
Scipion-EM-ProDy: A Graphical Interface for the ProDy Python Package within the Scipion Workflow Engine Enabling Integration of Databases, Simulations and Cryo-Electron Microscopy Image Processing


Abstract
1. Introduction
1.1. The Scipion Flexibility Hub Solves Recent Solutions to Challenges with Cryo-EM Continuous Heterogeneity Analysis
1.2. Scipion-EM-ProDy: A New Scipion Plugin for Better Interpretation and Simulation of Atomic Structures through Rapid Computational Biophysics
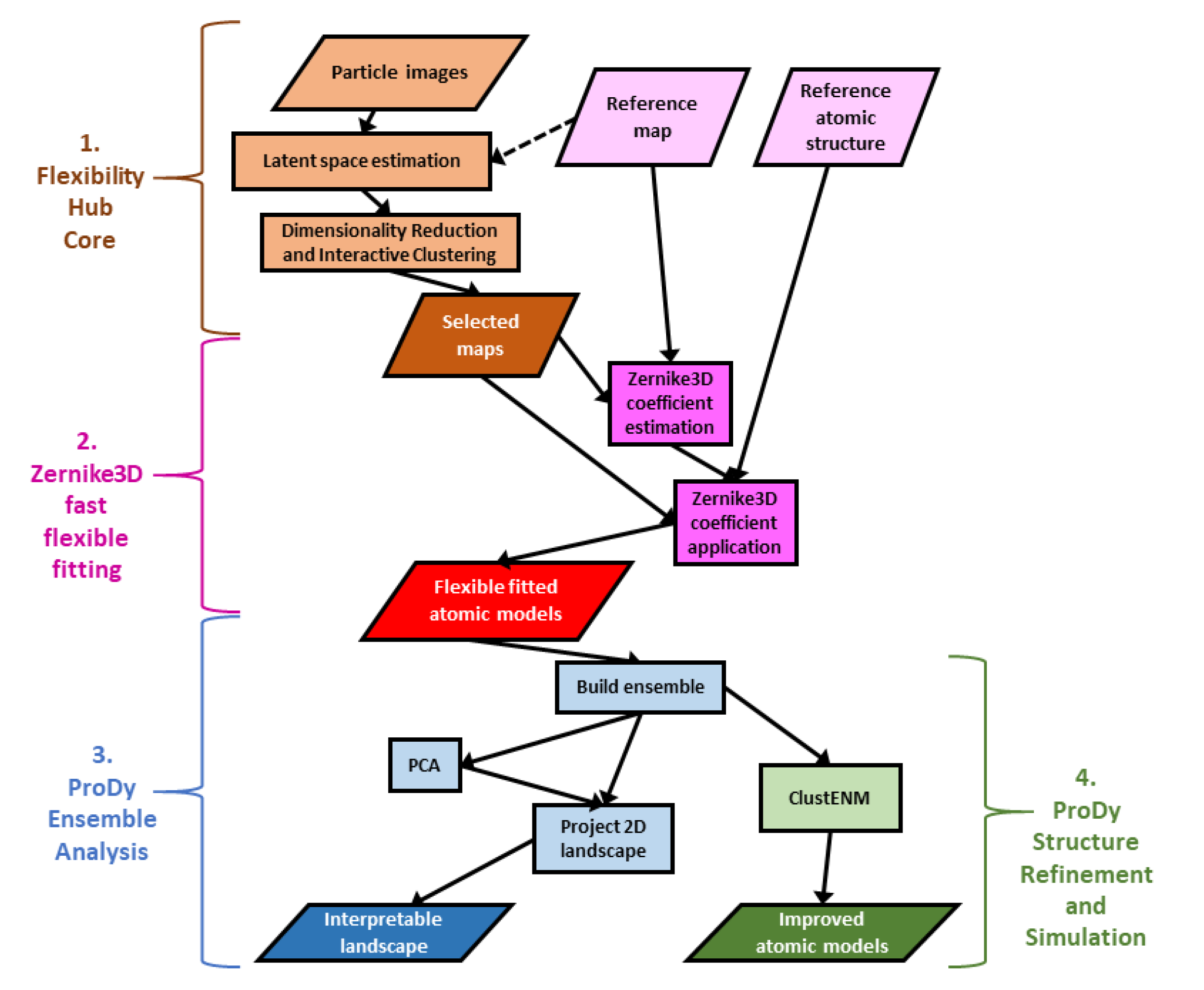
2. Results
2.1. General Overview of Scipion and Scipion-EM-ProDy for Building Workflows for Computational Biophysics
2.2. Ensemble Analysis via PCA Downstream of Flexibility Hub Enables Interpretation of Cryo-EM Conformational Landscapes
2.3. A New ClustENM(D) Protocol for Refining Atomic Models and Hybrid Simulations
2.4. A Combined Workflow for Comparing Structures from Experiments and Simulations
3. Discussion
4. Materials and Methods
4.1. Integration of ProDy Pipelines into Scipion Workflows
4.1.1. Building upon ProDy Classes, Functions and Apps to Create Scipion Protocols and Workflows
4.1.2. Protocols for Atomic Structure Operations
4.1.2.1. Reconstructing Biological Molecular Assemblies
4.1.2.2. Atom Selection
4.1.3. Pairwise Alignment and Ensemble Construction
4.1.4. Protocols for Calculating Global Modes of Motion
4.1.4.1. Deformation Vector Analysis
4.1.4.2. Principal Component Analysis
4.1.4.3. Normal Mode Analysis Protocols
4.2. Gaussian Network Model Analysis and Domain Decomposition
4.2.1. Protocols for Downstream Analysis
4.2.1.1. Mode Editing
4.2.1.2.Mode Comparison
4.2.1.3.Landscape Projection
4.2.2. ClustENM(D) Hybrid Simulations
4.2.3. Protocols for Imports
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ANM | Anisotropic network analysis |
| API | Application programming interface |
| PDB | Protein data bank |
| CE | Combinatorial extension |
| Cryo-EM | Cryo-electron microscopy |
| GNM | Gaussian network analysis |
| I | Intermediate state for RBD |
| NMA | Normal mode analysis |
| NM | Normal mode |
| NMWiz | Normal mode wizard |
| PCA | Principal component analysis |
| PC | Principal component |
| ps | Picoseconds |
| PDB | Protein data bank |
| RBD | Receptor-binding domain |
| RMSD | Root-mean-square deviation |
| RTB | Rotating and translating blocks |
| SPA | Single particle analysis |
| VMD | Visual molecular dynamics |
References
- Henzler-Wildman, K.; Kern, D. Dynamic personalities of proteins. Nature 2007, 450, 964–972. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Cheng, M.H.; Lee, J.Y.; Kaya, C.; Zhang, S. Structure-Encoded Global Motions and Their Role in Mediating Protein-Substrate Interactions. Biophys. J. 2015, 109, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Harpole, T.; Delemotte, L. Conformational landscapes of membrane proteins delineated by enhanced sampling molecular dynamics simulations. Biochim. Biophys Acta Biomembr. 2018, 1860, 909–926. [Google Scholar] [CrossRef] [PubMed]
- Van den Bedem, H.; Fraser, J. Integrative, dynamic structural biology at atomic resolution—It is about time. Nat. Methods 2015, 12, 307–318. [Google Scholar] [CrossRef]
- Srivastava, A.; Tiwari, S.; Miyashita, O.; Tama, F. Integrative/Hybrid Modeling Approaches for Studying Biomolecules. J. Mol. Biol. 2020, 432, 2846–2860. [Google Scholar] [CrossRef] [PubMed]
- Toader, B.; Sigworth, F.J.; Lederman, R.R. Methods for Cryo-EM Single Particle Reconstruction of Macromolecules Having Continuous Heterogeneity. J. Mol. Biol. 2023, 435, 168020. [Google Scholar] [CrossRef] [PubMed]
- Bonomi, M.; Vendruscolo, M. Determination of protein structural ensembles using cryo-electron microscopy. Curr. Opin. Struct. Biol. 2019, 56, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.; Jiménez, A.; Mota, J.; Vilas, J.; Maluenda, D.; Martínez, M.; Ramírez-Aportela, E.; Majtner, T.; Segura, J.; Sánchez-García, R.; et al. Survey of the analysis of continuous conformational variability of biological macromolecules by electron microscopy. Acta Crystallogr. F 2019, 75, 19–32. [Google Scholar] [CrossRef]
- Donnat, C.; Levy, A.; Poitevin, F.; Zhong, E.; Miolane, N. Deep generative modeling for volume reconstruction in cryo-electron microscopy. J. Struct. Biol. 2022, 214, 107920. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.S.; Zhong, E.D.; Hanson, S.M.; Thiede, E.H.; Cossio, P. Conformational heterogeneity and probability distributions from single-particle cryo-electron microscopy. Curr. Opin. Struct. Biol. 2023, 81, 102626. [Google Scholar] [CrossRef]
- Zhong, E.; Bepler, T.; Berger, B.; Davis, J. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Herreros, D.; Lederman, R.; Krieger, J.; Jiménez-Moreno, A.; Martínez, M.; Myška, D.; Strelak, D.; Filipovic, J.; Bahar, I.; Carazo, J.; et al. Approximating deformation fields for the analysis of continuous heterogeneity of biological macromolecules by 3D Zernike polynomials. IUCrJ 2021, 8, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Harastani, M.; Vuillemot, R.; Hamitouche, I.; Moghadam, N.B.; Jonic, S. ContinuousFlex: Software package for analyzing continuous conformational variability of macromolecules in cryo electron microscopy and tomography data. J. Struct. Biol. 2022, 214, 107906. [Google Scholar] [CrossRef] [PubMed]
- Punjani, A.; Fleet, D. 3DFlex: Determining structure and motion of flexible proteins from cryo-EM. Nat. Methods 2023, 20, 860–870. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.; Grubmüller, H. Effects of cryo-EM cooling on structural ensembles. Nat. Commun. 2022, 13, 1709. [Google Scholar] [CrossRef]
- Klebl, D.; Gravett, M.; Kontziampasis, D.; Wright, D.; Bon, R.; Monteiro, D.; Trebbin, M.; Sobott, F.; White, H.; Darrow, M.; et al. Need for Speed: Examining Protein Behavior during CryoEM Grid Preparation at Different Timescales. Structure 2020, 28, 1238–1248.e4. [Google Scholar] [CrossRef] [PubMed]
- Sanchez Sorzano, C.O.; Alvarez-Cabrera, A.L.; Kazemi, M.; Carazo, J.M.; Jonić, S. StructMap: Elastic Distance Analysis of Electron Microscopy Maps for Studying Conformational Changes. Biophys. J. 2016, 110, 1753–1765. [Google Scholar] [CrossRef] [PubMed]
- Sorzano, C.; Martin-Ramos, A.; Prieto, F.; Melero, R.; Martin-Benito, J.; Jonic, S.; Navas-Calvente, J.; Vargas, J.; Oton, J.; Abrishami, V.; et al. Local analysis of strains and rotations for macromolecular electron microscopy maps. J. Struct. Biol. 2016, 195, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.; Goddard, T.; Huang, C.; Meng, E.; Couch, G.; Croll, T.; Morris, J.; Ferrin, T. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Ecoffet, A.; Poitevin, F.; Dao Duc, K. MorphOT: Transport-based interpolation between EM maps with UCSF ChimeraX. Bioinformatics 2021, 36, 5528–5529. [Google Scholar] [CrossRef] [PubMed]
- Kinman, L.; Powell, B.; Zhong, E.; Berger, B.; Davis, J. Uncovering structural ensembles from single-particle cryo-EM data using cryoDRGN. Nat. Protoc. 2023, 18, 319–339. [Google Scholar] [CrossRef]
- Malhotra, S.; Träger, S.; Dal Peraro, M.; Topf, M. Modelling structures in cryo-EM maps. Curr. Opin. Struct. Biol. 2019, 58, 105–114. [Google Scholar] [CrossRef]
- Afonine, P.; Gobet, A.; Moissonnier, L.; Martin, J.; Poon, B.; Chaptal, V. Conformational space exploration of cryo-EM structures by variability refinement. Biochim. Biophys. Acta Biomembr. 2023, 1865, 184133. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Sorzano, C.; de la Rosa-Trevín, J.; Bilbao-Castro, J.; Núñez Ramírez, R.; Llorca, O.; Tama, F.; Jonić, S. Iterative Elastic 3D-to-2D Alignment Method Using Normal Modes for Studying Structural Dynamics of Large Macromolecular Complexes. Structure 2014, 22, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Harastani, M.; Eltsov, M.; Leforestier, A.; Jonić, S. HEMNMA-3D: Cryo Electron Tomography Method Based on Normal Mode Analysis to Study Continuous Conformational Variability of Macromolecular Complexes. Front. Mol. Biosci. 2021, 8, 663121. [Google Scholar] [CrossRef]
- Vuillemot, R.; Miyashita, O.; Tama, F.; Rouiller, I.; Jonic, S. NMMD: Efficient Cryo-EM Flexible Fitting Based on Simultaneous Normal Mode and Molecular Dynamics atomic displacements. J. Mol. Biol. 2022, 434, 167483. [Google Scholar] [CrossRef]
- Vuillemot, R.; Mirzaei, A.; Harastani, M.; Hamitouche, I.; Fréchin, L.; Klaholz, B.; Miyashita, O.; Tama, F.; Rouiller, I.; Jonic, S. MDSPACE: Extracting Continuous Conformational Landscapes from Cryo-EM Single Particle Datasets Using 3D-to-2D Flexible Fitting based on Molecular Dynamics Simulation. J. Mol. Biol. 2023, 435, 167951. [Google Scholar] [CrossRef]
- Cossio, P.; Hummer, G. Bayesian analysis of individual electron microscopy images: Towards structures of dynamic and heterogeneous biomolecular assemblies. J. Struct. Biol. 2013, 184, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Giraldo-Barreto, J.; Ortiz, S.; Thiede, E.; Palacio-Rodriguez, K.; Carpenter, B.; Barnett, A.; Cossio, P. A Bayesian approach to extracting free-energy profiles from cryo-electron microscopy experiments. Sci. Rep. 2021, 11, 13657. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Silva-Sánchez, D.; Giraldo-Barreto, J.; Carpenter, B.; Hanson, S.; Barnett, A.; Thiede, E.; Cossio, P. Ensemble Reweighting Using Cryo-EM Particle Images. J. Phys. Chem. B 2023, 127, 5410–5421. [Google Scholar] [CrossRef] [PubMed]
- Hamitouche, I.; Jonić, S. DeepHEMNMA: ResNet-based hybrid analysis of continuous conformational heterogeneity in cryo-EM single particle images. Front. Mol. Biosci. 2022, 9, 965645. [Google Scholar] [CrossRef] [PubMed]
- Herreros, D.; Krieger, J.M.; Fonseca, Y.; Conesa, P.; Harastani, M.; Vuillemot, R.; Hamitouche, I.; Serrano Gutiérrez, R.; Gragera, M.; Melero, R.; et al. Scipion Flexibility Hub: An integrative framework for advanced analysis of conformational heterogeneity in cryoEM. Acta Crystallogr. Sect. D 2023, 79, 569–584. [Google Scholar] [CrossRef]
- Conesa, P.; Fonseca, Y.; Jiménez de la Morena, J.; Sharov, G.; de la Rosa-Trevín, J.; Cuervo, A.; García Mena, A.; Rodríguez de Francisco, B.; del Hoyo, D.; Herreros, D.; et al. Scipion3: A workflow engine for cryo-electron microscopy image processing and structural biology. Biol. Imaging 2023, 3, e13. [Google Scholar] [CrossRef]
- Jimenez-Moreno, A.; Del Cano, L.; Martinez, M.; Ramirez-Aportela, E.; Cuervo, A.; Melero, R.; Sanchez-Garcia, R.; Strelak, D.; Fernandez-Gimenez, E.; de Isidro-Gomez, F.P.; et al. Cryo-EM and Single-Particle Analysis with Scipion. J. Vis. Exp. 2021, 171, e62261. [Google Scholar]
- Harastani, M.; Sorzano, C.O.S.; Jonic, S. Hybrid Electron Microscopy Normal Mode Analysis with Scipion. Protein Sci. 2020, 29, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Herreros, D.; Lederman, R.; Krieger, J.; Jiménez-Moreno, A.; Martínez, M.; Myška, D.; Strelak, D.; Filipovic, J.; Sorzano, C.; Carazo, J. Estimating conformational landscapes from Cryo-EM particles by 3D Zernike polynomials. Nat. Commun. 2023, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Orozco, M. A theoretical view of protein dynamics. Chem. Soc. Rev. 2014, 43, 5051–5066. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, S.; Dror, R. Molecular Dynamics Simulation for All. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.; Piggot, T.; Allison, J. Molecular Dynamics Simulation of Proteins. Methods Mol. Biol. 2020, 2073, 311–327. [Google Scholar] [CrossRef] [PubMed]
- Hénin, J.; Lelièvre, T.; Shirts, M.R.; Valsson, O.; Delemotte, L. Enhanced Sampling Methods for Molecular Dynamics Simulations [Article v1.0]. Living J. Comput. Mol. Sci. 2022, 4, 1583. [Google Scholar] [CrossRef]
- Krieger, J.; Doruker, P.; Scott, A.; Perahia, D.; Bahar, I. Towards gaining sight of multiscale events: Utilizing network models and normal modes in hybrid methods. Curr. Opin. Struct. Biol. 2020, 64, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, C. Chapter Two—Computational Methods for Efficient Sampling of Protein Landscapes and Disclosing Allosteric Regions. In Advances in Protein Chemistry and Structural Biology; Karabencheva-Christova, T.G., Christov, C.Z., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 113, pp. 33–63. [Google Scholar] [CrossRef]
- Dill, K.; Jernigan, R.L.; Bahar, I. Protein Actions: Principles and Modeling; Garland Science: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased Scale and Scope after 10 Years of Protein Dynamics Modelling with Python. Bioinformatics 2021, 37, 3657–3659. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.R.; Bakan, A.; Shrivastava, I.H. Normal Mode Analysis of Biomolecular Structures: Functional Mechanisms of Membrane Proteins. Chem. Rev. 2010, 110, 1463–1497. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Skjaerven, L.; Yao, X.Q. The Bio3D packages for structural bioinformatics. Protein Sci. 2021, 30, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Sanejouand, Y.H. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef]
- Tiwari, S.P.; Fuglebakk, E.; Hollup, S.M.; Skjaerven, L.; Cragnolini, T.; Grindhaug, S.H.; Tekle, K.M.; Reuter, N. WEBnm@ v2.0: Web server and services for comparing protein flexibility. BMC Bioinform. 2014, 15, 427. [Google Scholar] [CrossRef]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar] [CrossRef]
- Kaynak, B.T.; Zhang, S.; Bahar, I.; Doruker, P. ClustENMD: Efficient sampling of biomolecular conformational space at atomic resolution. Bioinformatics 2021, 37, 3956–3958. [Google Scholar] [CrossRef] [PubMed]
- Bakan, A.; Dutta, A.; Mao, W.; Liu, Y.; Chennubhotla, C.; Lezon, T.R.; Bahar, I. Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gur, M.; Madura, J.; Bahar, I. Global transitions of proteins explored by a multiscale hybrid methodology: Application to adenylate kinase. Biophys. J. 2013, 105, 1643–1652. [Google Scholar] [CrossRef]
- Phillips, J.; Hardy, D.; Maia, J.; Stone, J.; Ribeiro, J.; Bernardi, R.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, B.; Roel-Touris, J.; Romero-Durana, M.; Vidal, M.; Jiménez-González, D.; Fernández-Recio, J. LightDock: A new multi-scale approach to protein–protein docking. Bioinformatics 2018, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Atilgan, A.R.; Erman, B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential. Fold. Des. 1997, 2, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Lezon, T.R.; Bahar, I. Using entropy maximization to understand the determinants of structural dynamics beyond native contact topology. PLoS Comput. Biol. 2010, 6, e1000816. [Google Scholar] [CrossRef] [PubMed]
- Orellana, L.; Rueda, M.; Ferrer-Costa, C.; Lopez-Blanco, J.; Chacón, P.; Orozco, M. Approaching Elastic Network Models to Molecular Dynamics Flexibility. J. Chem. Theory Comput. 2010, 6, 2910–2923. [Google Scholar] [CrossRef]
- Durand, P.; Trinquier, G.; Sanejouand, Y.H. A new approach for determining low-frequency normal modes in macromolecules. Biopolymers 1994, 34, 759–771. [Google Scholar] [CrossRef]
- Tama, F.; Gadea, F.X.; Marques, O.; Sanejouand, Y.H. Building-block approach for determining low-frequency normal modes of macromolecules. Proteins 2000, 41, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Krieger, J.; Mikulska-Ruminska, K.; Kaynak, B.; Sorzano, C.O.S.; Carazo, J.; Xing, J.; Bahar, I. State-dependent sequential allostery exhibited by chaperonin TRiC/CCT revealed by network analysis of Cryo-EM maps. Prog. Biophys. Mol. Biol. 2021, 160, 104–120. [Google Scholar] [CrossRef]
- Kurkcuoglu, Z.; Bahar, I.; Doruker, P. ClustENM: ENM-Based Sampling of Essential Conformational Space at Full Atomic Resolution. J. Chem. Theory Comput. 2016, 12, 4549–4562. [Google Scholar] [CrossRef]
- Ginex, T.; Marco-Marín, C.; Wieczór, M.; Mata, C.; Krieger, J.; Ruiz-Rodriguez, P.; López-Redondo, M.; Francés-Gómez, C.; Melero, R.; Sánchez-Sorzano, C.; et al. The structural role of SARS-CoV-2 genetic background in the emergence and success of spike mutations: The case of the spike A222V mutation. PLoS Pathog. 2022, 18, e1010631. [Google Scholar] [CrossRef]
- De la Rosa-Trevín, J.; Otón, J.; Marabini, R.; Zaldívar, A.; Vargas, J.; Carazo, J.; Sorzano, C. Xmipp 3.0: An improved software suite for image processing in electron microscopy. J. Struct. Biol. 2013, 184, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Benton, D.; Wrobel, A.; Roustan, C.; Borg, A.; Xu, P.; Martin, S.; Rosenthal, P.; Skehel, J.; Gamblin, S. The effect of the D614G substitution on the structure of the spike glycoprotein of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2021, 118, e2022586118. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.; McGibbon, R.; Zhao, Y.; Beauchamp, K.; Wang, L.; Simmonett, A.; Harrigan, M.; Stern, C.; et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Li, H.; Krieger, J.M.; Bahar, I. Shared Signature Dynamics Tempered by Local Fluctuations Enables Fold Adaptability and Specificity. Mol. Biol. Evol. 2019, 36, 2053–2068. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.; Janowska, K.; McDowell, S.; Mansouri, K.; Parks, R.; Manne, K.; Stalls, V.; Kopp, M.; Henderson, R.; Edwards, R.; et al. D614G Mutation Alters SARS-CoV-2 Spike Conformation and Enhances Protease Cleavage at the S1/S2 Junction. Cell. Rep. 2021, 34, 108630. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.; Sorzano, C.; Carazo, J.; Bahar, I. Protein dynamics developments for the large scale and cryoEM: Case study of ProDy 2.0. Acta Crystallogr. D Struct. Biol. 2022, 78, 399–409. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.; Millman, K.; van der Walt, S.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Krieger, J.; Greger, I.; Bahar, I. Structure, Dynamics, and Allosteric Potential of Ionotropic Glutamate Receptor N-Terminal Domains. Biophys. J. 2015, 109, 1136–1148. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. A solution for the best rotation to relate two sets of vectors. Acta Cryst. 1976, A32, 922–923. [Google Scholar] [CrossRef]
- Holm, L.; Laakso, L. Dali server update. Nucleic Acids Res. 2016, 44, W351–W355. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.; Antao, T.; Chang, J.T.; Chapman, B.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Shindyalov, I.; Bourne, P. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998, 11, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.; Goldsmith, J.; Hsieh, C.; Abiona, O.; Graham, B.; McLellan, J. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.; Park, Y.; Tortorici, M.; Wall, A.; McGuire, A.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Yu, P.; Chang, Y.; Hsu, S. D614G mutation in the SARS-CoV-2 spike protein enhances viral fitness by desensitizing it to temperature-dependent denaturation. J. Biol. Chem. 2021, 297, 101238. [Google Scholar] [CrossRef] [PubMed]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Bahar, I.; Lezon, T.R.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef]
- Tirion, M.M. Low-amplitude elastic motions in proteins from a single-parameter atomic analysis. Phys. Rev. Lett. 1996, 77, 1905–1908. [Google Scholar] [CrossRef] [PubMed]
- Eyal, E.; Yang, L.W.; Bahar, I. Anisotropic network model: Systematic evaluation and a new web interface. Bioinformatics 2006, 22, 2619–2627. [Google Scholar] [CrossRef]
- Doruker, P.; Jernigan, R.L.; Bahar, I. Dynamics of large proteins through hierarchical levels of coarse-grained structures. J. Comput. Chem. 2002, 23, 119–127. [Google Scholar] [CrossRef]
- Sauerwald, N.; Zhang, S.; Kingsford, C.; Bahar, I. Chromosomal dynamics predicted by an elastic network model explains genome-wide accessibility and long-range couplings. Nucleic Acids Res. 2017, 45, 3663–3673. [Google Scholar] [CrossRef] [PubMed]
- Hinsen, K. Analysis of domain motions by approximate normal mode calculations. Proteins 1998, 33, 417–429. [Google Scholar] [CrossRef]
- Ming, D.; Wall, M.E. Allostery in a coarse-grained model of protein dynamics. Phys. Rev. Lett. 2005, 95, 198103. [Google Scholar] [CrossRef]
- Woodcock, H.L.; Zheng, W.; Ghysels, A.; Shao, Y.; Kong, J.; Brooks, B.R. Vibrational subsystem analysis: A method for probing free energies and correlations in the harmonic limit. J. Chem. Phys. 2008, 129, 214109. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, Y.; Zhang, S.; Xing, J.; Bahar, I. Normal mode analysis of membrane protein dynamics using the vibrational subsystem analysis. J. Chem. Phys. 2021, 154, 195102. [Google Scholar] [CrossRef] [PubMed]
- Lezon, T.; Bahar, I. Constraints imposed by the membrane selectively guide the alternating access dynamics of the glutamate transporter GltPh. Biophys. J. 2021, 102, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Stember, J.; Wriggers, W. Bend-twist-stretch model for coarse elastic network simulation of biomolecular motion. J. Chem. Phys. 2009, 131, 074112. [Google Scholar] [CrossRef]
- Fuglebakk, E.; Tiwari, S.; Reuter, N. Comparing the intrinsic dynamics of multiple protein structures using elastic network models. Biochim. Biophys. Acta 2015, 1850, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Marques, O.; Sanejouand, Y. Hinge-bending motion in citrate synthase arising from normal mode calculations. Proteins 1995, 23, 557–560. [Google Scholar] [CrossRef] [PubMed]
- Carnevale, V.; Pontiggia, F.; Micheletti, C. Structural and Dynamical Alignment of Enzymes with Partial Structural Similarity. J. Phys. Condens. Matter 2007, 19, 285206. [Google Scholar] [CrossRef]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E 2002, 65, 031910. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Problem | Solution | Existing Tools | Scipion-EM-ProDy 1 |
|---|---|---|---|
| Most analyses of continuous heterogeneity lack biological interpretability and physical meaning (often separate images and maps by non-structural factors) |
|
|
|
| Large numbers of lower quality maps from continuous heterogeneity are challenging to fit with good structures |
|
|
|
| Need to compare standard cryo-EM and continuous heterogeneity outputs to existing structures and those from simulations, and make sense of results in broader context |
|
|
|
| Scipion Object | ProDy Objects |
|---|---|
| AtomStruct, SetOfAtomStructs | Atomic 1, AtomGroup, Selection |
| NormalMode | VectorBase 1, Vector, Mode |
| SetOfNormalModes | NMA 1, ANM, RTB, GNM |
| SetOfPrincipalComponents 2 | PCA |
| TrajFrame 3 | Conformation, Frame |
| SetOfTrajFrames 3 | Ensemble, Trajectory |
| ProDyNpzEnsemble 3 | PDBEnsemble |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krieger, J.M.; Sorzano, C.O.S.; Carazo, J.M. Scipion-EM-ProDy: A Graphical Interface for the ProDy Python Package within the Scipion Workflow Engine Enabling Integration of Databases, Simulations and Cryo-Electron Microscopy Image Processing. Int. J. Mol. Sci. 2023, 24, 14245. https://doi.org/10.3390/ijms241814245
Krieger JM, Sorzano COS, Carazo JM. Scipion-EM-ProDy: A Graphical Interface for the ProDy Python Package within the Scipion Workflow Engine Enabling Integration of Databases, Simulations and Cryo-Electron Microscopy Image Processing. International Journal of Molecular Sciences. 2023; 24(18):14245. https://doi.org/10.3390/ijms241814245
Chicago/Turabian StyleKrieger, James M., Carlos Oscar S. Sorzano, and Jose Maria Carazo. 2023. "Scipion-EM-ProDy: A Graphical Interface for the ProDy Python Package within the Scipion Workflow Engine Enabling Integration of Databases, Simulations and Cryo-Electron Microscopy Image Processing" International Journal of Molecular Sciences 24, no. 18: 14245. https://doi.org/10.3390/ijms241814245
APA StyleKrieger, J. M., Sorzano, C. O. S., & Carazo, J. M. (2023). Scipion-EM-ProDy: A Graphical Interface for the ProDy Python Package within the Scipion Workflow Engine Enabling Integration of Databases, Simulations and Cryo-Electron Microscopy Image Processing. International Journal of Molecular Sciences, 24(18), 14245. https://doi.org/10.3390/ijms241814245