

Brain-Derived 11S Regulator (PA28αβ) Promotes Proteasomal Hydrolysis of Elongated Oligoglutamine-Containing Peptides


Abstract
1. Introduction
2. Results
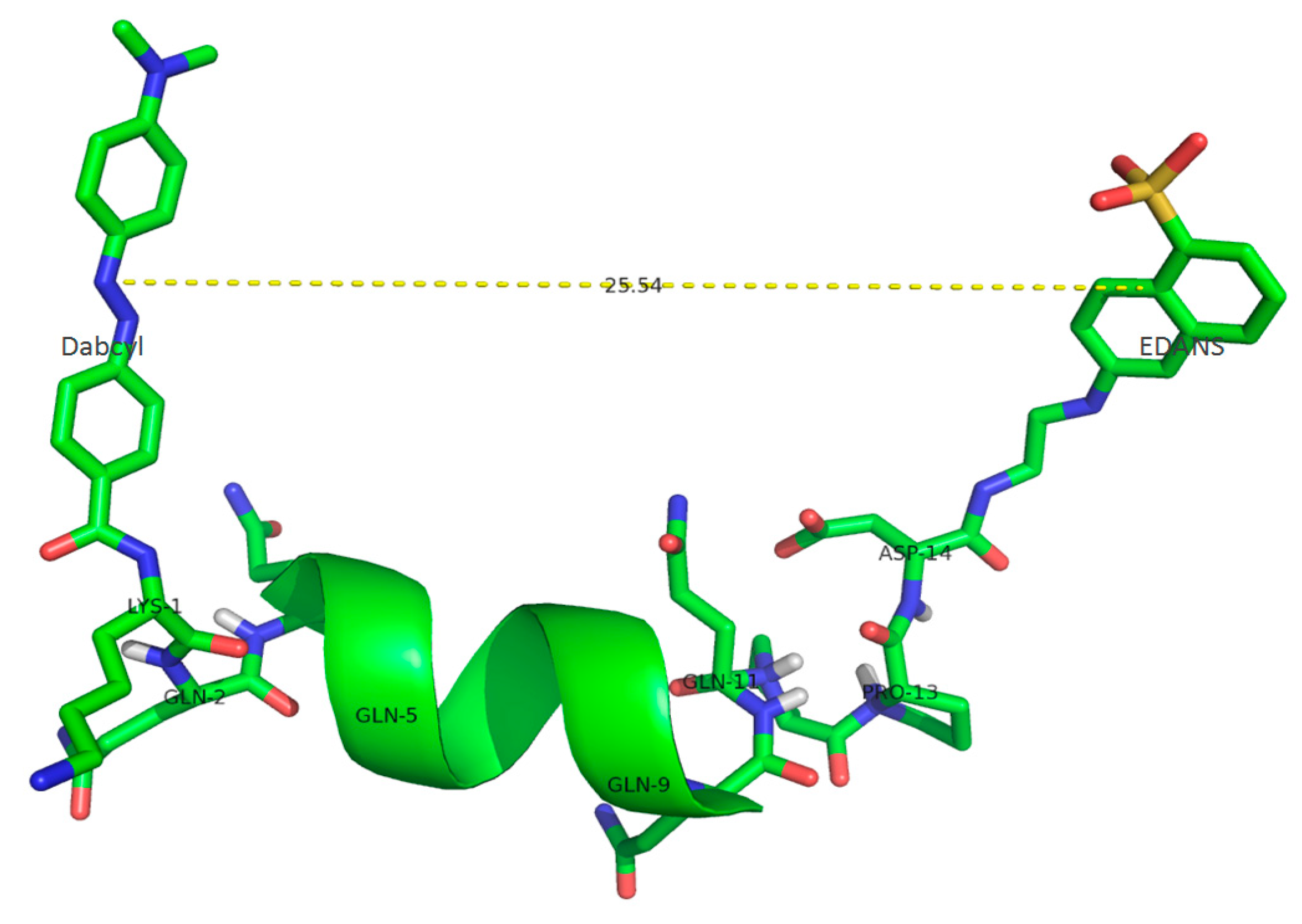
2.1. Substrate Characterization
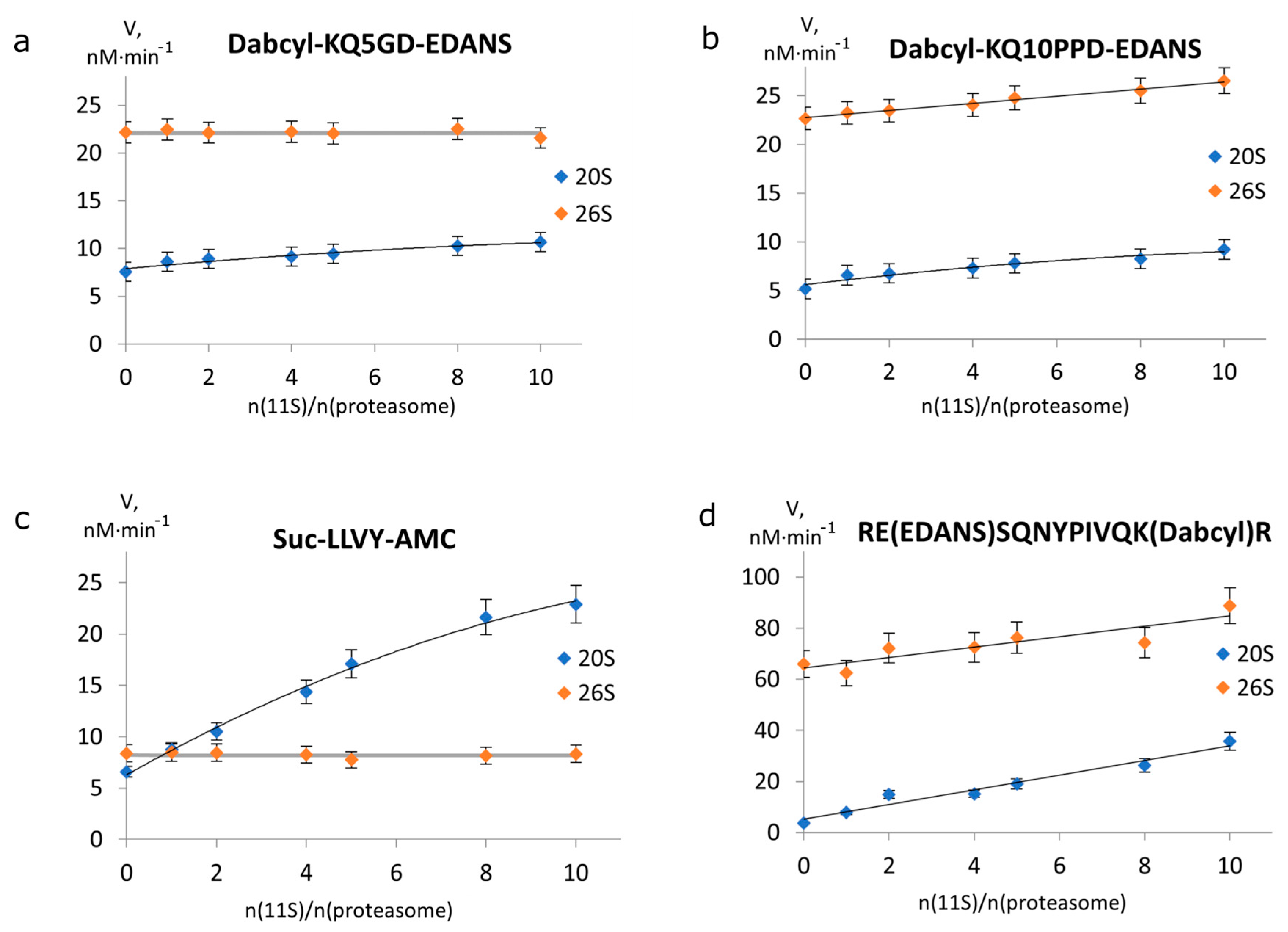
2.2. 11S Protein Increases 26S Proteasomal Activity towards Longer Peptides
2.3. The Effect of 11S on the Kinetic Parameters of Degradation
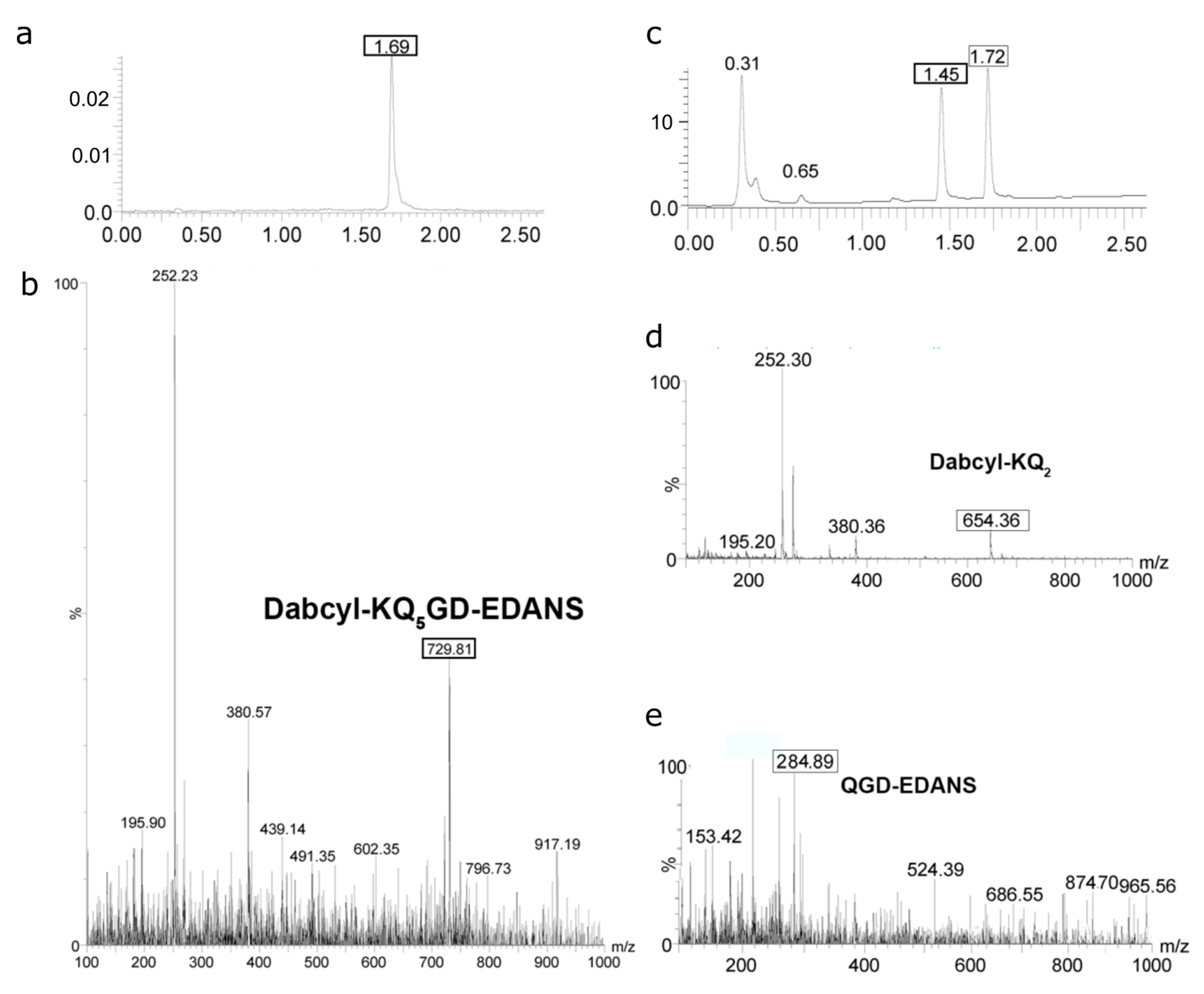
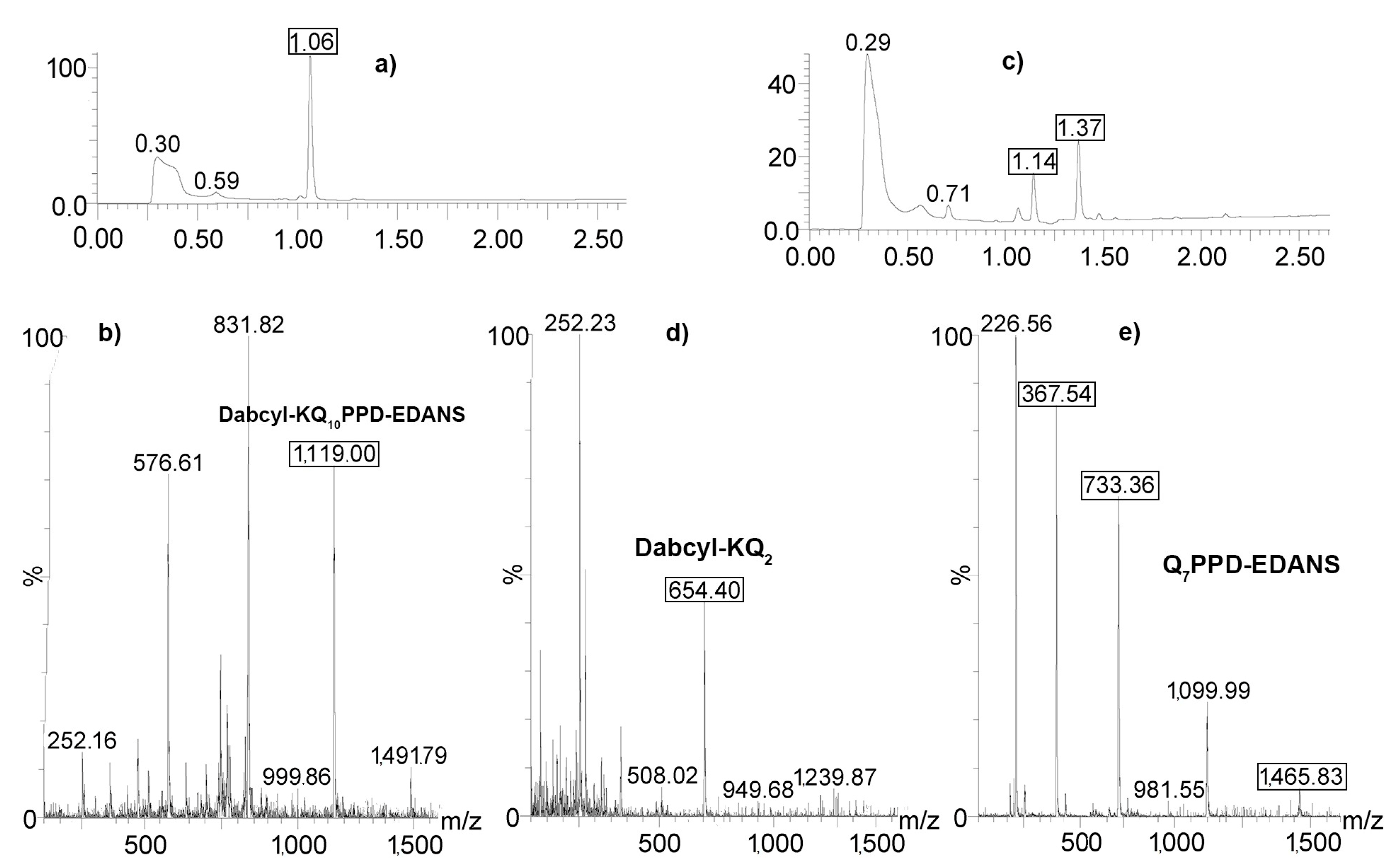
2.4. Determination of Proteolysis Sites
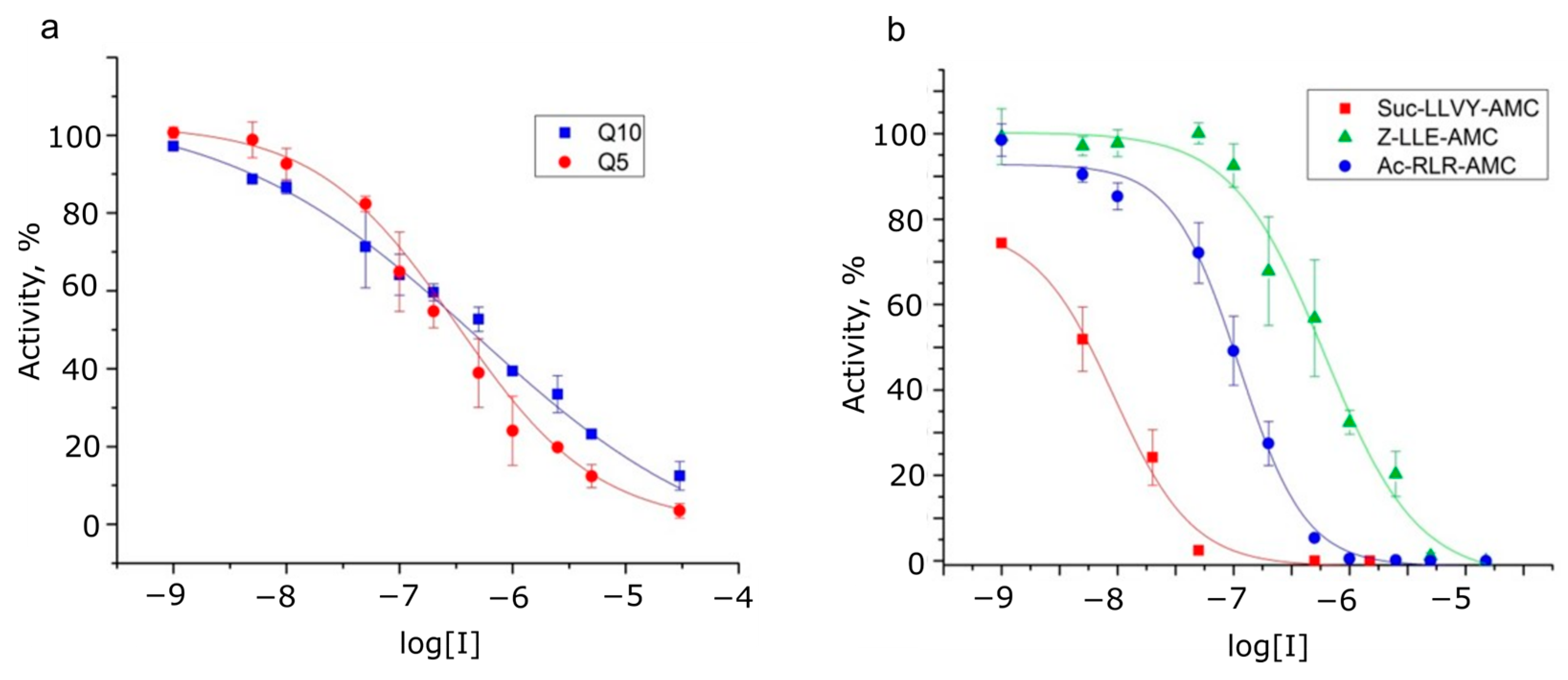
2.5. The Identification of Proteasome Catalytic Subunit That Digests Oligoglutamine Substrates
3. Discussion
4. Materials and Methods
4.1. Peptide Substrates and Inhibitors
4.2. Isolation of Proteasome Particles
4.3. Kinetic Measurements
4.4. Determination of Apparent Kinetic Parameters
4.5. Determination of Cleavage Sites by LC/Mass Spectrometry
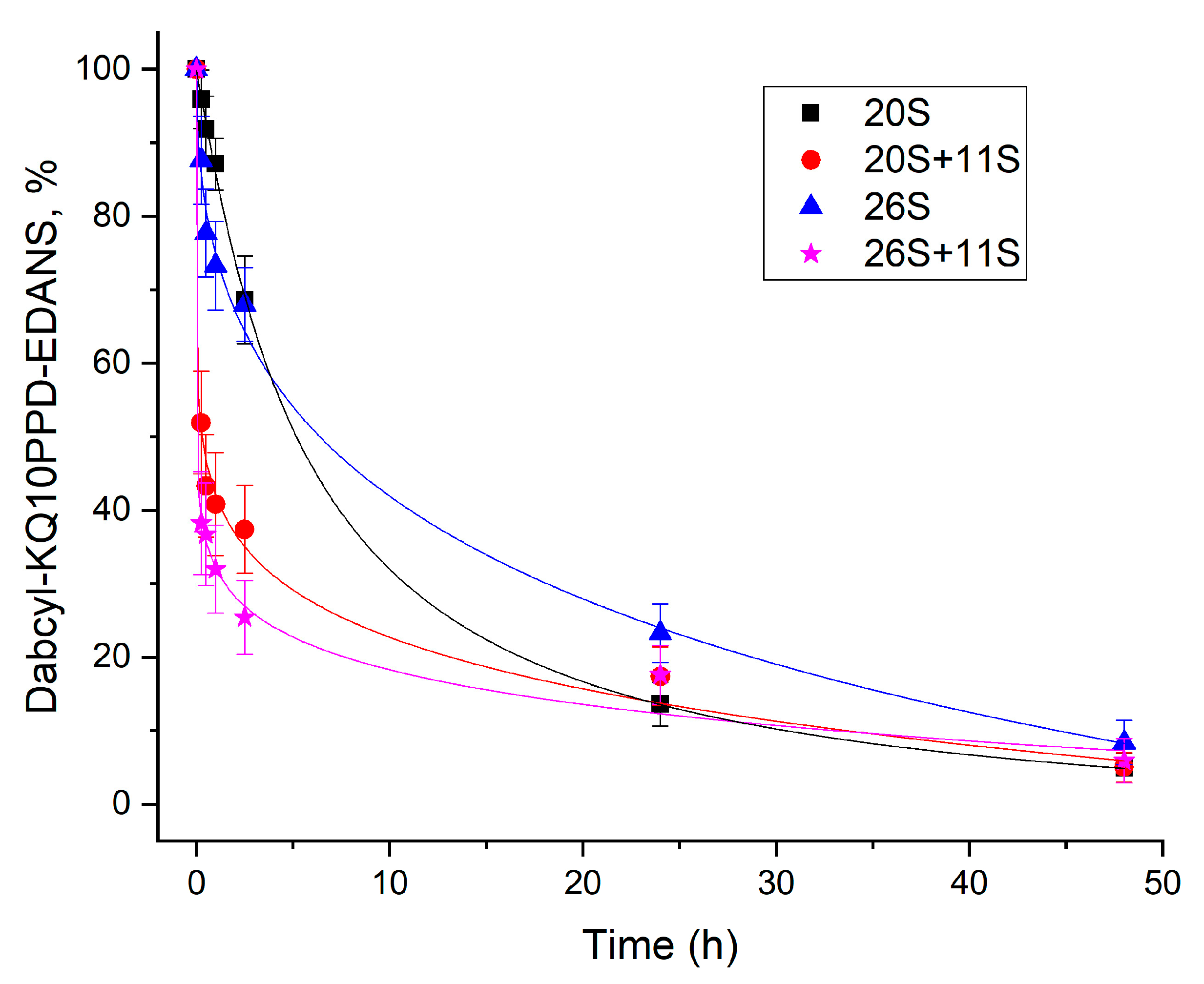
4.6. Time-Course of Dabcyl-KQ10PPD-EDANS Cleavage by Different Proteasome Complexes
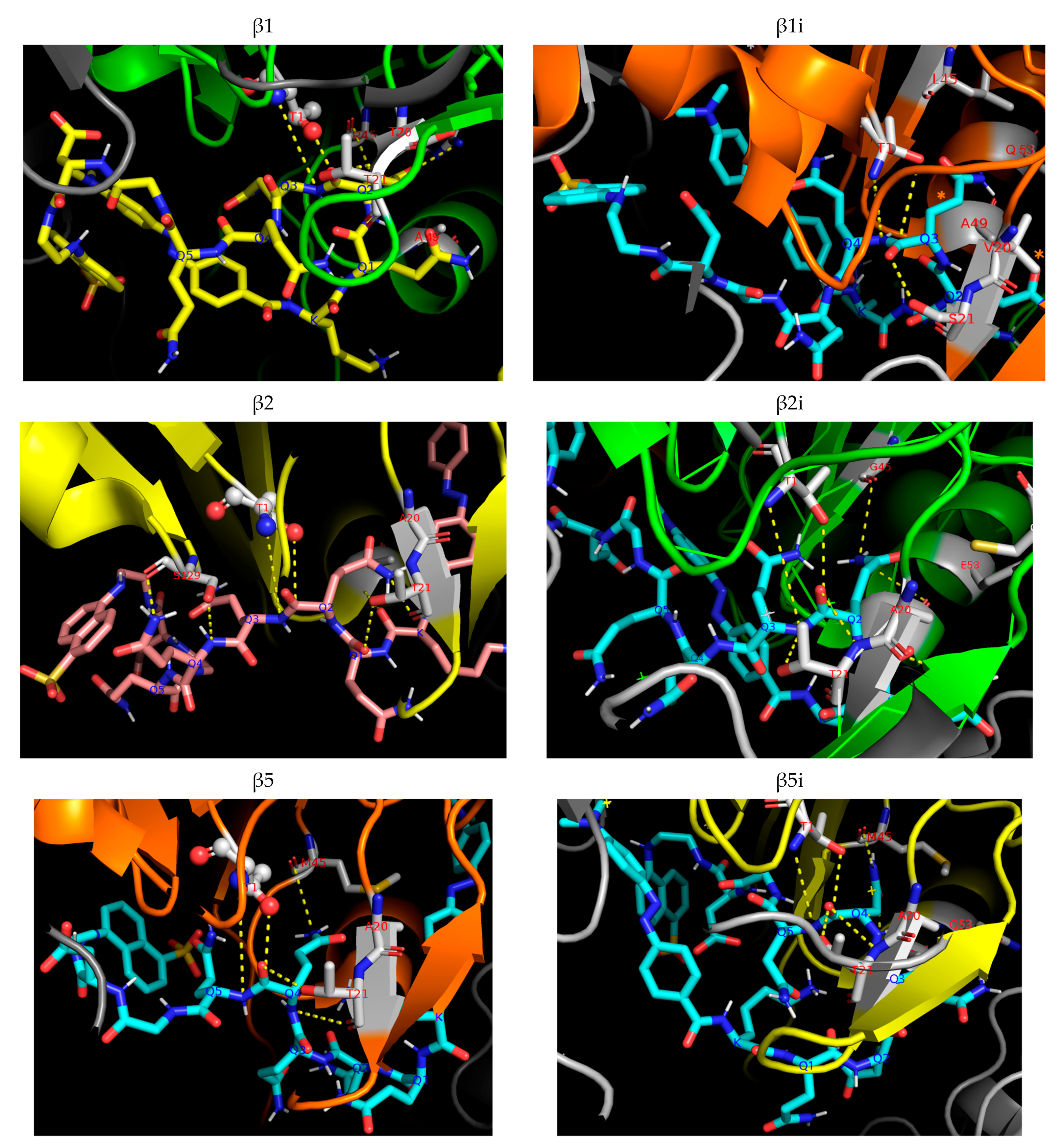
4.7. Docking Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ben-Nissan, G.; Sharon, M. Regulating the 20S Proteasome Ubiquitin-Independent Degradation Pathway. Biomolecules 2014, 4, 862–884. [Google Scholar] [CrossRef] [PubMed]
- Ciechanover, A.; Kwon, Y.T. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front. Neurosci. 2017, 11, 185. [Google Scholar] [CrossRef]
- Fabre, B.; Lambour, T.; Garrigues, L.; Amalric, F.; Vigneron, N.; Menneteau, T.; Stella, A.; Monsarrat, B.; Eynde, B.V.D.; Burlet-Schiltz, O.; et al. Deciphering preferential interactions within supramolecular protein complexes: The proteasome case. Mol. Syst. Biol. 2015, 11, 771. [Google Scholar] [CrossRef] [PubMed]
- Yerbury, J.J.; Ooi, L.; Dillin, A.; Saunders, D.N.; Hatters, D.M.; Beart, P.M.; Cashman, N.R.; Wilson, M.R.; Ecroyd, H. Walking the tightrope: Proteostasis and neurodegenerative disease. J. Neurochem. 2016, 137, 489–505. [Google Scholar] [CrossRef]
- Chai, Y.; Koppenhafer, S.L.; Shoesmith, S.J.; Perez, M.K.; Paulson, H.L. Evidence for proteasome involvement in polyglutamine disease: Localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Genet. 1999, 8, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.R.; Lieberman, A.P. The Ubiquitination, Disaggregation and Proteasomal Degradation Machineries in Polyglutamine Disease. Front. Mol. Neurosci. 2017, 10, 78. [Google Scholar] [CrossRef]
- Schmidt, T.; Lindenberg, K.S.; Krebs, A.; Schöls, L.; Laccone, F.; Herms, J.; Rechsteiner, M.; Riess, O.; Landwehrmeyer, G.B. Protein surveillance machinery in brains with spinocerebellar ataxia type 3: Redistribution and differential recruitment of 26S proteasome subunits and chaperones to neuronal intranuclear inclusions. Ann. Neurol. 2002, 51, 302–310. [Google Scholar] [CrossRef]
- Petrasch-Parwez, E.; Nguyen, H.P.; Löbbecke-Schumacher, M.; Habbes, H.W.; Wieczorek, S.; Riess, O.; Andres, K.H.; Dermietzel, R.; Von Hörsten, S. Cellular and subcellular localization of Huntingtin [corrected] aggregates in the brain of a rat transgenic for Huntington disease. J. Comp. Neurol. 2007, 501, 716–730. [Google Scholar] [CrossRef]
- Weiss, A.; Klein, C.; Woodman, B.; Sathasivam, K.; Bibel, M.; Régulier, E.; Bates, G.P.; Paganetti, P. Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington’s disease. J. Neurochem. 2007, 104, 846–858. [Google Scholar] [CrossRef]
- Sethi, R.; Tripathi, N.; Pallapati, A.R.; Gaikar, A.; Bharatam, P.V.; Roy, I. Does N-terminal huntingtin function as a ‘holdase’ for inhibiting cellular protein aggregation? FEBS J. 2018, 285, 1791–1811. [Google Scholar] [CrossRef]
- Venkatraman, P.; Wetzel, R.; Tanaka, M.; Nukina, N.; Goldberg, A.L. Eukaryotic Proteasomes Cannot Digest Polyglutamine Sequences and Release Them during Degradation of Polyglutamine-Containing Proteins. Mol. Cell 2004, 14, 95–104. [Google Scholar] [CrossRef]
- Holmberg, C.I.; Staniszewski, K.E.; Mensah, K.N.; Matouschek, A.; Morimoto, R.I. Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 2004, 23, 4307–4318. [Google Scholar] [CrossRef]
- Seo, H.; Sonntag, K.C.; Isacson, O. Generalized brain and skin proteasome inhibition in Huntington’s disease. Ann. Neurol. 2004, 56, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Hernández, M.; Valera, A.G.; Morán, M.A.; Gómez-Ramos, P.; Alvarez-Castelao, B.; Castaño, J.G.; Hernández, F.; Lucas, J.J. Inhibition of 26S proteasome activity by huntingtin filaments but not inclusion bodies isolated from mouse and human brain. J. Neurochem. 2006, 98, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Bennett, E.J.; Shaler, T.A.; Woodman, B.; Ryu, K.Y.; Zaitseva, T.S.; Becker, C.H.; Bates, G.P.; Schulman, H.; Kopito, R.R. Global changes to the ubiquitin system in Huntington’s disease. Nature 2007, 448, 704–708. [Google Scholar] [CrossRef]
- Hipp, M.S.; Patel, C.N.; Bersuker, K.; Riley, B.E.; Kaiser, S.E.; Shaler, T.A.; Brandeis, M.; Kopito, R.R. Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol. 2012, 196, 573–587. [Google Scholar] [CrossRef]
- Michalik, A.; Van Broeckhoven, C. Proteasome degrades soluble expanded polyglutamine completely and efficiently. Neurobiol. Dis. 2004, 16, 202–211. [Google Scholar] [CrossRef]
- Bowman, A.B.; Yoo, S.-Y.; Dantuma, N.P.; Zoghbi, H.Y. Neuronal dysfunction in a polyglutamine disease model occurs in the absence of ubiquitin–proteasome system impairment and inversely correlates with the degree of nuclear inclusion formation. Hum. Mol. Genet. 2005, 14, 679–691. [Google Scholar] [CrossRef]
- Pratt, G.; Rechsteiner, M. Proteasomes cleave at multiple sites within polyglutamine tracts: Activation by PA28gamma(K188E). J. Biol. Chem. 2008, 283, 12919–12925. [Google Scholar] [CrossRef]
- Maynard, C.J.; Böttcher, C.; Ortega, Z.; Smith, R.; Florea, B.I.; Díaz-Hernández, M.; Brundin, P.; Overkleeft, H.S.; Li, J.-Y.; Lucas, J.J.; et al. Accumulation of ubiquitin conjugates in a polyglutamine disease model occurs without global ubiquitin/proteasome system impairment. Proc. Natl. Acad. Sci. USA 2009, 106, 13986–13991. [Google Scholar] [CrossRef]
- Juenemann, K.; Schipper-Krom, S.; Wiemhoefer, A.; Kloss, A.; Sanz, A.S.; Reits, E.A. Expanded Polyglutamine-containing N-terminal Huntingtin Fragments Are Entirely Degraded by Mammalian Proteasomes. J. Biol. Chem. 2013, 288, 27068–27084. [Google Scholar] [CrossRef]
- Chapiro, J.; Claverol, S.; Piette, F.; Ma, W.; Stroobant, V.; Guillaume, B.; Gairin, J.-E.; Morel, S.; Burlet-Schiltz, O.; Monsarrat, B.; et al. Destructive Cleavage of Antigenic Peptides Either by the Immunoproteasome or by the Standard Proteasome Results in Differential Antigen Presentation. J. Immunol. 2006, 176, 1053–1061. [Google Scholar] [CrossRef] [PubMed]
- Deol, P.; Zaiss, D.M.W.; Monaco, J.J.; Sijts, A.J.A.M. Rates of Processing Determine the Immunogenicity of Immunoproteasome-Generated Epitopes. J. Immunol. 2007, 178, 7557–7562. [Google Scholar] [CrossRef]
- Wetzel, R. Physical Chemistry of Polyglutamine: Intriguing Tales of a Monotonous Sequence. J. Mol. Biol. 2012, 421, 466–490. [Google Scholar] [CrossRef]
- Li, M.; Reddy, L.G.; Bennett, R.; Silva, N.D.; Jones, L.R.; Thomas, D.D. A Fluorescence Energy Transfer Method for Analyzing Protein Oligomeric Structure: Application to Phospholamban. Biophys. J. 1999, 76, 2587–2599. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucleic Acids Res. 2005, 33, W72–W76. [Google Scholar] [CrossRef] [PubMed]
- Kryachkov, V.A.; Tashlitsky, V.N.; Bacheva, A.V. Design and Properties of Novel Proteasome Substrates Containing a Polyglutamine Sequence. Mosc. Univ. Chem. Bull. 2018, 73, 166–172. [Google Scholar] [CrossRef]
- Zhang, Z.; Clawson, A.; Rechsteiner, M. The proteasome activator 11 S regulator or PA28. Contribution By both alpha and beta subunits to proteasome activation. J. Biol. Chem. 1998, 273, 30660–30668. [Google Scholar] [CrossRef]
- Bacheva, A.V.; Korobkina, O.V.; Nesterova, P.S.; Kryachkov, V.A.; Gabibov, A.G. The 11S proteasome activator: Isolation from mouse brain and the influence on peptide substrate hydrolysis of the 20S and 26S proteasomes. Mosc. Univ. Chem. Bull. 2016, 71, 97–103. [Google Scholar] [CrossRef]
- Cascio, P.; Call, M.; Petre, B.M.; Walz, T.; Goldberg, A.L. Properties of the hybrid form of the 26S proteasome containing both 19S and PA28 complexes. EMBO J. 2002, 21, 2636–2645. [Google Scholar] [CrossRef]
- Kopp, F.; Dahlmann, B.; Kuehn, L. Reconstitution of hybrid proteasomes from purified PA700-20 S complexes and PA28alphabeta activator: Ultrastructure and peptidase activities. J. Mol. Biol. 2001, 313, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Huber, E.M.; Basler, M.; Schwab, R.; Heinemeyer, W.; Kirk, C.J.; Groettrup, M.; Groll, M. Immuno- and Constitutive Proteasome Crystal Structures Reveal Differences in Substrate and Inhibitor Specificity. Cell 2012, 148, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Macherla, V.R.; Mitchell, S.S.; Manam, R.R.; Reed, K.A.; Chao, T.-H.; Nicholson, B.; Deyanat-Yazdi, G.; Mai, B.; Jensen, P.R.; Fenical, W.F.; et al. Structure-activity relationship studies of salinosporamide A (NPI-0052), a novel marine derived proteasome inhibitor. J. Med. Chem. 2005, 48, 3684–3687. [Google Scholar] [CrossRef]
- Potts, B.C.; Albitar, M.X.; Anderson, K.C.; Baritaki, S.; Berkers, C.; Bonavida, B.; Chandra, J.; Chauhan, D.; Cusack, J.C.; Fenical, W.; et al. Marizomib, a Proteasome Inhibitor for All Seasons: Preclinical Profile and a Framework for Clinical Trials. Curr. Cancer Drug Targets 2011, 11, 254–284. [Google Scholar] [CrossRef]
- Lee, M.J.; Bhattarai, D.; Yoo, J.; Miller, Z.; Park, J.E.; Lee, S.; Lee, W.; Driscoll, J.J.; Kim, K.B. Development of Novel Epoxyketone-Based Proteasome Inhibitors as a Strategy To Overcome Cancer Resistance to Carfilzomib and Bortezomib. J. Med. Chem. 2019, 62, 4444–4455. [Google Scholar] [CrossRef]
- Bhattacharyya, A.; Thakur, A.K.; Chellgren, V.M.; Thiagarajan, G.; Williams, A.D.; Chellgren, B.W.; Creamer, T.P.; Wetzel, R. Oligoproline Effects on Polyglutamine Conformation and Aggregation. J. Mol. Biol. 2006, 355, 524–535. [Google Scholar] [CrossRef]
- Bajorek, M.; Finley, D.; Glickman, M.H. Proteasome Disassembly and Downregulation Is Correlated with Viability during Stationary Phase. Curr. Biol. 2003, 13, 1140–1144. [Google Scholar] [CrossRef]
- Huber, E.M.; de Bruin, G.; Heinemeyer, W.; Soriano, G.P.; Overkleeft, H.S.; Groll, M. Systematic Analyses of Substrate Preferences of 20S Proteasomes Using Peptidic Epoxyketone Inhibitors. J. Am. Chem. Soc. 2015, 137, 7835–7842. [Google Scholar] [CrossRef] [PubMed]
- Shanley, K.L.; Hu, C.-L.; Bizzozero, O.A. Proteasome Composition in Cytokine-Treated Neurons and Astrocytes is Determined Mainly by Subunit Displacement. Neurochem. Res. 2020, 45, 860–871. [Google Scholar] [CrossRef]
- Thomas, T.A.; Smith, D.M. Proteasome activator 28gamma (PA28gamma) allosterically activates trypsin-like proteolysis by binding to the alpha-ring of the 20S proteasome. J. Biol. Chem. 2022, 298, 102140. [Google Scholar] [CrossRef]
- Cascio, P. PA28alphabeta: The enigmatic magic ring of the proteasome? Biomolecules 2014, 4, 566–584. [Google Scholar] [CrossRef] [PubMed]
- Kuzina, E.S.; Chernolovskaya, E.L.; Kudriaeva, A.A.; Zenkova, M.A.; Knorre, V.D.; Surina, E.A.; Ponomarenko, N.A.; Bobik, T.V.; Smirnov, I.V.; Bacheva, A.V.; et al. Immunoproteasome enhances intracellular proteolysis of myelin basic protein. Dokl. Biochem. Biophys. 2013, 453, 300–303. [Google Scholar] [CrossRef] [PubMed]
- Bacheva, A.V.; Belogurov, A.A.; Ponomarenko, N.A.; Knorre, V.D.; Govorun, V.M.; Serebryakova, M.V.; Gabibov, A.G. Analysis of myelin basic protein fragmentation by proteasome. Acta Nat. 2009, 1, 84–87. [Google Scholar] [CrossRef]
- Bacheva, A.V.; Belogurov, A.A.; Kuzina, E.S.; Serebryakova, M.V.; Ponomarenko, N.A.; Knorre, V.D.; Govorun, V.M.; Gabibov, A.G. Functional degradation of myelin basic protein. The proteomic approach. Russ. J. Bioorganic Chem. 2011, 37, 39–47. [Google Scholar] [CrossRef] [PubMed]
- VerPlank, J.J.S.; Tyrkalska, S.D.; Fleming, A.; Rubinsztein, D.C.; Goldberg, A.L. cGMP via PKG activates 26S proteasomes and enhances degradation of proteins, including ones that cause neurodegenerative diseases. Proc. Natl. Acad. Sci. USA 2020, 117, 14220–14230. [Google Scholar] [CrossRef]
- Goldberg, A.L.; Kim, H.T.; Lee, D.; Collins, G.A. Mechanisms That Activate 26S Proteasomes and Enhance Protein Degradation. Biomolecules 2021, 11, 779. [Google Scholar] [CrossRef]
- Yersak, J.M.; Montie, H.L.; Chevalier-Larsen, E.S.; Liu, Y.; Huang, L.; Rechsteiner, M.; Merry, D.E. The 11S Proteasomal Activator REGgamma Impacts Polyglutamine-Expanded Androgen Receptor Aggregation and Motor Neuron Viability through Distinct Mechanisms. Front. Mol. Neurosci. 2017, 10, 159. [Google Scholar] [CrossRef]
- Crawford, L.J.; Walker, B.; Ovaa, H.; Chauhan, D.; Anderson, K.C.; Morris, T.C.; Irvine, A.E. Comparative Selectivity and Specificity of the Proteasome Inhibitors BzLLLCOCHO, PS-341, and MG-132. Cancer Res 2006, 66, 6379–6386. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tomko, R.J., Jr.; Hochstrasser, M. Proteasomes: Isolation and Activity Assays. Curr. Protoc. Cell Biol. 2015, 67, 3.43.1–43.43.203. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Sequence | Mr/Z Calculated/Detected | r, Å | FRET Efficiency (E), % |
|---|---|---|---|
| Dabcyl-KQ5GD-EDANS | 729.34/729.81 (Z = +2) | 20.0 | 95.3 |
| Dabcyl-KQ10GD-EDANS | 700.31/700.7 (Z = +3) | 24.9 | 84.4 |
| Dabcyl-KQ10PPD-EDANS | 1118.51/1119.24 (Z = +2) | 25.5 | 82.4 |
| Dabcyl-KQ5GD-EDANS | |||
| Proteasome complex | Km, μM | Vmax·10−2, μM·min−1 | Vmax (+11S)/Vmax |
| 20S | 52 ± 8 | 2.3 ± 0.3 | 2.7 |
| 20S+11S | 60 ± 8 | 6.1 ± 1.4 | |
| 26S | 54 ± 10 | 2.0 ± 0.3 | 1.0 |
| 26S+11S | 52 ± 10 | 2.1 ± 0.3 | |
| Dabcyl-KQ10GD-EDANS | |||
| Proteasome complex | Km, μM | Vmax·10−2, μM·min−1 | Vmax (+11S)/Vmax |
| 20S | 40 ± 3 | 0.61 ± 0.02 | 1.5 |
| 20S+11S | 64 ± 14 | 0.92 ± 0.15 | |
| 26S | 18 ± 2 | 0.09 ± 0.01 | 1.4 |
| 26S+11S | 38 ± 9 | 0.12 ± 0.03 | |
| Dabcyl-KQ10PPD-EDANS | |||
| Proteasome complex | Km, μM | Vmax·10−2, μM·min−1 | Vmax (+11S)/Vmax |
| 20S | 8.2 ± 1.5 | 0.73 ± 0.03 | 2.3 |
| 20S+11S | 5.3 ± 1.3 | 1.66 ± 0.07 | |
| 26S | 24.5 ± 10 | 2.3 ± 0.2 | 2.0 |
| 26S+11S | 20.0 ± 4.6 | 4.73 ± 0.35 | |
| Inhibitor | IC50 Values of Peptide Substrates Hydrolysis by 20S Brain Proteasome | ||||
|---|---|---|---|---|---|
| Suc-LLVY-AMC (Substrate for Chymotrypsin-like Activity) | Ac-RLR-AMC (Substrate for Trypsin-like Activity) | Z-LLE-AMC (Substrate for Caspase-like Activity) | Dabcyl-KQ5 PPD-EDANS | Dabcyl-KQ10 PPD-EDANS | |
| Bortezomib (chymotrypsin-like activity) | 8.1 ± 0.8 nM | 2.6 ± 0.7 µM | 23 ± 3 nM | 5.3 ± 2.1 µM | 4.1 ± 2 µM |
| Z-LLL-CHO (MG132) (chymotrypsin-like activity) | 12 ± 3 nM | 2.8 ± 0.3 µM | 155 ± 5 nM | 50 ± 6 µM | 72 ± 8 µM |
| Z-P-nLeu-D-CHO (caspase-like activity) | 100 ± 10 µM | >150 µM | 5.1 ± 0.3 µM | n.d. | n.d. |
| Marizomib (inhibits all 3 activities) | 9.3 ± 1.5 nM | 111.0 ± 8.0 nM | 610 ± 40 nM | 323 ± 15 nM | 480 ± 20 nM |
| (Hill coefficient) | (−1.28) | (−1.51) | (−1.1) | (−0.69) | (−0.39) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kriachkov, V.A.; Gotmanova, N.N.; Tashlitsky, V.N.; Bacheva, A.V. Brain-Derived 11S Regulator (PA28αβ) Promotes Proteasomal Hydrolysis of Elongated Oligoglutamine-Containing Peptides. Int. J. Mol. Sci. 2023, 24, 13275. https://doi.org/10.3390/ijms241713275
Kriachkov VA, Gotmanova NN, Tashlitsky VN, Bacheva AV. Brain-Derived 11S Regulator (PA28αβ) Promotes Proteasomal Hydrolysis of Elongated Oligoglutamine-Containing Peptides. International Journal of Molecular Sciences. 2023; 24(17):13275. https://doi.org/10.3390/ijms241713275
Chicago/Turabian StyleKriachkov, Viacheslav A., Natalia N. Gotmanova, Vadim N. Tashlitsky, and Anna V. Bacheva. 2023. "Brain-Derived 11S Regulator (PA28αβ) Promotes Proteasomal Hydrolysis of Elongated Oligoglutamine-Containing Peptides" International Journal of Molecular Sciences 24, no. 17: 13275. https://doi.org/10.3390/ijms241713275
APA StyleKriachkov, V. A., Gotmanova, N. N., Tashlitsky, V. N., & Bacheva, A. V. (2023). Brain-Derived 11S Regulator (PA28αβ) Promotes Proteasomal Hydrolysis of Elongated Oligoglutamine-Containing Peptides. International Journal of Molecular Sciences, 24(17), 13275. https://doi.org/10.3390/ijms241713275