
Extensive Angular Sampling Enables the Sensitive Localization of Macromolecules in Electron Tomograms

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
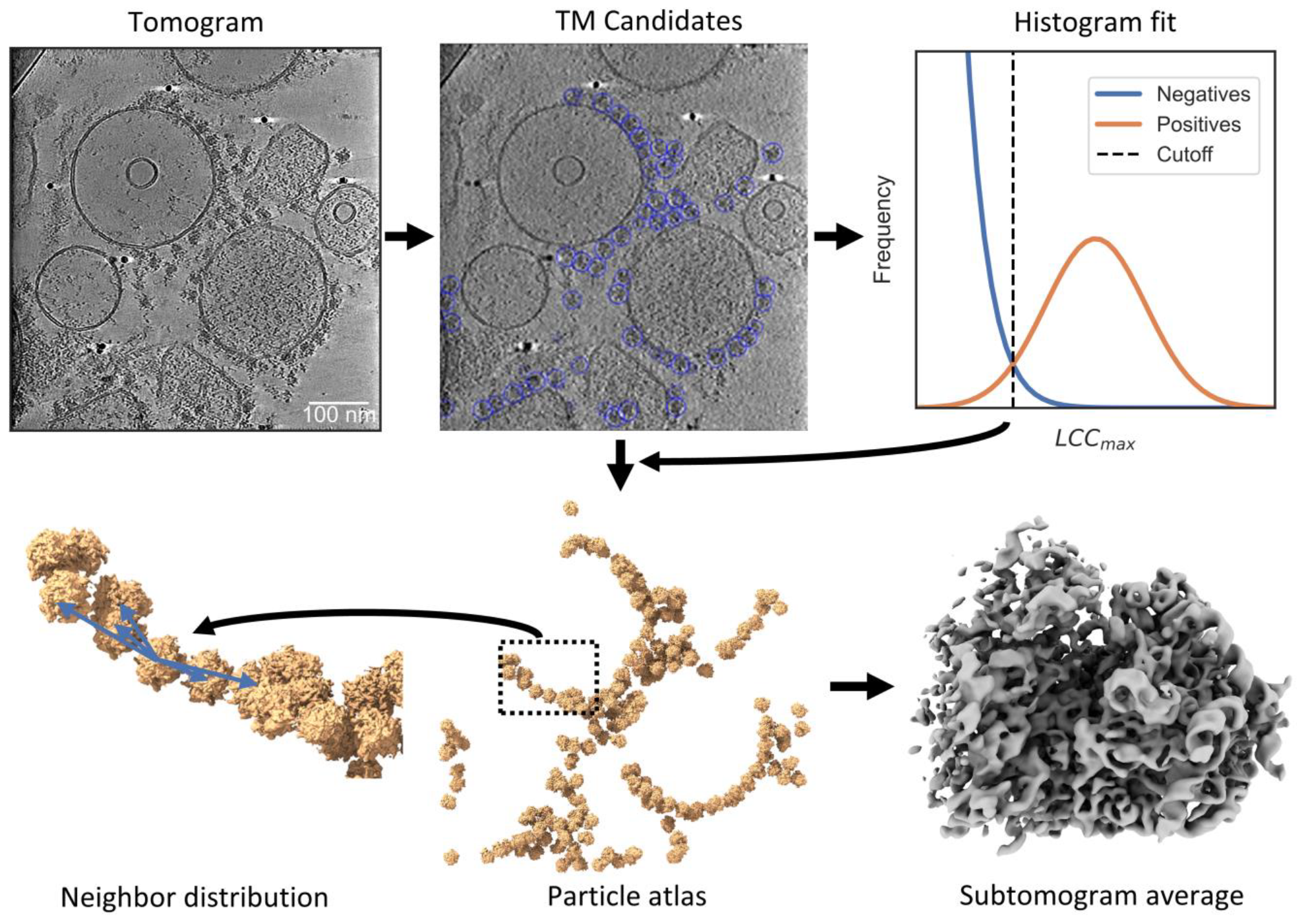
2.1. PyTOM Integrates with Common Tomography Software in a Workflow
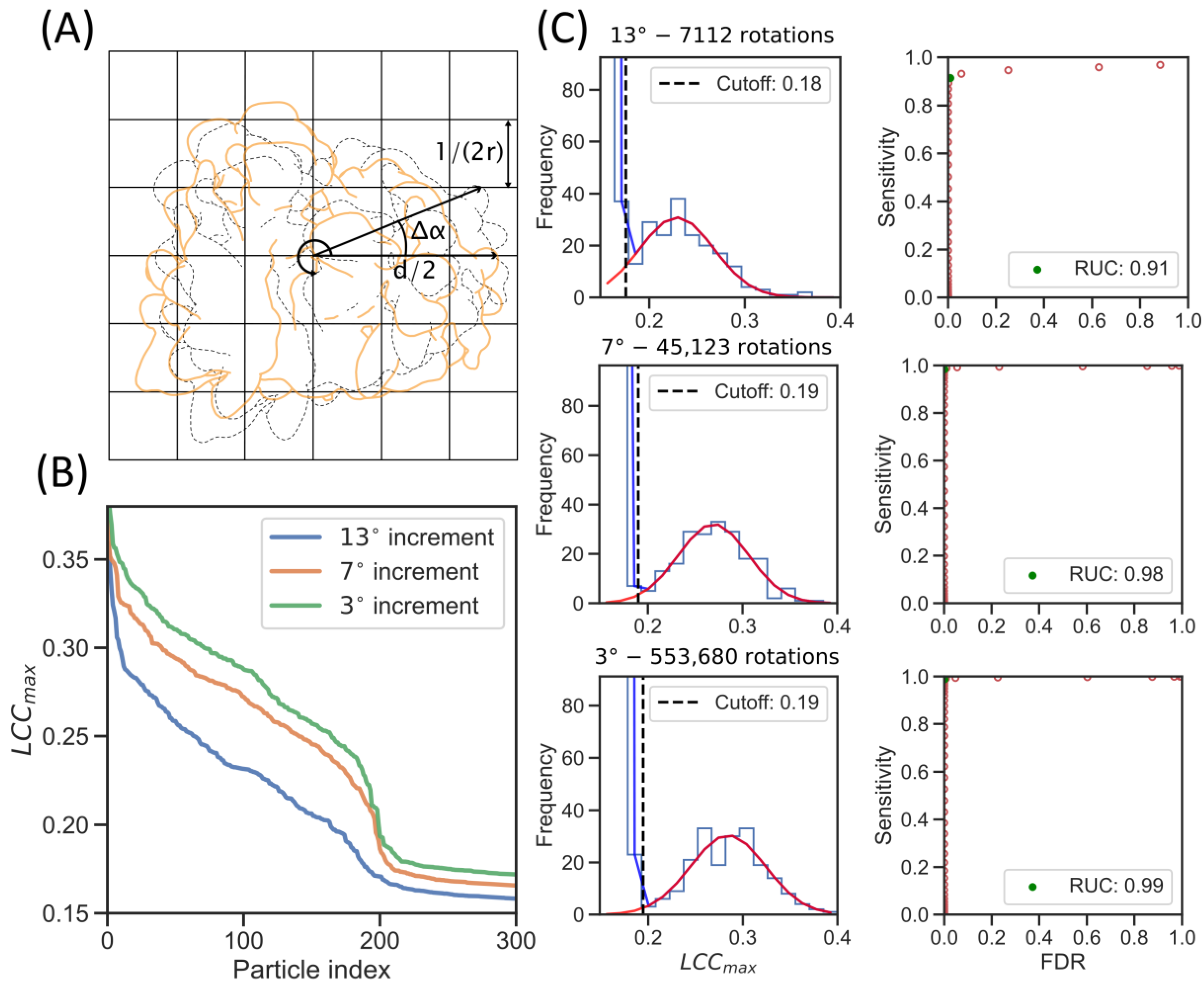
2.2. GPU Acceleration Enables Enhanced Rotational Sampling in TM
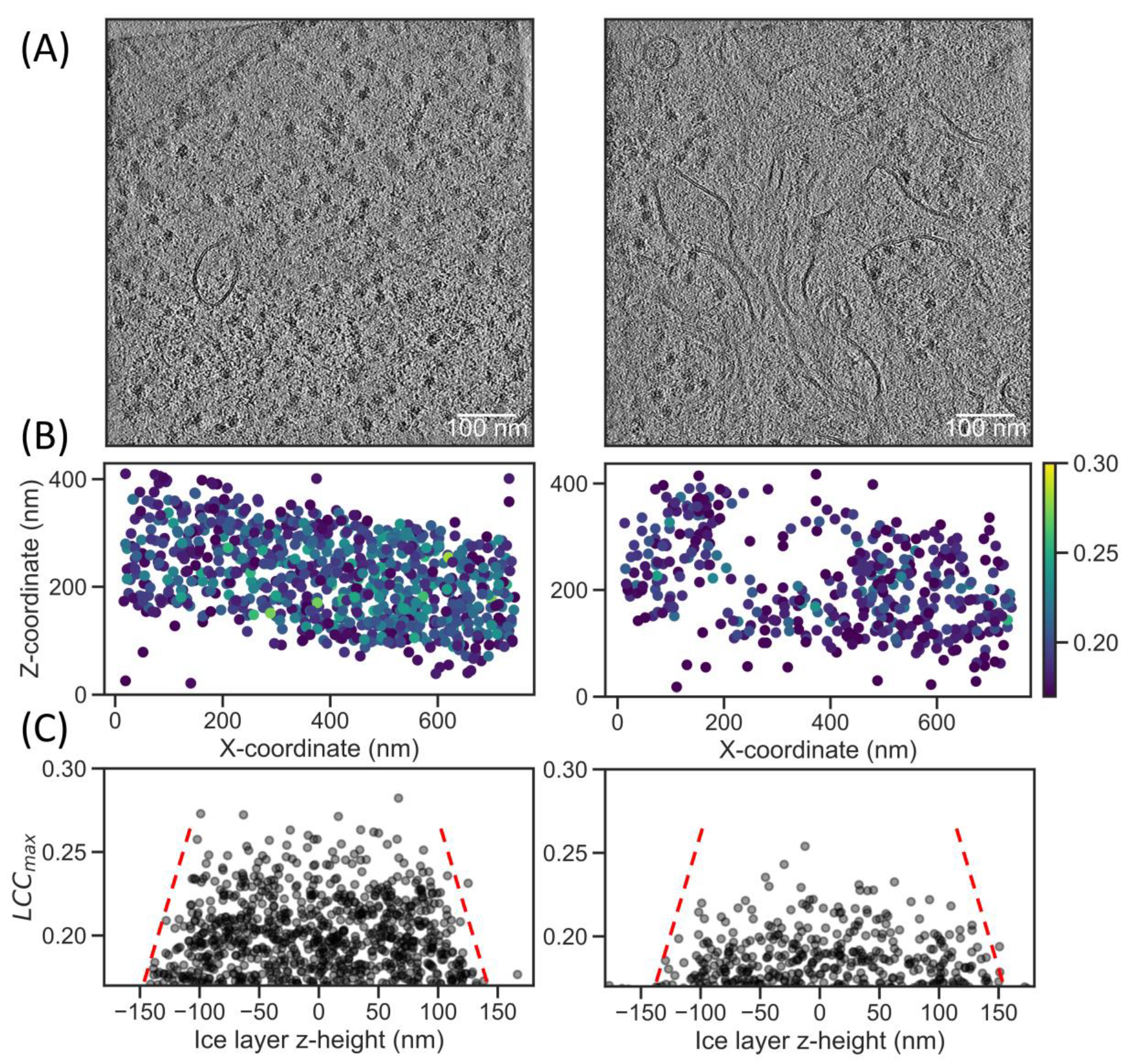
2.3. Increased Angular Sampling Increases Detection Specificity
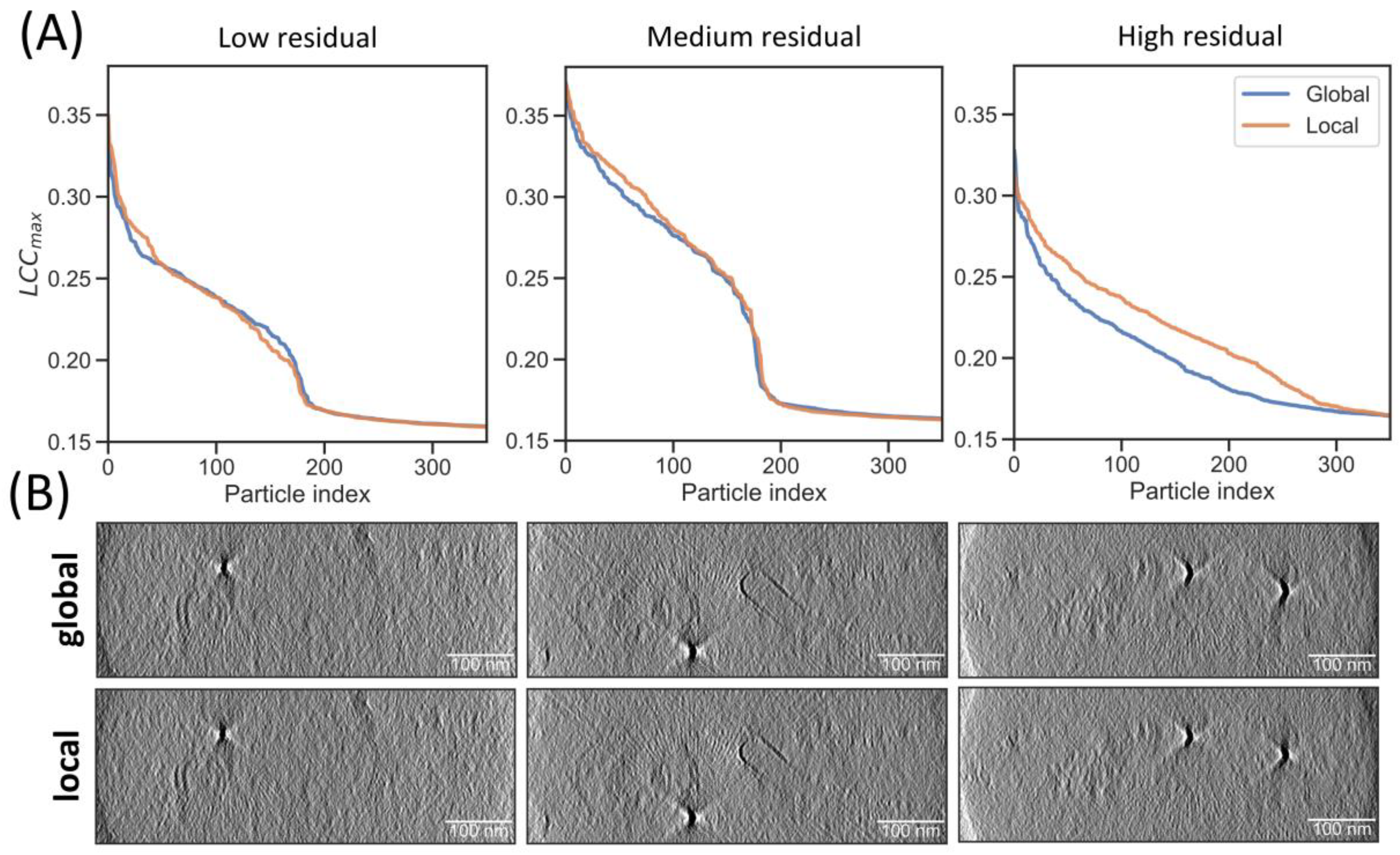
2.4. Local Tilt Series Alignment Increases Particle Detection Fidelity
2.5. Correlation Scores in Lamella Correlate with FIB Beam Damage
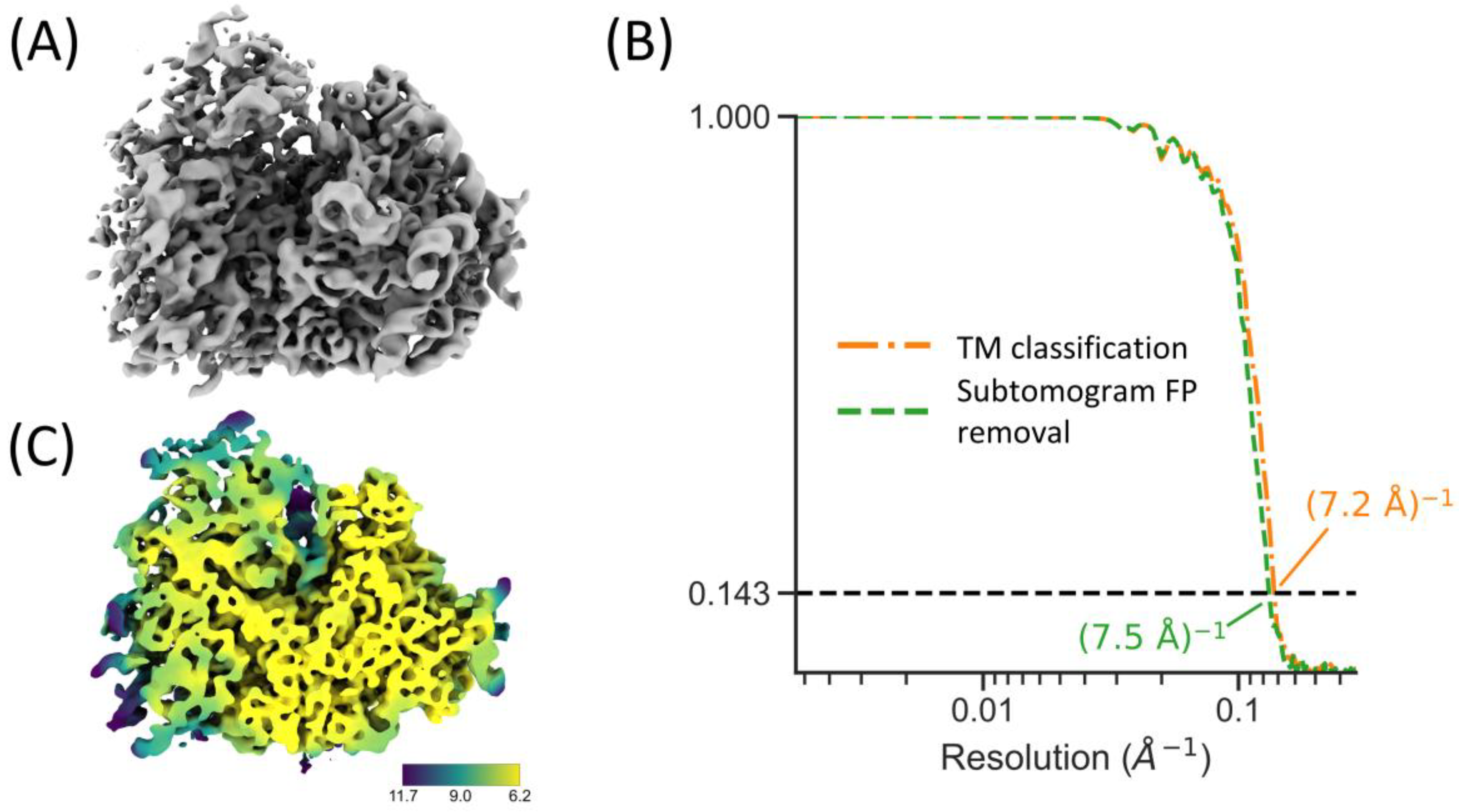
2.6. Integration with RELION and M for High-Resolution STA
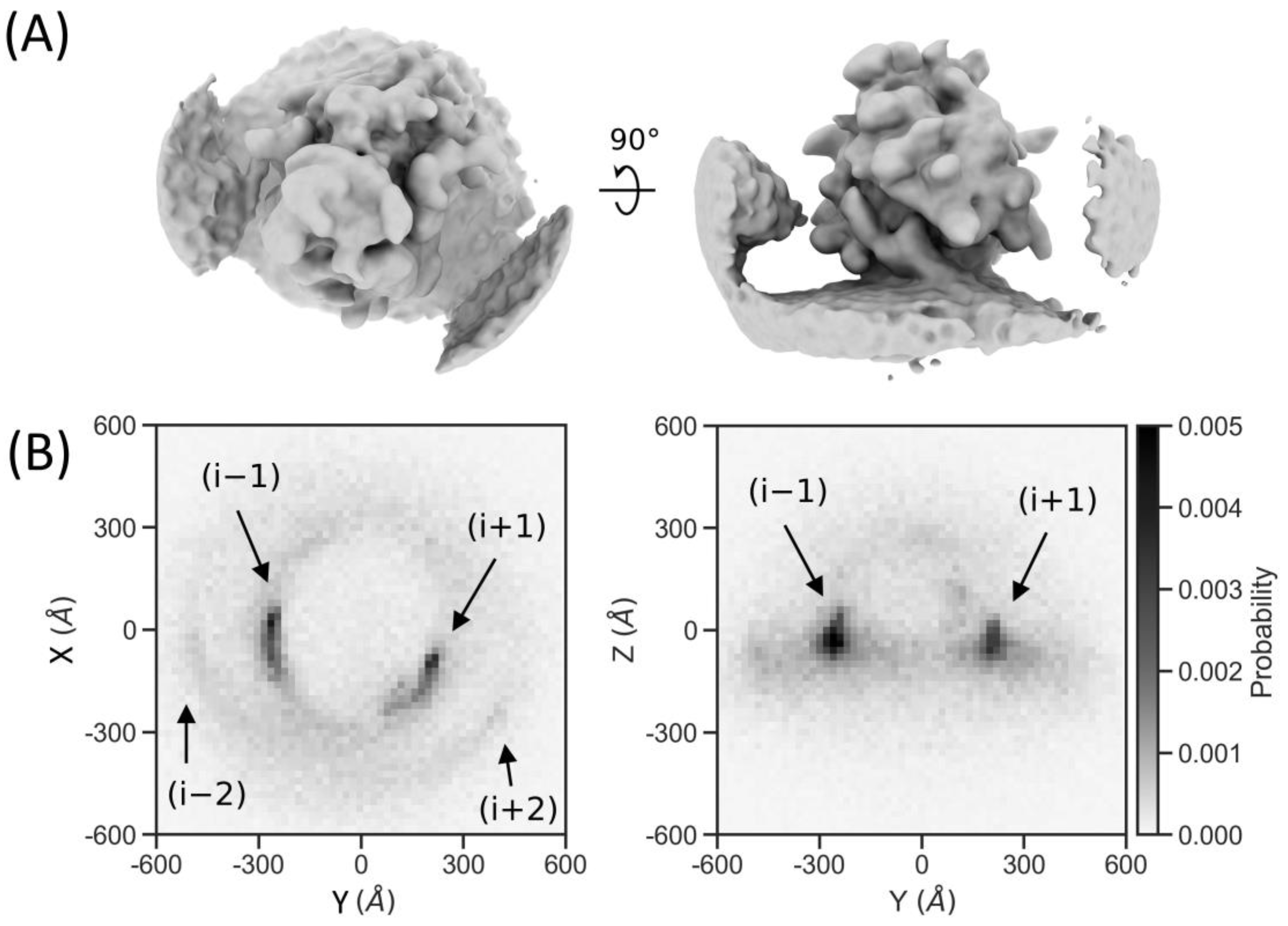
2.7. Particle Detection Is Sufficiently Sensitive to Study Molecular Sociology
3. Discussion
3.1. Extensive Rotation Sampling and Integration into Tomography Workflows
3.2. Sensitivity of TM to Tomogram Quality
3.3. TM with 3D CTF and Dose Weighting
3.4. 2D vs. 3D TM
3.5. Outlook in Relation to Deep-Learning-Based Particle Picking
4. Materials and Methods
4.1. Preprocessing
4.2. Tomogram Alignment and Reconstruction
4.3. Tomogram Denoising
4.4. GPU Template Matching
4.5. Particle Localization
4.6. True Positive Estimation
4.7. Particle List File Conversion
4.8. Subtomogram Averaging
4.9. Neighbor Density Plotting
4.10. Particle Atlas Visualization
4.11. Code Availability
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lucic, V.; Forster, F.; Baumeister, W. Structural studies by electron tomography: From cells to molecules. Annu. Rev. Biochem. 2005, 74, 833–865. [Google Scholar] [CrossRef]
- Mahamid, J.; Pfeffer, S.; Schaffer, M.; Villa, E.; Danev, R.; Cuellar, L.K.; Förster, F.; Hyman, A.A.; Plitzko, J.M.; Baumeister, W. Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science 2016, 351, 969–972. [Google Scholar] [CrossRef] [PubMed]
- Gemmer, M.; Chaillet, M.L.; van Loenhout, J.; Cuevas Arenas, R.; Vismpas, D.; Grollers-Mulderij, M.; Koh, F.A.; Albanese, P.; Scheltema, R.A.; Howes, S.C.; et al. Visualization of translation and protein biogenesis at the ER membrane. Nature 2023, 614, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Baumeister, W. Cryo-Electron Tomography: Can it Reveal the Molecular Sociology of Cells in Atomic Detail? Trends Cell Biol. 2016, 26, 825–837. [Google Scholar] [CrossRef]
- Asano, S.; Fukuda, Y.; Beck, F.; Aufderheide, A.; Förster, F.; Danev, R.; Baumeister, W. Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 2015, 347, 439–442. [Google Scholar] [CrossRef]
- Förster, F. Subtomogram analysis: The sum of a tomogram’s particles reveals molecular structure in situ. J. Struct. Biol. X 2022, 6, 100063. [Google Scholar] [CrossRef]
- Zivanov, J.; Oton, J.; Ke, Z.; von Kugelgen, A.; Pyle, E.; Qu, K.; Morado, D.; Castano-Diez, D.; Zanetti, G.; Bharat, T.A.M.; et al. A Bayesian approach to single-particle electron cryo-tomography in RELION-4.0. Elife 2022, 11, e83724. [Google Scholar] [CrossRef]
- Tegunov, D.; Xue, L.; Dienemann, C.; Cramer, P.; Mahamid, J. Multi-particle cryo-EM refinement with M visualizes ribosome-antibiotic complex at 3.5 A in cells. Nat. Methods 2021, 18, 186–193. [Google Scholar] [CrossRef]
- Himes, B.A.; Zhang, P. emClarity: Software for high-resolution cryo-electron tomography and subtomogram averaging. Nat. Methods 2018, 15, 955–961. [Google Scholar] [CrossRef] [PubMed]
- Förster, F.; Han, B.G.; Beck, M. Visual Proteomics. Meth. Enzymol. 2010, 483, 215–243. [Google Scholar]
- Frangakis, A.S.; Bohm, J.; Forster, F.; Nickell, S.; Nicastro, D.; Typke, D.; Hegerl, R.; Baumeister, W. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. Proc. Natl. Acad. Sci. USA 2002, 99, 14153–14158. [Google Scholar] [CrossRef]
- Roseman, A.M. Particle finding in electron micrographs using a fast local correlation algorithm. Ultramicroscopy 2003, 94, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Förster, F.; Medalia, O.; Zauberman, N.; Baumeister, W.; Fass, D. Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2005, 102, 4729–4734. [Google Scholar] [CrossRef] [PubMed]
- Gubins, I.; Chaillet, M.L.; van der Schot, G.; Veltkamp, R.C.; Förster, F.; Hao, Y.; Wan, X.; Cui, X.; Zhang, F.; Moebel, E.; et al. SHREC 2020: Classification in cryo-electron tomograms. Comput. Graph. 2020, 91, 279–289. [Google Scholar] [CrossRef]
- Gubins, I.; Chaillet, M.L.; van Der Schot, G.; Trueba, M.C.; Veltkamp, R.C.; Förster, F.; Wang, X.; Kihara, D.; Moebel, E.; Nguyen, N.P.; et al. SHREC 2021: Classification in Cryo-Electron Tomograms; Eurographics Workshop on 3D Object Retrieval, 2021; Biasotti, S.A.D., Roberto, M., Yukun, L., Rosin, P.L., Veltkamp, R.C., Eds.; The Eurographics Association: Saarbrücken, Germany, 2021. [Google Scholar]
- Kimanius, D.; Forsberg, B.O.; Scheres, S.H.; Lindahl, E. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife 2016, 5, e18722. [Google Scholar] [CrossRef] [PubMed]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Vilas, J.L.; Oton, J.; Messaoudi, C.; Melero, R.; Conesa, P.; Ramirez-Aportela, E.; Mota, J.; Martinez, M.; Jimenez, A.; Marabini, R.; et al. Measurement of local resolution in electron tomography. J. Struct. Biol. X 2020, 4, 100016. [Google Scholar] [CrossRef]
- Brilot, A.F.; Chen, J.Z.; Cheng, A.; Pan, J.; Harrison, S.C.; Potter, C.S.; Carragher, B.; Henderson, R.; Grigorieff, N. Beam-induced motion of vitrified specimen on holey carbon film. J. Struct. Biol. 2012, 177, 630–637. [Google Scholar] [CrossRef]
- Fernandez, J.J.; Li, S.; Bharat, T.A.M.; Agard, D.A. Cryo-tomography tilt-series alignment with consideration of the beam-induced sample motion. J. Struct. Biol. 2018, 202, 200–209. [Google Scholar] [CrossRef]
- Fernandez, J.J.; Li, S. TomoAlign: A novel approach to correcting sample motion and 3D CTF in CryoET. J. Struct. Biol. 2021, 213, 107778. [Google Scholar] [CrossRef]
- Zheng, S.W.; Wolff, G.; Greenan, G.; Chen, Z.; Faas, F.G.A.; Barcena, M.; Koster, A.J.; Cheng, Y.F.; Agard, D.A. AreTomo: An integrated software package for automated marker-free, motion-corrected cryo-electron tomographic alignment and reconstruction. J. Struct. Biol.-X 2022, 6, 100068. [Google Scholar] [CrossRef] [PubMed]
- Hrabe, T.; Chen, Y.; Pfeffer, S.; Cuellar, L.K.; Mangold, A.V.; Forster, F. PyTom: A python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. J. Struct. Biol. 2012, 178, 177–188. [Google Scholar] [CrossRef]
- Tegunov, D.; Cramer, P. Real-time cryo-electron microscopy data preprocessing with Warp. Nat. Methods 2019, 16, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hrabe, T.; Pfeffer, S.; Pauly, O.; Mateus, D.; Navab, N.; Förster, F. Detection and identification of macromolecular complexes in cryo-electron tomograms using support vector machines. In Proceedings of the 2012 9th IEEE International Symposium on Biomedical Imaging (ISBI), Barcelona, Spain, 2–5 May 2012; pp. 1373–1376. [Google Scholar]
- Crowther, R.A.; Derosier, D.J.; Klug, A. Reconstruction of 3 Dimensional Structure from Projections and Its Application to Electron Microscopy. Proc. R. Soc. Lond. Ser. A 1970, 317, 319–340. [Google Scholar]
- Mastronarde, D.N. Dual-axis tomography: An approach with alignment methods that preserve resolution. J. Struct. Biol. 1997, 120, 343–352. [Google Scholar] [CrossRef]
- Lucas, B.A.; Grigorieff, N. Quantification of gallium cryo-FIB milling damage in biological lamellae. Proc. Natl. Acad. Sci. USA 2023, 120, e2301852120. [Google Scholar] [CrossRef]
- Berger, C.; Dumoux, M.; Glen, T.; Yee, N.B.; Mitchels, J.M.; Patakova, Z.; Darrow, M.C.; Naismith, J.H.; Grange, M. Plasma FIB milling for the determination of structures in situ. Nat. Commun. 2023, 14, 629. [Google Scholar] [CrossRef]
- Jiang, W.; Wagner, J.; Du, W.; Plitzko, J.; Baumeister, W.; Beck, F.; Guo, Q. A transformation clustering algorithm and its application in polyribosomes structural profiling. Nucleic Acids Res. 2022, 50, 9001–9011. [Google Scholar] [CrossRef] [PubMed]
- Fedry, J.; Silva, J.; Vanevic, M.; Fronik, S.; Mechulam, Y.; Schmitt, E.; des Georges, A.; Faller, W.; Forster, F. Visualization of translation reorganization upon persistent collision stress in mammalian cells. bioRxiv 2023. [Google Scholar] [CrossRef]
- Xue, L.; Lenz, S.; Zimmermann-Kogadeeva, M.; Tegunov, D.; Cramer, P.; Bork, P.; Rappsilber, J.; Mahamid, J. Visualizing translation dynamics at atomic detail inside a bacterial cell. Nature 2022, 610, 205–211. [Google Scholar] [CrossRef]
- Xing, H.; Taniguchi, R.; Khusainov, I.; Kreysing, J.P.; Welsch, S.; Turoňová, B.; Beck, M. Translation dynamics in human cells visualized at high-resolution reveal cancer drug action. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hoffmann, P.C.; Kreysing, J.P.; Khusainov, I.; Tuijtel, M.W.; Welsch, S.; Beck, M. Structures of the eukaryotic ribosome and its translational states in situ. Nat. Commun. 2022, 13, 7435. [Google Scholar] [CrossRef] [PubMed]
- Rickgauer, J.P.; Grigorieff, N.; Denk, W. Single-protein detection in crowded molecular environments in cryo-EM images. Elife 2017, 6, e25648. [Google Scholar] [CrossRef]
- Lucas, B.A.; Himes, B.A.; Xue, L.; Grant, T.; Mahamid, J.; Grigorieff, N. Locating macromolecular assemblies in cells by 2D template matching with cisTEM. Elife 2021, 10, e68946. [Google Scholar] [CrossRef] [PubMed]
- Lucas, B.A.; Zhang, K.; Loerch, S.; Grigorieff, N. In situ single particle classification reveals distinct 60S maturation intermediates in cells. Elife 2022, 11, e79272. [Google Scholar] [CrossRef] [PubMed]
- Moebel, E.; Martinez-Sanchez, A.; Lamm, L.; Righetto, R.D.; Wietrzynski, W.; Albert, S.; Lariviere, D.; Fourmentin, E.; Pfeffer, S.; Ortiz, J.; et al. Deep learning improves macromolecule identification in 3D cellular cryo-electron tomograms. Nat. Methods 2021, 18, 1386–1394. [Google Scholar] [CrossRef]
- Rice, G.; Wagner, T.; Stabrin, M.; Sitsel, O.; Prumbaum, D.; Raunser, S. TomoTwin: Generalized 3D localization of macromolecules in cryo-electron tomograms with structural data mining. Nat. Methods 2023, 20, 871–880. [Google Scholar] [CrossRef]
- de Teresa-Trueba, I.; Goetz, S.K.; Mattausch, A.; Stojanovska, F.; Zimmerli, C.E.; Toro-Nahuelpan, M.; Cheng, D.W.C.; Tollervey, F.; Pape, C.; Beck, M.; et al. Convolutional networks for supervised mining of molecular patterns within cellular context. Nat. Methods 2023, 20, 284–294. [Google Scholar] [CrossRef]
- Purnell, C.; Heebner, J.; Swulius, M.T.; Hylton, R.; Kabonick, S.; Grillo, M. Rapid Synthesis of Cryo-ET Data for Training Deep Learning Models. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hagen, W.J.; Wan, W.; Briggs, J.A. Implementation of a cryo-electron tomography tilt-scheme optimized for high resolution subtomogram averaging. J. Struct. Biol. 2016, 197, 191–198. [Google Scholar] [CrossRef]
- Zheng, S.Q.; Palovcak, E.; Armache, J.P.; Verba, K.A.; Cheng, Y.; Agard, D.A. MotionCor2: Anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 2017, 14, 331–332. [Google Scholar] [CrossRef]
- Buchholz, T.O.; Krull, A.; Shahidi, R.; Pigino, G.; Jekely, G.; Jug, F. Content-aware image restoration for electron microscopy. Methods Cell Biol. 2019, 152, 277–289. [Google Scholar] [PubMed]
- Okuta, R.; Unno, Y.; Nishino, D.; Hido, S.; Loomis, C. CuPy: A NumPy-Compatible Library for NVIDIA GPU Calculations. In Proceedings of the 31st Conference on Neural Information Processing Systems, Tokyo, Japan, 8 December 2017. [Google Scholar]
- Khatter, H.; Myasnikov, A.G.; Natchiar, S.K.; Klaholz, B.P. Structure of the human 80S ribosome. Nature 2015, 520, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.M.; Russo, C.J.; Lowe, J.; Passmore, L.A.; Scheres, S.H.W. Advances in Single-Particle Electron Cryomicroscopy Structure Determination applied to Sub-tomogram Averaging. Structure 2015, 23, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Ermel, U.H.; Arghittu, S.M.; Frangakis, A.S. ArtiaX: An electron tomography toolbox for the interactive handling of sub-tomograms in UCSF ChimeraX. Protein Sci. 2022, 31, e4472. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Collection Parameters | Angular Increment (°) | Cut-Off (LCCmax) | Median (LCCmax) | Sensitivity |
|---|---|---|---|---|
| ~160 nm ice layers, 200 keV—K2 Summit, ER microsomes of HEK cells | 13 | 0.18 | 0.23 | 0.91 |
| 7 | 0.19 | 0.27 | 0.99 | |
| 3 | 0.19 | 0.29 | 0.99 |
| Collection Parameters | Angular Increment (°) | Cut-Off (LCCmax) | Median (LCCmax) | Sensitivity |
|---|---|---|---|---|
| ~300 nm ice layers, 300 keV—Falcon 4, lamellae of HeLa cells | 3 | 0.17 | 0.19 | 0.79 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaillet, M.L.; van der Schot, G.; Gubins, I.; Roet, S.; Veltkamp, R.C.; Förster, F. Extensive Angular Sampling Enables the Sensitive Localization of Macromolecules in Electron Tomograms. Int. J. Mol. Sci. 2023, 24, 13375. https://doi.org/10.3390/ijms241713375
Chaillet ML, van der Schot G, Gubins I, Roet S, Veltkamp RC, Förster F. Extensive Angular Sampling Enables the Sensitive Localization of Macromolecules in Electron Tomograms. International Journal of Molecular Sciences. 2023; 24(17):13375. https://doi.org/10.3390/ijms241713375
Chicago/Turabian StyleChaillet, Marten L., Gijs van der Schot, Ilja Gubins, Sander Roet, Remco C. Veltkamp, and Friedrich Förster. 2023. "Extensive Angular Sampling Enables the Sensitive Localization of Macromolecules in Electron Tomograms" International Journal of Molecular Sciences 24, no. 17: 13375. https://doi.org/10.3390/ijms241713375
APA StyleChaillet, M. L., van der Schot, G., Gubins, I., Roet, S., Veltkamp, R. C., & Förster, F. (2023). Extensive Angular Sampling Enables the Sensitive Localization of Macromolecules in Electron Tomograms. International Journal of Molecular Sciences, 24(17), 13375. https://doi.org/10.3390/ijms241713375