ansari, M.S.; Li, R.; Marks-Shulman, P.; Abumrad, N.N. Relationship of Dopamine Type 2 Receptor Binding Potential With Fasting Neuroendocrine Hormones and Insulin Sensitivity in Human Obesity. Diabetes Care 2012, 35, 1105–1111. [Google Scholar] [CrossRef]
- Wang, G.-J.; Volkow, N.D.; Logan, J.; Pappas, N.R.; Wong, C.T.; Zhu, W.; Netusll, N.; Fowler, J.S. Brain dopamine and obesity. Lancet 2001, 357, 354–357. [Google Scholar] [CrossRef]
- Volkow, N.D.; Wang, G.-J.; Telang, F.; Fowler, J.S.; Thanos, P.K.; Logan, J.; Alexoff, D.; Ding, Y.-S.; Wong, C.; Ma, Y.; et al. Low dopamine striatal D2 receptors are associated with prefrontal metabolism in obese subjects: Possible contributing factors. Neuroimage 2008, 42, 1537–1543. [Google Scholar] [CrossRef]
- Haltia, L.T.; Rinne, J.O.; Merisaari, H.; Maguire, R.P.; Savontaus, E.; Helin, S.; Någren, K.; Kaasinen, V. Effects of intravenous glucose on dopaminergic function in the human brain in vivo. Synapse 2007, 61, 748–756. [Google Scholar] [CrossRef]
- Berland, C.; Montalban, E.; Perrin, E.; Di Miceli, M.; Nakamura, Y.; Martinat, M.; Sullivan, M.; Davis, X.S.; Shenasa, M.A.; Martin, C.; et al. Circulating Triglycerides Gate Dopamine-Associated Behaviors through DRD2-Expressing Neurons. Cell Metab. 2020, 31, 773–790.e11. [Google Scholar] [CrossRef]
- Caravaggio, F.; Borlido, C.; Hahn, M.; Feng, Z.; Fervaha, G.; Gerretsen, P.; Nakajima, S.; Plitman, E.; Chung, J.K.; Iwata, Y.; et al. Reduced insulin sensitivity is related to less endogenous dopamine at D2/3 receptors in the ventral striatum of healthy nonobese humans. Int. J. Neuropsychopharmacol. 2015, 18, pyv014. [Google Scholar] [CrossRef]
- Ter Horst, K.W.; Lammers, N.M.; Trinko, R.; Opland, D.M.; Figee, M.; Ackermans, M.T.; Booij, J.; van den Munckhof, P.; Schuurman, P.R.; Fliers, E.; et al. Striatal dopamine regulates systemic glucose metabolism in humans and mice. Sci. Transl. Med. 2018, 10, eaar3752. [Google Scholar] [CrossRef]
- Bahler, L.; Verberne, H.J.; Brakema, E.; Tepaske, R.; Booij, J.; Hoekstra, J.B.; Holleman, F. Bromocriptine and insulin sensitivity in lean and obese subjects. Endocr. Connect. 2016, 5, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.P.; Kohn, P.D.; Baller, E.B.; Bronstein, J.A.; Masdeu, J.C.; Berman, K.F. Seasonal effects on human striatal presynaptic dopamine synthesis. J. Neurosci. 2010, 30, 14691–14694. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Raote, I.; Bhattacharya, A.; Miledi, R.; Panicker, M.M. Activation, internalization, and recycling of the serotonin 2A receptor by dopamine. Proc. Natl. Acad. Sci. USA 2006, 103, 15248–15253. [Google Scholar] [CrossRef] [PubMed]
- Esposito, E.; Di Matteo, V.; Di Giovanni, G. Serotonin–dopamine interaction: An overview. Prog. Brain Res. 2008, 172, 3–6. [Google Scholar] [PubMed]
- Roelfsema, F.; Pijl, H. Phase Difference between Serum Prolactin and Cortisol Rhythms Is Related to Body Mass Index in Humans. J. Clin. Endocrinol. Metab. 2012, 97, E2293–E2296. [Google Scholar] [CrossRef][Green Version]
- Deibert, D.C.; Defronzo, R.A. Epinephrine-induced Insulin Resistance in Man. J. Clin. Investig. 1980, 65, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.S.; Stoohs, R.A.; Reaven, G.M. Enhanced sympathetic nervous system activity*The linchpin between insulin resistance, hyperinsulinemia, and heart rate. Am. J. Hypertens. 1996, 9 Pt 1, 1013–1017. [Google Scholar] [CrossRef]
- Peltonen, G.L.; Scalzo, R.L.; Schweder, M.M.; Larson, D.G.; Luckasen, G.J.; Irwin, D.; Hamilton, K.L.; Schroeder, T.; Bell, C. Sympathetic inhibition attenuates hypoxia induced insulin resistance in healthy adult humans. J. Physiol. 2012, 590, 2801–2809. [Google Scholar] [CrossRef]
- Gamboa, A.; Okamoto, L.E.; Arnold, A.C.; Figueroa, R.A.; Diedrich, A.; Raj, S.R.; Paranjape, S.Y.; Farley, G.; Abumrad, N.; Biaggioni, I. Autonomic blockade improves insulin sensitivity in obese subjects. Hypertension 2014, 64, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Yeckel, C.W.; Gulanski, B.; Zgorski, M.L.; Dziura, J.; Parish, R.; Sherwin, R.S. Simple Exercise Recovery Index for Sympathetic Overactivity Is Linked to Insulin Resistance. Med. Sci. Sports Exerc. 2009, 41, 505–515. [Google Scholar] [CrossRef]
- Saito, I.; Maruyama, K.; Eguchi, E.; Kato, T.; Kawamura, R.; Takata, Y.; Onuma, H.; Osawa, H.; Tanigawa, T. Low Heart Rate Variability and Sympathetic Dominance Modifies the Association Between Insulin Resistance and Metabolic Syndrome―The Toon Health Study. Circ. J. 2017, 81, 1447–1453. [Google Scholar] [CrossRef]
- Brunner, E.J.; Hemingway, H.; Walker, B.R.; Page, M.; Clarke, P.; Juneja, M.; Shipley, M.J.; Kumari, M.; Andrew, R.; Seckl, J.R.; et al. Adrenocortical, autonomic, and inflammatory causes of the metabolic syndrome: Nested case-control study. Circulation 2002, 106, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Wulsin, L.R.; Horn, P.S.; Perry, J.L.; Massaro, J.M.; D’Agostino, R.B. Autonomic Imbalance as a Predictor of Metabolic Risks, Cardiovascular Disease, Diabetes, and Mortality. J. Clin. Endocrinol. Metab. 2015, 100, 2443–2448. [Google Scholar] [CrossRef]
- Egan, B.M. Insulin resistance and the sympathetic nervous system. Curr. Hypertens. Rep. 2003, 5, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Bjorntorp, P.; Rosmond, R. Neuroendocrine abnormalities in visceral obesity. Int. J. Obes. Relat. Metab. Disord. 2000, 24 (Suppl. 2), S80–S85. [Google Scholar] [CrossRef]
- Chiodini, I.; Adda, G.; Scillitani, A.; Coletti, F.; Morelli, V.; Di Lembo, S.; Epaminonda, P.; Masserini, B.; Beck-Peccoz, P.; Orsi, E.; et al. Cortisol secretion in patients with type 2 diabetes: Relationship with chronic complications. Diabetes Care 2007, 30, 83–88. [Google Scholar] [CrossRef]
- Kann, P.H.; Munzel, M.; Hadji, P.; Daniel, H.; Flache, S.; Nyarango, P.; Wilhelm, A. Alterations of cortisol homeostasis may link changes of the sociocultural environment to an increased diabetes and metabolic risk in developing countries: A prospective diagnostic study performed in cooperation with the Ovahimba people of the Kunene region/northwestern Namibia. J. Clin. Endocrinol. Metab. 2015, 100, E482–E486. [Google Scholar] [PubMed]
- Ortiz, R.; Kluwe, B.; Odei, J.B.; Tcheugui, J.B.E.; Sims, M.; Kalyani, R.R.; Bertoni, A.G.; Golden, S.H.; Joseph, J.J. The association of morning serum cortisol with glucose metabolism and diabetes: The Jackson Heart Study. Psychoneuroendocrinology 2018, 103, 25–32. [Google Scholar] [CrossRef]
- Joseph, J.J.; Golden, S.H. Cortisol dysregulation: The bidirectional link between stress, depression, and type 2 diabetes mellitus. Ann. N. Y. Acad. Sci. 2017, 1391, 20–34. [Google Scholar] [CrossRef]
- Rizza, R.A.; Mandarino, L.J.; Gerich, J.E. Cortisol-induced insulin resistance in man: Impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J. Clin. Endocrinol. Metab. 1982, 54, 131–138. [Google Scholar] [CrossRef]
- Adam, T.C.; Hasson, R.E.; Ventura, E.E.; Toledo-Corral, C.; Le, K.-A.; Mahurkar, S.; Lane, C.J.; Weigensberg, M.J.; Goran, M.I. Cortisol Is Negatively Associated with Insulin Sensitivity in Overweight Latino Youth. J. Clin. Endocrinol. Metab. 2010, 95, 4729–4735. [Google Scholar] [CrossRef]
- Manenschijn, L.; Schaap, L.; van Schoor, N.M.; van der Pas, S.; Peeters, G.M.E.E.; Lips, P.; Koper, J.W.; van Rossum, E.F.C. High Long-Term Cortisol Levels, Measured in Scalp Hair, Are Associated With a History of Cardiovascular Disease. J. Clin. Endocrinol. Metab. 2013, 98, 2078–2083. [Google Scholar] [CrossRef] [PubMed]
- Bjorntorp, P.; Holm, G.; Rosmond, R. Hypothalamic arousal, insulin resistance and Type 2 diabetes mellitus. Diabet. Med. 1999, 16, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Russo, B.; Menduni, M.; Borboni, P.; Picconi, F.; Frontoni, S. Autonomic Nervous System in Obesity and Insulin-Resistance—The Complex Interplay between Leptin and Central Nervous System. Int. J. Mol. Sci. 2021, 22, 5187. [Google Scholar] [CrossRef]
- Lim, K.; Jackson, K.L.; Sata, Y.; Head, G.A. Factors Responsible for Obesity-Related Hypertension. Curr. Hypertens. Rep. 2017, 19, 53. [Google Scholar] [CrossRef]
- Hosoi, T.; Ozawa, K. Possible Pharmacological Approach Targeting Endoplasmic Reticulum Stress to Ameliorate Leptin Resistance in Obesity. Front. Endocrinol. 2016, 7, 59. [Google Scholar] [CrossRef]
- Kalil, G.Z.; Haynes, W.G. Sympathetic nervous system in obesity-related hypertension: Mechanisms and clinical implications. Hypertens. Res. 2012, 35, 4–16. [Google Scholar] [CrossRef]
- Guarino, D.; Nannipieri, M.; Iervasi, G.; Taddei, S.; Bruno, R.M. The Role of the Autonomic Nervous System in the Pathophysiology of Obesity. Front. Physiol. 2017, 8, 665. [Google Scholar] [CrossRef]
- Mark, A.L. Selective leptin resistance revisited. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 305, R566–R581. [Google Scholar] [CrossRef] [PubMed]
- Henneicke, H.; Kim, S.; Swarbrick, M.M.; Li, J.; Gasparini, S.J.; Thai, J.; Foong, D.; Cavanagh, L.L.; Fong-Yee, C.; Karsten, E.; et al. Skeletal glucocorticoid signalling determines leptin resistance and obesity in aging mice. Mol. Metab. 2020, 42, 101098. [Google Scholar] [CrossRef] [PubMed]
- Oishi, K.; Hashimoto, C. Short-term time-restricted feeding during the resting phase is sufficient to induce leptin resistance that contributes to development of obesity and metabolic disorders in mice. Chrono-Int. 2018, 35, 1576–1594. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, U.; Sartori, C. Defective nitric oxide synthesis: A link between metabolic insulin resistance, sympathetic overactivity and cardiovascular morbidity. Eur. J. Endocrinol. 2000, 142, 315–323. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kraft, G.; Vrba, A.; Scott, M.; Allen, E.; Edgerton, D.S.; Williams, P.E.; Vafai, S.B.; Azamian, B.R.; Cherrington, A.D. Sympathetic Denervation of the Common Hepatic Artery Lessens Glucose Intolerance in the Fat- and Fructose-Fed Dog. Diabetes 2019, 68, 1143–1155. [Google Scholar] [CrossRef]
- Huggett, R.J.; Scott, E.M.; Gilbey, S.G.; Stoker, J.B.; Mackintosh, A.F.; Mary, D.A. Impact of Type 2 Diabetes Mellitus on Sympathetic Neural Mechanisms in Hypertension. Circulation 2003, 108, 3097–3101. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.J.; Polotsky, V.Y. Carotid Body and Metabolic Syndrome: Mechanisms and Potential Therapeutic Targets. Int. J. Mol. Sci. 2020, 21, 5117. [Google Scholar] [CrossRef]
- Fossum, E.; Høieggen, A.; Reims, H.; Moan, A.; Rostrup, M.; Eide, I.; Kjeldsen, S. High screening blood pressure is related to sympathetic nervous system activity and insulin resistance in healthy young men. Blood Press. 2004, 13, 89–94. [Google Scholar] [CrossRef]
- Murabayashi, M.; Daimon, M.; Murakami, H.; Fujita, T.; Sato, E.; Tanabe, J.; Matsuhashi, Y.; Takayasu, S.; Yanagimachi, M.; Terui, K.; et al. Association between higher urinary normetanephrine and insulin resistance in a Japanese population. PLoS ONE 2020, 15, e0228787. [Google Scholar] [CrossRef]
- Wang, W.; Mu, L.; Su, T.; Ye, L.; Jiang, Y.; Jiang, L.; Zhou, W. Plasma Metanephrines Are Associated With Glucose Metabolism in Patients With Essential Hypertension. Medicine 2015, 94, e1496. [Google Scholar] [CrossRef]
- Carnagarin, R.; Matthews, V.B.; Herat, L.Y.; Ho, J.K.; Schlaich, M.P. Autonomic Regulation of Glucose Homeostasis: A Specific Role for Sympathetic Nervous System Activation. Curr. Diabetes Rep. 2018, 18, 107. [Google Scholar] [CrossRef]
- Paton, J.F.; Sobotka, P.A.; Fudim, M.; Engelman, Z.J.; Hart, E.C.; McBryde, F.D.; Abdala, A.P.; Marina, N.; Gourine, A.V.; Lobo, M.; et al. The carotid body as a therapeutic target for the treatment of sympathetically mediated diseases. Hypertension 2013, 61, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Masuo, K.; Mikami, H.; Ogihara, T.; Tuck, M.L. Sympathetic Nerve Hyperactivity Precedes Hyperinsulinemia and Blood Pressure Elevation in a Young, Nonobese Japanese Population. Am. J. Hypertens. 1997, 10, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.K.; Lindmark, S.; Wiklund, U.; Rask, P.; Karlsson, M.; Myrin, J.; Kullberg, J.; Johansson, L.; Eriksson, J.W. Alterations in heart rate variability during everyday life are linked to insulin resistance. A role of dominating sympathetic over parasympathetic nerve activity? Cardiovasc. Diabetol. 2016, 15, 91. [Google Scholar] [CrossRef]
- Licht, C.M.; de Geus, E.J.; Penninx, B.W. Dysregulation of the autonomic nervous system predicts the development of the metabolic syndrome. J. Clin. Endocrinol. Metab. 2013, 98, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, H.; Pauli, L.K.; Janssen, L.K.; Huhn, S.; Ceglarek, U.; Horstmann, A. Preliminary evidence for an association between intake of high-fat high-sugar diet, variations in peripheral dopamine precursor availability and dopamine-dependent cognition in humans. J. Neuroendocrinol. 2020, 32, e12917. [Google Scholar] [CrossRef]
- Burger, K.S.; Stice, E. Frequent ice cream consumption is associated with reduced striatal response to receipt of an ice cream–based milkshake. Am. J. Clin. Nutr. 2012, 95, 810–817. [Google Scholar] [CrossRef]
- Cansell, C.; Luquet, S. Triglyceride sensing in the reward circuitry: A new insight in feeding behaviour regulation. Biochimie 2016, 120, 75–80. [Google Scholar] [CrossRef]
- Rivera, H.M.; Kievit, P.; Kirigiti, M.A.; Bauman, L.A.; Baquero, K.; Blundell, P.; Dean, T.A.; Valleau, J.C.; Takahashi, D.L.; Frazee, T.; et al. Maternal high-fat diet and obesity impact palatable food intake and dopamine signaling in nonhuman primate offspring. Obesity 2015, 23, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.W.; Fordahl, S.C. Obesity and dietary fat influence dopamine neurotransmission: Exploring the convergence of metabolic state, physiological stress, and inflammation on dopaminergic control of food intake. Nutr. Res. Rev. 2022, 35, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Leite, F.; Leite, Â.; Rasini, E.; Gaiazzi, M.; Ribeiro, L.; Marino, F.; Cosentino, M. Dopaminergic pathways in obesity-associated immuno-metabolic depression. Psychol. Med. 2018, 48, 2273–2275. [Google Scholar] [CrossRef] [PubMed]
- Volkow, N.D.; Tomasi, D.; Wang, G.-J.; Telang, F.; Fowler, J.S.; Logan, J.; Benveniste, H.; Kim, R.; Thanos, P.K.; Ferré, S. Evidence That Sleep Deprivation Downregulates Dopamine D2R in Ventral Striatum in the Human Brain. J. Neurosci. 2012, 32, 6711–6717. [Google Scholar] [CrossRef]
- Baik, J.-H. Stress and the dopaminergic reward system. Exp. Mol. Med. 2020, 52, 1879–1890. [Google Scholar] [CrossRef]
- Alghasham, A.; Rasheed, N. Stress-mediated modulations in dopaminergic system and their subsequent impact on behavioral and oxidative alterations: An update. Pharm. Biol. 2014, 52, 368–377. [Google Scholar] [CrossRef]
- Wing, Y.K.; Lam, S.P.; Zhang, J.; Leung, E.; Chen, S.; Cheung, M.K.; Li, S.X.; Chan, J.W.Y.; Mok, V.; Tsoh, J.; et al. Reduced striatal dopamine transmission in REM sleep behavior disorder comorbid with depression. Neurology 2015, 84, 516–522. [Google Scholar] [CrossRef]
- Yadid, G.; Friedman, A. Dynamics of the dopaminergic system as a key component to the understanding of depression. Prog. Brain Res. 2008, 172, 265–286. [Google Scholar]
- Hernaus, D.; Collip, D.; Lataster, J.; Viechtbauer, W.; Myin, E.; Ceccarini, J.; Van Laere, K.; van Os, J.; Myin-Germeys, I. Psychotic reactivity to daily life stress and the dopamine system: A study combining experience sampling and [¹⁸F]fallypride positron emission tomography. J. Abnorm. Psychol. 2015, 124, 27–37. [Google Scholar] [CrossRef]
- Furuyashiki, T. Roles of Dopamine and Inflammation-Related Molecules in Behavioral Alterations Caused by Repeated Stress. J. Pharmacol. Sci. 2012, 120, 63–69. [Google Scholar] [CrossRef]
- Zipursky, R.T.; Press, M.C.; Srikanthan, P.; Gornbein, J.; McClelland, R.; Watson, K.; Horwich, T.B. Relation of Stress Hormones (Urinary Catecholamines/Cortisol) to Coronary Artery Calcium in Men Versus Women (from the Multi-Ethnic Study of Atherosclerosis [MESA]). Am. J. Cardiol. 2017, 119, 1963–1971. [Google Scholar] [CrossRef] [PubMed]
- Hering, D.; Lachowska, K.; Schlaich, M. Role of the Sympathetic Nervous System in Stress-Mediated Cardiovascular Disease. Curr. Hypertens Rep. 2015, 17, 80. [Google Scholar] [CrossRef]
- Merz, C.N.B.; Dwyer, J.; Nordstrom, C.K.; Walton, K.G.; Salerno, J.W.; Schneider, R.H. Psychosocial Stress and Cardiovascular Disease: Pathophysiological Links. Behav. Med. 2002, 27, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H.; Garbutt, L.D. Stress, inflammation and cardiovascular disease. J. Psychosom. Res. 2002, 52, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Mądry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Kuo, L.E.; Czarnecka, M.; Kitlinska, J.B.; Tilan, J.U.; Zukowska, Z.; Kvetňanský, R. Chronic Stress, Combined with a High-Fat/High-Sugar Diet, Shifts Sympathetic Signaling toward Neuropeptide Y and Leads to Obesity and the Metabolic Syndrome. Ann. N.Y. Acad. Sci. 2008, 1148, 232–237. [Google Scholar] [CrossRef]
- Pervanidou, P.; Chrousos, G.P. Metabolic consequences of stress during childhood and adolescence. Metabolism 2012, 61, 611–619. [Google Scholar] [CrossRef]
- Drager, L.F.; Togeiro, S.M.; Polotsky, V.Y.; Lorenzi-Filho, G. Obstructive sleep apnea: A cardiometabolic risk in obesity and the metabolic syndrome. J. Am. Coll. Cardiol. 2013, 62, 569–576. [Google Scholar] [CrossRef]
- Gold, P.W.; Chrousos, G.P. The Endocrinology of Melancholic and Atypical Depression: Relation to Neurocircuitry and Somatic Consequences. Proc. Assoc. Am. Physicians 1999, 111, 22–34. [Google Scholar] [CrossRef]
- Sanchez-Gonzalez, M.A.; May, R.W.; Koutnik, A.P.; Kabbaj, M.; Fincham, F.D. Sympathetic Vasomotor Tone Is Associated With Depressive Symptoms in Young Females: A Potential Link Between Depression and Cardiovascular Disease. Am. J. Hypertens. 2013, 26, 1389–1397. [Google Scholar] [CrossRef]
- Black, P.H. The inflammatory consequences of psychologic stress: Relationship to insulin resistance, obesity, atherosclerosis and diabetes mellitus, type II. Med. Hypotheses 2006, 67, 879–891. [Google Scholar] [CrossRef]
- Zhao, Z.; Wang, L.; Gao, W.; Hu, F.; Zhang, J.; Ren, Y.; Lin, R.; Feng, Q.; Cheng, M.; Ju, D.; et al. A Central Catecholaminergic Circuit Controls Blood Glucose Levels during Stress. Neuron 2017, 95, 138–152.e5. [Google Scholar] [CrossRef] [PubMed]
- Scheer, F.A.; Hilton, M.F.; Mantzoros, C.S.; Shea, S.A. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc. Natl. Acad. Sci. USA 2009, 106, 4453–4458. [Google Scholar] [CrossRef] [PubMed]
- Knutson, K.L.; Van Cauter, E.; Rathouz, P.J.; Yan, L.L.; Hulley, S.B.; Liu, K.; Lauderdale, D.S. Association between sleep and blood pressure in midlife: The CARDIA sleep study. Arch. Intern. Med. 2009, 169, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Suka, M.; Yoshida, K.; Sugimori, H. Persistent Insomnia is a Predictor of Hypertension in Japanese Male Workers. J. Occup. Health 2003, 45, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Narang, I.; Manlhiot, C.; Davies-Shaw, J.; Gibson, D.; Chahal, N.; Stearne, K.; Fisher, A.; Dobbin, S.; McCrindle, B.W. Sleep disturbance and cardiovascular risk in adolescents. Can. Med. Assoc. J. 2012, 184, E913–E920. [Google Scholar] [CrossRef]
- Meisinger, C.; Heier, M.; Löwel, H.; Schneider, A.; Döring, A. Sleep Duration and Sleep Complaints and Risk of Myocardial Infarction in Middle-aged Men and Women from the General Population: The MONICA/KORA Augsburg Cohort Study. Sleep 2007, 30, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Narkiewicz, K.; Somers, V.K. Sympathetic nerve activity in obstructive sleep apnoea. Acta Physiol. Scand. 2003, 177, 385–390. [Google Scholar] [CrossRef]
- Zhong, P.; Vickstrom, C.R.; Liu, X.; Hu, Y.; Yu, L.; Yu, H.G.; Liu, Q.S. HCN2 channels in the ventral tegmental area regulate behavioral responses to chronic stress. Elife 2018, 7, e32420. [Google Scholar] [CrossRef]
- Davis, J.F.; Tracy, A.L.; Schurdak, J.D.; Tschöp, M.H.; Lipton, J.W.; Clegg, D.J.; Benoit, S.C. Exposure to elevated levels of dietary fat attenuates psychostimulant reward and mesolimbic dopamine turnover in the rat. Behav. Neurosci. 2008, 122, 1257–1263. [Google Scholar] [CrossRef]
- Rada, P.; Bocarsly, M.E.; Barson, J.R.; Hoebel, B.G.; Leibowitz, S.F. Reduced accumbens dopamine in Sprague–Dawley rats prone to overeating a fat-rich diet. Physiol. Behav. 2010, 101, 394–400. [Google Scholar] [CrossRef]
- Geiger, B.M.; Behr, G.G.; Frank, L.E.; Caldera-Siu, A.D.; Beinfeld, M.C.; Kokkotou, E.G.; Pothos, E.N. Evidence for defective mesolimbic dopamine exocytosis in obesity-prone rats. FASEB J. 2008, 22, 2740–2746. [Google Scholar] [CrossRef] [PubMed]
- van de Giessen, E.; la Fleur, S.E.; Eggels, L.; de Bruin, K.; van den Brink, W.; Booij, J. High fat/carbohydrate ratio but not total energy intake induces lower striatal dopamine D2/3 receptor availability in diet-induced obesity. Int. J. Obes. 2013, 37, 754–757. [Google Scholar] [CrossRef]
- York, D.A.; Teng, L.; Park-York, M. Effects of dietary fat and enterostatin on dopamine and 5-hydroxytrytamine release from rat striatal slices. Brain Res. 2010, 1349, 48–55. [Google Scholar] [CrossRef]
- Raskin, P.; Cincotta, A.H. Bromocriptine-QR therapy for the management of type 2 diabetes mellitus: Developmental basis and therapeutic profile summary. Expert Rev. Endocrinol. Metab. 2016, 11, 113–148. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Luo, S.; Cincotta, A.H. Long-term infusion of norepinephrine plus serotonin into the ventromedial hypothalamus impairs pancreatic islet function. Metabolism 1999, 48, 1287–1289. [Google Scholar] [CrossRef]
- Grassi, G.; Dell’oro, R.; Quarti-Trevano, F.; Scopelliti, F.; Seravalle, G.; Paleari, F.; Gamba, P.L.; Mancia, G. Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia 2005, 48, 1359–1365. [Google Scholar] [CrossRef]
- Beske, S.D.; Alvarez, G.E.; Ballard, T.P.; Davy, K.P. Reduced cardiovagal baroreflex gain in visceral obesity: Implications for the metabolic syndrome. Am. J. Physiol. Circ. Physiol. 2002, 282, H630–H635. [Google Scholar] [CrossRef] [PubMed]
- Straznicky, N.E.; Lambert, E.A.; Lambert, G.W.; Masuo, K.; Esler, M.D.; Nestel, P.J. Effects of Dietary Weight Loss on Sympathetic Activity and Cardiac Risk Factors Associated with the Metabolic Syndrome. J. Clin. Endocrinol. Metab. 2005, 90, 5998–6005. [Google Scholar] [CrossRef]
- Esler, M.; Rumantir, M.; Wiesner, G.; Kaye, D.; Hastings, J.; Lambert, G. Sympathetic nervous system and insulin resistance: From obesity to diabetes. Am. J. Hypertens. 2001, 14, S304–S309. [Google Scholar] [CrossRef]
- Flaa, A.; Aksnes, T.A.; Kjeldsen, S.E.; Eide, I.; Rostrup, M. Increased sympathetic reactivity may predict insulin resistance: An 18-year follow-up study. Metabolism 2008, 57, 1422–1427. [Google Scholar] [CrossRef]
- Carnethon, M.R.; Golden, S.H.; Folsom, A.R.; Haskell, W.; Liao, D. Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: The Atherosclerosis Risk In Communities study, 1987–1998. Circulation 2003, 107, 2190–2195. [Google Scholar] [CrossRef]
- Carnethon, M.R.; Jacobs, D.R., Jr.; Sidney, S.; Liu, K. Influence of autonomic nervous system dysfunction on the development of type 2 diabetes: The CARDIA study. Diabetes Care 2003, 26, 3035–3041. [Google Scholar] [CrossRef]
- Fiorentini, A.; Perciaccante, A.; Paris, A.; Serra, P.; Tubani, L. Circadian rhythm of autonomic activity in non diabetic offsprings of type 2 diabetic patients. Cardiovasc. Diabetol. 2005, 4, 15. [Google Scholar] [CrossRef][Green Version]
- Frontoni, S.; Bracaglia, D.; Baroni, A.; Pellegrini, F.; Perna, M.; Cicconetti, E.; Ciampittiello, G.; Menzinger, G.; Gambardella, S. Early Autonomic Dysfunction in Glucose-Tolerant but Insulin-Resistant Offspring of Type 2 Diabetic Patients. Hypertension 2003, 41, 1223–1227. [Google Scholar] [CrossRef]
- Mancia, G.; Bousquet, P.; Elghozi, J.L.; Esler, M.; Grassi, G.; Julius, S.; Reid, J.; Van Zwieten, P.A. The sympathetic nervous system and the metabolic syndrome. J. Hypertens. 2007, 25, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Canale, M.P.; di Villahermosa, S.M.; Martino, G.; Rovella, V.; Noce, A.; De Lorenzo, A.; Di Daniele, N. Obesity-Related Metabolic Syndrome: Mechanisms of Sympathetic Overactivity. Int. J. Endocrinol. 2013, 2013, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T. Central nervous system regulation of liver and adipose tissue metabolism. Diabetologia 1981, 20 (Suppl. 1), 343–356. [Google Scholar] [CrossRef]
- Moore, M.C.; Coate, K.C.; Winnick, J.J.; An, Z.; Cherrington, A.D. Regulation of Hepatic Glucose Uptake and Storage In Vivo. Adv. Nutr. Int. Rev. J. 2012, 3, 286–294. [Google Scholar] [CrossRef]
- DiCostanzo, C.A.; Dardevet, D.P.; Neal, D.W.; Lautz, M.; Allen, E.; Snead, W.; Cherrington, A.D. Role of the hepatic sympathetic nerves in the regulation of net hepatic glucose uptake and the mediation of the portal glucose signal. Am. J. Physiol. Metab. 2006, 290, E9–E16. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, T. Neuronal regulation of hepatic glucose metabolism in mammals. Diabetes/Metabolism Res. Rev. 1987, 3, 185–206. [Google Scholar] [CrossRef] [PubMed]
- Hoek, A.M.v.D.; van Heijningen, C.; der Elst, J.P.S.-V.; Ouwens, D.M.; Havekes, L.M.; Romijn, J.A.; Kalsbeek, A.; Pijl, H. Intracerebroventricular Administration of Neuropeptide Y Induces Hepatic Insulin Resistance via Sympathetic Innervation. Diabetes 2008, 57, 2304–2310. [Google Scholar] [CrossRef]
- Shimazu, T. Reciprocal Innervation of the Liver: Its Significance in Metabolic Control. Adv. Metab. Disord. 1983, 10, 355–384. [Google Scholar]
- Lembo, G.; Capaldo, B.; Rendina, V.; Iaccarino, G.; Napoli, R.; Guida, R.; Trimarco, B.; Sacca, L. Acute noradrenergic activation induces insulin resistance in human skeletal muscle. Am. J. Physiol. Metab. 1994, 266, E242–E247. [Google Scholar] [CrossRef] [PubMed]
- Jamerson, K.A.; Julius, S.; Gudbrandsson, T.; Andersson, O.; Brant, D.O. Reflex sympathetic activation induces acute insulin resistance in the human forearm. Hypertension 1993, 21, 618–623. [Google Scholar] [CrossRef]
- Rojas, J.M.; Bruinstroop, E.; Printz, R.L.; Alijagic-Boers, A.; Foppen, E.; Turney, M.K.; George, L.; Beck-Sickinger, A.G.; Kalsbeek, A.; Niswender, K.D. Central nervous system neuropeptide Y regulates mediators of hepatic phospholipid remodeling and very low-density lipoprotein triglyceride secretion via sympathetic innervation. Mol. Metab. 2015, 4, 210–221. [Google Scholar] [CrossRef]
- Bruinstroop, E.; Pei, L.; Ackermans, M.T.; Foppen, E.; Borgers, A.J.; Kwakkel, J.; Alkemade, A.; Fliers, E.; Kalsbeek, A. Hypothalamic Neuropeptide Y (NPY) Controls Hepatic VLDL-Triglyceride Secretion in Rats via the Sympathetic Nervous System. Diabetes 2012, 61, 1043–1050. [Google Scholar] [CrossRef]
- Geerling, J.J.; Boon, M.R.; Kooijman, S.; Parlevliet, E.T.; Havekes, L.M.; Romijn, J.A.; Meurs, I.M.; Rensen, P.C.N. Sympathetic nervous system control of triglyceride metabolism: Novel concepts derived from recent studies. J. Lipid Res. 2014, 55, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Steffens, A.; Scheurink, A.; Luiten, P.; Bohus, B. Hypothalamic food intake regulating areas are involved in the homeostasis of blood glucose and plasma FFA levels. Physiol. Behav. 1988, 44, 581–589. [Google Scholar] [CrossRef]
- Bonnet, F.; Empana, J.P.; Natali, A.; Monti, L.; Golay, A.; Lalic, K.; Dekker, J.; Mari, A.; Balkau, B.; Group, R.S. Elevated heart rate predicts beta cell function in non-diabetic individuals: The RISC cohort. Eur. J. Endocrinol. 2015, 173, 409–415. [Google Scholar] [CrossRef]
- Defronzo, R.A. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef]
- Bajaj, M.; Pratipanawatr, T.; Berria, R.; Pratipanawatr, W.; Kashyap, S.; Cusi, K.; Mandarino, L.; DeFronzo, R.A. Free Fatty Acids Reduce Splanchnic and Peripheral Glucose Uptake in Patients With Type 2 Diabetes. Diabetes 2002, 51, 3043–3048. [Google Scholar] [CrossRef][Green Version]
- Daniele, G.; Eldor, R.; Merovci, A.; Clarke, G.D.; Xiong, J.; Tripathy, D.; Taranova, A.; Abdul-Ghani, M.; DeFronzo, R.A. Chronic Reduction of Plasma Free Fatty Acid Improves Mitochondrial Function and Whole-Body Insulin Sensitivity in Obese and Type 2 Diabetic Individuals. Diabetes 2014, 63, 2812–2820. [Google Scholar] [CrossRef] [PubMed]
- Belfort, R.; Mandarino, L.; Kashyap, S.; Wirfel, K.; Pratipanawatr, T.; Berria, R.; DeFronzo, R.A.; Cusi, K. Dose-Response Effect of Elevated Plasma Free Fatty Acid on Insulin Signaling. Diabetes 2005, 54, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Keung, W.; Samokhvalov, V.; Wang, W.; Lopaschuk, G.D. Role of fatty acid uptake and fatty acid beta-oxidation in mediating insulin resistance in heart and skeletal muscle. Biochim. Biophys. Acta 2010, 1801, 1–22. [Google Scholar] [CrossRef]
- Tumova, J.; Andel, M.; Trnka, J. Excess of Free Fatty Acids as a Cause of Metabolic Dysfunction in Skeletal Muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar] [CrossRef]
- Gadegbeku, C.A.; Dhandayuthapani, A.; Sadler, Z.E.; Egan, B.M. Raising lipids acutely reduces baroreflex sensitivity. Am. J. Hypertens. 2002, 15, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.; Bomhoff, G.; Gorres, B.; Davis, V.; Kim, J.; Lee, P.-P.; Brooks, W.; Gerhardt, G.; Geiger, P.; Stanford, J. Insulin resistance impairs nigrostriatal dopamine function. Exp. Neurol. 2011, 231, 171–180. [Google Scholar] [CrossRef]
- Stouffer, M.A.; Woods, C.A.; Patel, J.C.; Lee, C.R.; Witkovsky, P.; Bao, L.; Machold, R.P.; Jones, K.T.; De Vaca, S.C.; Reith, M.E.A.; et al. Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward. Nat. Commun. 2015, 6, 8543. [Google Scholar] [CrossRef]
- Heni, M.; Wagner, R.; Kullmann, S.; Gancheva, S.; Roden, M.; Peter, A.; Stefan, N.; Preissl, H.; Häring, H.-U.; Fritsche, A. Hypothalamic and Striatal Insulin Action Suppresses Endogenous Glucose Production and May Stimulate Glucose Uptake During Hyperinsulinemia in Lean but Not in Overweight Men. Diabetes 2017, 66, 1797–1806. [Google Scholar] [CrossRef]
- Ong, Z.Y.; Muhlhausler, B.S. Maternal “junk-food” feeding of rat dams alters food choices and development of the mesolimbic reward pathway in the offspring. FASEB J. 2011, 25, 2167–2179. [Google Scholar] [CrossRef]
- Grassi, G.; Biffi, A.; Seravalle, G.; Trevano, F.Q.; Dell’oro, R.; Corrao, G.; Mancia, G. Sympathetic Neural Overdrive in the Obese and Overweight State. Hypertension 2019, 74, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Farr, S.A.; Salameh, T.S.; Niehoff, M.L.; Rhea, E.M.; Morley, J.E.; Hanson, A.J.; Hansen, K.M.; Craft, S. Triglycerides cross the blood–brain barrier and induce central leptin and insulin receptor resistance. Int. J. Obes. 2017, 42, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Galletti, F.; D’Elia, L.; Barba, G.; Siani, A.; Cappuccio, F.P.; Farinaro, E.; Iacone, R.; Russo, O.; De Palma, D.; Ippolito, R.; et al. High-Circulating Leptin Levels Are Associated with Greater Risk of Hypertension in Men Independently of Body Mass and Insulin Resistance: Results of an Eight-Year Follow-Up Study. J. Clin. Endocrinol. Metab. 2008, 93, 3922–3926. [Google Scholar] [CrossRef] [PubMed]
- Eikelis, N.; Schlaich, M.; Aggarwal, A.; Kaye, D.; Esler, M. Interactions between leptin and the human sympathetic nervous system. Hypertension 2003, 41, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.J.; Fontes, M.A.; Killinger, S.; Pawlak, D.B.; Polson, J.W.; Dampney, R.A. Cardiovascular Responses Evoked by Leptin Acting on Neurons in the Ventromedial and Dorsomedial Hypothalamus. Hypertension 2003, 42, 488–493. [Google Scholar] [CrossRef]
- Ciriello, J. Leptin in nucleus of the solitary tract alters the cardiovascular responses to aortic baroreceptor activation. Peptides 2013, 44, 1–7. [Google Scholar] [CrossRef]
- Li, B.; Shi, Z.; Cassaglia, P.A.; Brooks, V.L. Leptin acts in the forebrain to differentially influence baroreflex control of lumbar, renal, and splanchnic sympathetic nerve activity and heart rate. Hypertension 2013, 61, 812–819. [Google Scholar] [CrossRef]
- da Silva, A.A.; Carmo, J.M.D.; Hall, J.E. Role of leptin and central nervous system melanocortins in obesity hypertension. Curr. Opin. Nephrol. Hypertens. 2013, 22, 135–140. [Google Scholar] [CrossRef]
- Tack, C.J.; Smits, P.; Willemsen, J.J.; Lenders, J.W.; Thien, T.; Lutterman, J.A. Effects of insulin on vascular tone and sympathetic nervous system in NIDDM. Diabetes 1996, 45, 15–22. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rowe, J.W.; Young, J.B.; Minaker, K.L.; Stevens, A.L.; Pallotta, J.; Landsberg, L. Effect of Insulin and Glucose Infusions on Sympathetic Nervous System Activity in Normal Man. Diabetes 1981, 30, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Yasunari, K.; Matsui, T.; Maeda, K.; Nakamura, M.; Watanabe, T.; Kiriike, N. Anxiety-Induced Plasma Norepinephrine Augmentation Increases Reactive Oxygen Species Formation by Monocytes in Essential Hypertension. Am. J. Hypertens. 2006, 19, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Lambert, G.W.; Straznicky, N.E.; Lambert, E.A.; Dixon, J.B.; Schlaich, M.P. Sympathetic nervous activation in obesity and the metabolic syndrome--causes, consequences and therapeutic implications. Pharmacol. Ther. 2010, 126, 159–172. [Google Scholar] [CrossRef]
- Moreira, M.C.; Pinto, I.S.; Mourao, A.A.; Fajemiroye, J.O.; Colombari, E.; Reis, A.A.; Freiria-Oliveira, A.H.; Ferreira-Neto, M.L.; Pedrino, G.R. Does the sympathetic nervous system contribute to the pathophysiology of metabolic syndrome? Front. Physiol. 2015, 6, 234. [Google Scholar] [CrossRef]
- Tentolouris, N.; Argyrakopoulou, G.; Katsilambros, N. Perturbed Autonomic Nervous System Function in Metabolic Syndrome. NeuroMolecular Med. 2008, 10, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Cabandugama, P.K.; Gardner, M.J.; Sowers, J.R. The Renin Angiotensin Aldosterone System in Obesity and Hypertension: Roles in the Cardiorenal Metabolic Syndrome. Med. Clin. N. Am. 2017, 101, 129–137. [Google Scholar] [CrossRef]
- Grassi, G.; Seravalle, G.; Mancia, G. Sympathetic activation in cardiovascular disease: Evidence, clinical impact and therapeutic implications. Eur. J. Clin. Investig. 2015, 45, 1367–1375. [Google Scholar] [CrossRef] [PubMed]
- Graham, L.N.; Smith, P.A.; Huggett, R.J.; Stoker, J.B.; Mackintosh, A.F.; Mary, D.A. Sympathetic Drive in Anterior and Inferior Uncomplicated Acute Myocardial Infarction. Circulation 2004, 109, 2285–2289. [Google Scholar] [CrossRef]
- Leimbach, W.N., Jr.; Wallin, B.G.; Victor, R.G.; Aylward, P.; Sundlöf, G.; Mark, A.L. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 1986, 73, 913–919. [Google Scholar] [CrossRef]
- Grassi, G.; Cattaneo, B.M.; Seravalle, G.; Lanfranchi, A.; Mancia, G. Baroreflex Control of Sympathetic Nerve Activity in Essential and Secondary Hypertension. Hypertension 1998, 31, 68–72. [Google Scholar] [CrossRef]
- Converse, R.L.; Jacobsen, T.N.; Toto, R.D.; Jost, C.M.; Cosentino, F.; Fouad-Tarazi, F.; Victor, R.G. Sympathetic Overactivity in Patients with Chronic Renal Failure. N. Engl. J. Med. 1992, 327, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Young, C.N.; Fadel, P.J. Central sympathetic overactivity: Maladies and mechanisms. Auton. Neurosci. 2009, 148, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, A.A.; Mircoli, L.; Giannattasio, C.; Mancia, G.; Ferrari, A.U. Effect of Sympathectomy on Mechanical Properties of Common Carotid and Femoral Arteries. Hypertension 1997, 30, 1085–1088. [Google Scholar] [CrossRef] [PubMed]
- Hijmering, M.L.; Stroes, E.S.; Olijhoek, J.; Hutten, B.A.; Blankestijn, P.J.; Rabelink, T.J. Sympathetic activation markedly reduces endothelium-dependent, flow-mediated vasodilation. J. Am. Coll. Cardiol. 2002, 39, 683–688. [Google Scholar] [CrossRef] [PubMed]
- DiBona, G.F. Sympathetic nervous system and the kidney in hypertension. Curr. Opin. Nephrol. Hypertens. 2002, 11, 197–200. [Google Scholar] [CrossRef]
- Hu, A.; Jiao, X.; Gao, E.; Koch, W.J.; Sharifi-Azad, S.; Grunwald, Z.; Ma, X.L.; Sun, J.Z. Chronic beta-adrenergic receptor stimulation induces cardiac apoptosis and aggravates myocardial ischemia/reperfusion injury by provoking inducible nitric-oxide synthase-mediated nitrative stress. J. Pharmacol. Exp. Ther. 2006, 318, 469–475. [Google Scholar] [CrossRef]
- Rohleder, N. Stimulation of Systemic Low-Grade Inflammation by Psychosocial Stress. Psychosom. Med. 2014, 76, 181–189. [Google Scholar] [CrossRef]
- Gu, H.F.; Tang, C.K.; Yang, Y.Z. Psychological stress, immune response, and atherosclerosis. Atherosclerosis 2012, 223, 69–77. [Google Scholar] [CrossRef]
- Shimojo, G.; Joseph, B.; Shah, R.; Consolim-Colombo, F.M.; De Angelis, K.; Ulloa, L. Exercise activates vagal induction of dopamine and attenuates systemic inflammation. Brain Behav. Immun. 2019, 75, 181–191. [Google Scholar] [CrossRef]
- Lambert, E.; Straznicky, N.; Sari, C.I.; Eikelis, N.; Hering, D.; Head, G.; Dixon, J.; Esler, M.; Schlaich, M.; Lambert, G. Dyslipidemia Is Associated With Sympathetic Nervous Activation and Impaired Endothelial Function in Young Females. Am. J. Hypertens. 2013, 26, 250–256. [Google Scholar] [CrossRef]
- Steven, S.; Frenis, K.; Oelze, M.; Kalinovic, S.; Kuntic, M.; Bayo Jimenez, M.T.; Vujacic-Mirski, K.; Helmstädter, J.; Kröller-Schön, S.; Münzel, T.; et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid. Med. Cell Longev. 2019, 2019, 7092151. [Google Scholar] [CrossRef]
- Daiber, A.; Steven, S.; Euler, G.; Schulz, R. Vascular and Cardiac Oxidative Stress and Inflammation as Targets for Cardioprotection. Curr. Pharm. Des. 2021, 27, 2112–2130. [Google Scholar] [CrossRef] [PubMed]
- Henein, M.Y.; Vancheri, S.; Longo, G.; Vancheri, F. The Role of Inflammation in Cardiovascular Disease. Int. J. Mol. Sci. 2022, 23, 12906. [Google Scholar] [CrossRef]
- Meyers, A.M.; Mourra, D.; Beeler, J.A. High fructose corn syrup induces metabolic dysregulation and altered dopamine signaling in the absence of obesity. PLoS ONE 2017, 12, e0190206. [Google Scholar] [CrossRef] [PubMed]
- Page, K.A.; Chan, O.; Arora, J.; Belfort-DeAguiar, R.; Dzuira, J.; Roehmholdt, B.; Cline, G.W.; Naik, S.; Sinha, R.; Constable, R.T.; et al. Effects of Fructose vs Glucose on Regional Cerebral Blood Flow in Brain Regions Involved with Appetite and Reward Pathways. JAMA 2013, 309, 63–70. [Google Scholar] [CrossRef]
- Witek, K.; Wydra, K.; Filip, M. A High-Sugar Diet Consumption, Metabolism and Health Impacts with a Focus on the Development of Substance Use Disorder: A Narrative Review. Nutrients 2022, 14, 2940. [Google Scholar] [CrossRef]
- Barnes, C.N.; Wallace, C.W.; Jacobowitz, B.S.; Fordahl, S.C. Reduced phasic dopamine release and slowed dopamine uptake occur in the nucleus accumbens after a diet high in saturated but not unsaturated fat. Nutr. Neurosci. 2022, 25, 33–45. [Google Scholar] [CrossRef]
- Palomer, X.; Pizarro-Delgado, J.; Barroso, E.; Vázquez-Carrera, M. Palmitic and Oleic Acid: The Yin and Yang of Fatty Acids in Type 2 Diabetes Mellitus. Trends Endocrinol. Metab. 2018, 29, 178–190. [Google Scholar] [CrossRef]
- Gehrmann, W.; Würdemann, W.; Plötz, T.; Jörns, A.; Lenzen, S.; Elsner, M. Antagonism Between Saturated and Unsaturated Fatty Acids in ROS Mediated Lipotoxicity in Rat Insulin-Producing Cells. Cell. Physiol. Biochem. 2015, 36, 852–865. [Google Scholar] [CrossRef] [PubMed]
- Alkhateeb, H.; Qnais, E. Preventive effect of oleate on palmitate-induced insulin resistance in skeletal muscle and its mechanism of action. J. Physiol. Biochem. 2017, 73, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Chamarthi, B.; Vinik, A.; Ezrokhi, M.; Cincotta, A.H. Circadian-timed quick-release bromocriptine lowers elevated resting heart rate in patients with type 2 diabetes mellitus. Endocrinol. Diabetes Metab. 2020, 3, e00101. [Google Scholar] [CrossRef]
- Franchi, F.; Lazzeri, C.; Barletta, G.; Ianni, L.; Mannelli, M. Centrally mediated effects of bromocriptine on cardiac sympathovagal balance. Hypertension 2001, 38, 123–129. [Google Scholar] [CrossRef]
- Friis, M.L.; Paulson, O.B.; Hertz, M.M. Transfer of bromocriptine across the blood-brain barrier in man. Acta Neurol. Scand. 2009, 59, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Cincotta, A.H.; Meier, A.H.; Cincotta, M., Jr. Bromocriptine improves glycaemic control and serum lipid profile in obese Type 2 diabetic subjects: A new approach in the treatment of diabetes. Expert Opin. Investig. Drugs 1999, 8, 1683–1707. [Google Scholar] [CrossRef]
- Pijl, H.; Ohashi, S.; Matsuda, M.; Miyazaki, Y.; Mahankali, A.; Kumar, V.; Pipek, R.; Iozzo, P.; Lancaster, J.L.; Cincotta, A.H.; et al. Bromocriptine: A novel approach to the treatment of type 2 diabetes. Diabetes Care 2000, 23, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Alatrach, M.; Agyin, C.; Adams, J.; Chilton, R.; Triplitt, C.; DeFronzo, R.A.; Cersosimo, E. Glucose lowering and vascular protective effects of cycloset added to GLP-1 receptor agonists in patients with type 2 diabetes. Endocrinol. Diabetes Metab. 2018, 1, e00034. [Google Scholar] [CrossRef] [PubMed]
- Vinik, A.I.; Cincotta, A.H.; Scranton, R.E.; Bohannon, N.; Ezrokhi, M.; Gaziano, J.M. Effect of Bromocriptine-Qr On Glycemic Control in Subjects with Uncontrolled Hyperglycemia On One or Two Oral Anti-Diabetes Agents. Endocr. Pract. 2012, 18, 931–943. [Google Scholar] [CrossRef]
- Florez, H.; Scranton, R.; Farwell, W.R.; DeFronzo, R.A.; Ezrokhi, M.; Gaziano, J.M.; Cincotta, A.H. Randomized Clinical Trial Assessing the Efficacy and Safety of Bromocriptine-QR when Added to Ongoing Thiazolidinedione Therapy in Patients with Type 2 Diabetes Mellitus. J. Diabetes Metab. 2011, 2, 142. [Google Scholar] [CrossRef]
- Roe, E.D.; Chamarthi, B.; Raskin, P. Impact of Bromocriptine-QR Therapy on Glycemic Control and Daily Insulin Requirement in Type 2 Diabetes Mellitus Subjects Whose Dysglycemia Is Poorly Controlled on High-Dose Insulin: A Pilot Study. J. Diabetes Res. 2015, 2015, 834903. [Google Scholar] [CrossRef]
- Chamarthi, B.; Cincotta, A.H. Effect of bromocriptine-QR therapy on glycemic control in subjects with type 2 diabetes mellitus whose dysglycemia is inadequately controlled on insulin. Postgrad. Med. 2017, 129, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Chamarthi, B.; Ezrokhi, M.; Cincotta, A.H. 1083-P: Bromocriptine-QR (BQR) Reduces Hypertriglyceridemia (HTG) in Hypertensive Type 2 Diabetes (T2D) Subjects. Diabetes 2020, 69, 1083-P. [Google Scholar] [CrossRef]
- Cincotta, A.H.; Ezrokhi, M. The anti-diabetes efficacy of Bromocriptine-QR in type 2 diabetes mellitus (T2DM) subjects is enhanced among those with elevated blood pressure and plasma triglyceride levels. Diabetes 2013, 62, A305. [Google Scholar]
- Scranton, R.; Cincotta, A. Bromocriptine—Unique formulation of a dopamine agonist for the treatment of type 2 diabetes. Expert Opin. Pharmacother. 2009, 11, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.G.; Lake, C.R.; Williams, A.C.; Teychenne, P.F.; Shoulson, I.; Steinsland, O. Bromocriptine inhibits norepinephrine release. Clin. Pharmacol. Ther. 1979, 25, 137–142. [Google Scholar] [CrossRef]
- Sowers, J.R.; Stern, N.; Nyby, M.D.; Jasberg, K. Dopaminergic regulation of circadian rhythms of blood pressure, renin and aldosterone in essential hypertension. Cardiovasc. Res. 1982, 16, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.K.; Ravussin, E.; Johannsen, D.L.; Stull, A.J.; Cefalu, W.T.; Johnson, W.D. Endothelial Dysfunction: An Early Cardiovascular Risk Marker in Asymptomatic Obese Individuals with Prediabetes. Br. J. Med. Res. 2012, 2, 413–423. [Google Scholar] [CrossRef]
- Williams, I.L.; Wheatcroft, S.B.; Shah, A.M.; Kearney, M.T. Obesity, atherosclerosis and the vascular endothelium: Mechanisms of reduced nitric oxide bioavailability in obese humans. Int. J. Obes. Relat. Metab. Disord. 2002, 26, 754–764. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ayajiki, K.; Kashiwagi, A.; Masada, M.; Okamura, T. Malfunction of Vascular Control in Lifestyle-Related Diseases: Mechanisms Underlying Endothelial Dysfunction in the Insulin-Resistant State. J. Pharmacol. Sci. 2004, 96, 401–405. [Google Scholar] [CrossRef]
- Xu, S.; Ilyas, I.; Little, P.J.; Li, H.; Kamato, D.; Zheng, X.; Luo, S.; Li, Z.; Liu, P.; Han, J.; et al. Endothelial Dysfunction in Atherosclerotic Cardiovascular Diseases and Beyond: From Mechanism to Pharmacotherapies. Pharmacol. Rev. 2021, 73, 924–967. [Google Scholar] [CrossRef]
- Shaito, A.; Aramouni, K.; Assaf, R.; Parenti, A.; Orekhov, A.; El Yazbi, A.; Pintus, G.; Eid, A.H. Oxidative Stress-Induced Endothelial Dysfunction in Cardiovascular Diseases. Front. Biosci. 2022, 27, 105. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial Nitric Oxide Synthase Uncoupling Impairs Endothelial Progenitor Cell Mobilization and Function in Diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Munzel, T.; Daiber, A.; Ullrich, V.; Mulsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arter. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function--role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef]
- Munzel, T.; Daiber, A. Environmental Stressors and Their Impact on Health and Disease with Focus on Oxidative Stress. Antioxid. Redox. Signal. 2018, 28, 735–740. [Google Scholar] [CrossRef]
- Masuo, K.; Rakugi, H.; Ogihara, T.; Esler, M.D.; Lambert, G.W. Cardiovascular and renal complications of type 2 diabetes in obesity: Role of sympathetic nerve activity and insulin resistance. Curr. Diabetes Rev. 2010, 6, 58–67. [Google Scholar] [CrossRef]
- Carnagarin, R.; Tan, K.; Adams, L.; Matthews, V.B.; Kiuchi, M.G.; Gavidia, L.M.L.; Lambert, G.W.; Lambert, E.A.; Herat, L.Y.; Schlaich, M.P. Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD)—A Condition Associated with Heightened Sympathetic Activation. Int. J. Mol. Sci. 2021, 22, 4241. [Google Scholar] [CrossRef] [PubMed]
- Hurr, C.; Simonyan, H.; Morgan, D.A.; Rahmouni, K.; Young, C.N. Liver sympathetic denervation reverses obesity-induced hepatic steatosis. J. Physiol. 2019, 597, 4565–4580. [Google Scholar] [CrossRef]
- Bartness, T.J.; Liu, Y.; Shrestha, Y.B.; Ryu, V. Neural innervation of white adipose tissue and the control of lipolysis. Front. Neuroendocr. 2014, 35, 473–493. [Google Scholar] [CrossRef] [PubMed]
- Geisler, C.E.; Renquist, B.J. Hepatic lipid accumulation: Cause and consequence of dysregulated glucoregulatory hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef]
- Tentolouris, N.; Liatis, S.; Katsilambros, N. Sympathetic System Activity in Obesity and Metabolic Syndrome. Ann. N. Y. Acad. Sci. 2006, 1083, 129–152. [Google Scholar] [CrossRef]
- Fliers, E.; Kreier, F.; Voshol, P.J.; Havekes, L.M.; Sauerwein, H.P.; Kalsbeek, A.; Buijs, R.M.; Romijn, J.A. White adipose tissue: Getting nervous. J. Neuroendocr. 2003, 15, 1005–101010. [Google Scholar] [CrossRef]
- Masi, S.; Uliana, M.; Virdis, A. Angiotensin II and vascular damage in hypertension: Role of oxidative stress and sympathetic activation. Vasc. Pharmacol. 2019, 115, 13–17. [Google Scholar] [CrossRef]
- Schweda, F.; Kurtz, A. Regulation of renin release by local and systemic factors. Rev. Physiol. Biochem. Pharmacol. 2011, 161, 1–44. [Google Scholar]
- van Zwieten, P.A.; de Jonge, A. Interaction between the adrenergic and renin-angiotensin-aldosterone-systems. Postgrad Med. J. 1986, 62 (Suppl. 1), 23–27. [Google Scholar]
- Kishi, T.; Hirooka, Y. Sympathoexcitation Associated with Renin-Angiotensin System in Metabolic Syndrome. Int. J. Hypertens. 2013, 2013, 406897. [Google Scholar] [CrossRef]
- Armando, I.; Konkalmatt, P.; Felder, R.A.; Jose, P.A. The renal dopaminergic system: Novel diagnostic and therapeutic approaches in hypertension and kidney disease. Transl. Res. 2015, 165, 505–511. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Missale, C.; Memo, M.; Liberini, P.; Spano, P. Dopamine selectively inhibits angiotensin II-induced aldosterone secretion by interacting with D-2 receptors. J. Pharmacol. Exp. Ther. 1988, 246, 1137–1143. [Google Scholar] [PubMed]
- Yang, J.; Villar, V.A.M.; Jose, P.A.; Zeng, C. Renal Dopamine Receptors and Oxidative Stress: Role in Hypertension. Antioxidants Redox Signal. 2021, 34, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Hernández, A.; Figuero-Pérez, L.; Cruz-Hernandez, J.J.; Sarmiento, R.G.; Usategui-Martin, R.; Miramontes-González, J.P. Dopamine Receptors and the Kidney: An Overview of Health- and Pharmacological-Targeted Implications. Biomolecules 2021, 11, 254. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, W.; Peng, X.; Liu, L.; Zhang, A.; Li, X.; Ma, K.; Wang, L. alpha(1) -Adrenoceptors activate the NLRP3 inflammasome through downregulation of Kir2.1 in cardiac inflammation. Exp. Physiol. 2022, 107, 589–600. [Google Scholar] [CrossRef]
- Higashikuni, Y.; Liu, W.; Numata, G.; Tanaka, K.; Fukuda, D.; Tanaka, Y.; Hirata, Y.; Imamura, T.; Takimoto, E.; Komuro, I.; et al. NLRP3 Inflammasome Activation Through Heart-Brain Interaction Initiates Cardiac Inflammation and Hypertrophy During Pressure Overload. Circulation 2023, 147, 338–355. [Google Scholar] [CrossRef]
- Holwerda, S.W.; Luehrs, R.E.; DuBose, L.; Collins, M.T.; Wooldridge, N.A.; Stroud, A.K.; Fadel, P.J.; Abboud, F.M.; Pierce, G.L. Elevated Muscle Sympathetic Nerve Activity Contributes to Central Artery Stiffness in Young and Middle-Age/Older Adults. Hypertension 2019, 73, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Nardone, M.; Floras, J.S.; Millar, P.J. Sympathetic neural modulation of arterial stiffness in humans. Am. J. Physiol. Circ. Physiol. 2020, 319, H1338–H1346. [Google Scholar] [CrossRef]
- Harvey, R.E.; Barnes, J.N.; Hart, E.C.J.; Nicholson, W.T.; Joyner, M.J.; Casey, D.P. Influence of sympathetic nerve activity on aortic hemodynamics and pulse wave velocity in women. Am. J. Physiol. Circ. Physiol. 2017, 312, H340–H346. [Google Scholar] [CrossRef]
- Singh, R.B.; Hristova, K.; Fedacko, J.; El-Kilany, G.; Cornelissen, G. Chronic heart failure: A disease of the brain. Hear Fail. Rev. 2019, 24, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Anderson, A.S. The Sympathetic Nervous System and Heart Failure. Cardiol. Clin. 2013, 32, 33–45. [Google Scholar] [CrossRef]
- Esler, M.; Lambert, G.; Rocca, H.P.B.-L.; Vaddadi, G.; Kaye, D. Sympathetic nerve activity and neurotransmitter release in humans: Translation from pathophysiology into clinical practice. Acta Physiol. Scand. 2003, 177, 275–284. [Google Scholar] [CrossRef]
- Schafer, M.; Browne, L.P.; Truong, U.; Bjornstad, P.; Tell, S.; Snell-Bergeon, J.; Baumgartner, A.; Hunter, K.S.; Reusch, J.E.B.; Barker, A.J.; et al. Bromocriptine Improves Central Aortic Stiffness in Adolescents With Type 1 Diabetes: Arterial Health Results From the BCQR-T1D Study. Hypertension 2023, 80, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Trongtorsak, A.; Kittipibul, V.; Mahabir, S.; Ibrahim, M.; Saint Croix, G.R.; Hernandez, G.A.; Chaparro, S. Effects of bromocriptine in peripartum cardiomyopathy: A systematic review and meta-analysis. Heart Fail Rev. 2022, 27, 533–543. [Google Scholar] [CrossRef]
- Tremblay-Gravel, M.; Marquis-Gravel, G.; Avram, R.; Desplantie, O.; Ducharme, A.; Bibas, L.; Pacheco, C.; Couture, E.; Simard, F.; Poulin, A.; et al. The effect of bromocriptine on left ventricular functional recovery in peripartum cardiomyopathy: Insights from the BRO-HF retrospective cohort study. ESC Hear Fail. 2018, 6, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S.; Parks, R.; Cohn, J.N. The effects of bromocriptine in patients with congestive heart failure. Am. Heart J. 1983, 106 Pt 1, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Mejía-Rodríguez, O.; Alvarez-Aguilar, C.; Vega-Gómez, H.E.; Belio-Caro, F.; Vargas-Espinosa, J.M.; Paniagua-Sierra, J.R. Bromocriptine induces regression of left ventricular hypertrophy in peritoneal dialysis patients. Proc. West. Pharmacol. Soc. 2005, 48, 122–125. [Google Scholar] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cincotta, A.H. Brain Dopamine–Clock Interactions Regulate Cardiometabolic Physiology: Mechanisms of the Observed Cardioprotective Effects of Circadian-Timed Bromocriptine-QR Therapy in Type 2 Diabetes Subjects. Int. J. Mol. Sci. 2023, 24, 13255. https://doi.org/10.3390/ijms241713255
Cincotta AH. Brain Dopamine–Clock Interactions Regulate Cardiometabolic Physiology: Mechanisms of the Observed Cardioprotective Effects of Circadian-Timed Bromocriptine-QR Therapy in Type 2 Diabetes Subjects. International Journal of Molecular Sciences. 2023; 24(17):13255. https://doi.org/10.3390/ijms241713255
Chicago/Turabian StyleCincotta, Anthony H. 2023. "Brain Dopamine–Clock Interactions Regulate Cardiometabolic Physiology: Mechanisms of the Observed Cardioprotective Effects of Circadian-Timed Bromocriptine-QR Therapy in Type 2 Diabetes Subjects" International Journal of Molecular Sciences 24, no. 17: 13255. https://doi.org/10.3390/ijms241713255
APA StyleCincotta, A. H. (2023). Brain Dopamine–Clock Interactions Regulate Cardiometabolic Physiology: Mechanisms of the Observed Cardioprotective Effects of Circadian-Timed Bromocriptine-QR Therapy in Type 2 Diabetes Subjects. International Journal of Molecular Sciences, 24(17), 13255. https://doi.org/10.3390/ijms241713255