
Evaluation of Human Hepatocyte Drug Metabolism Carrying High-Risk or Protection-Associated Liver Disease Genetic Variants

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Study Design
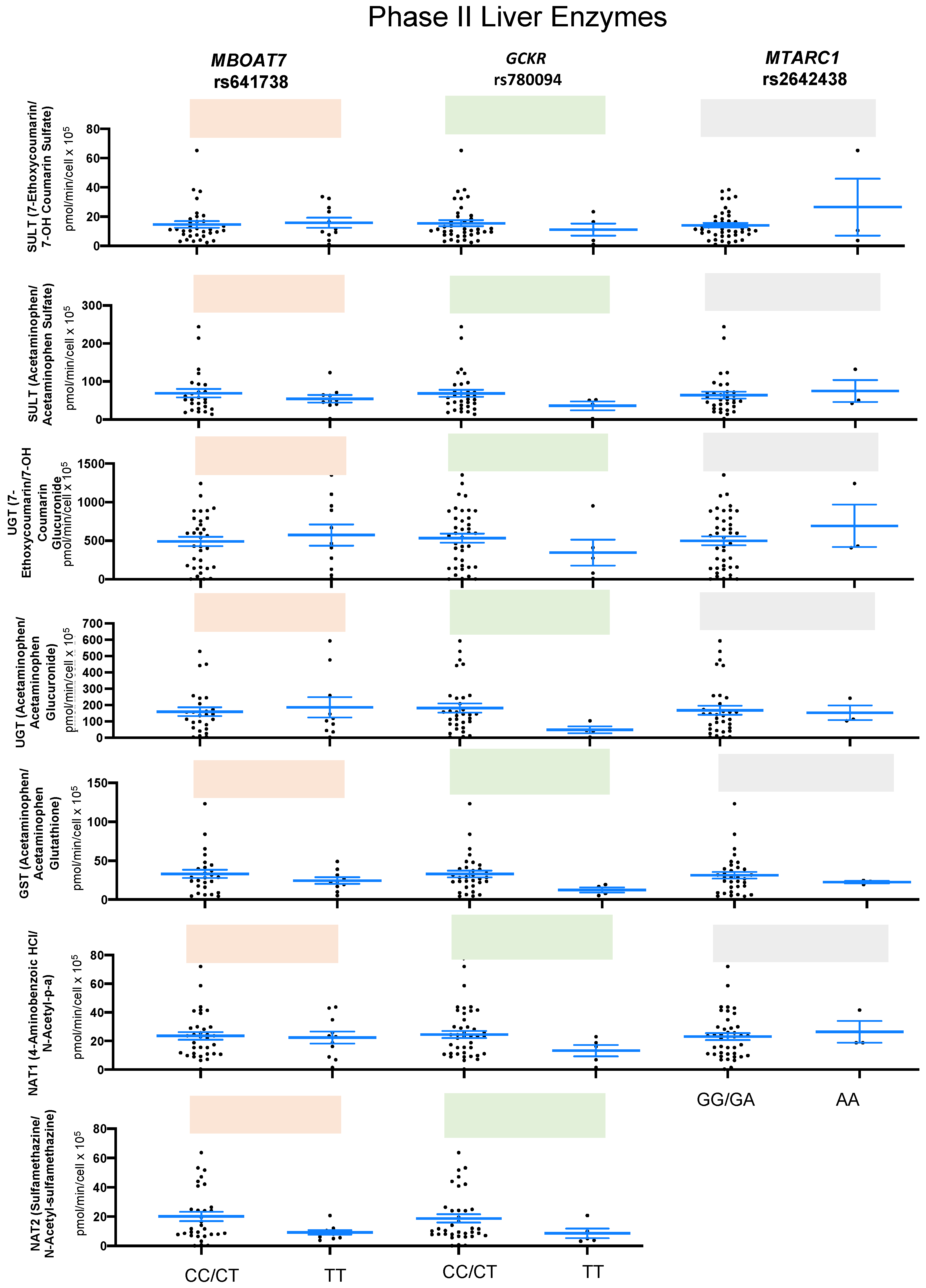
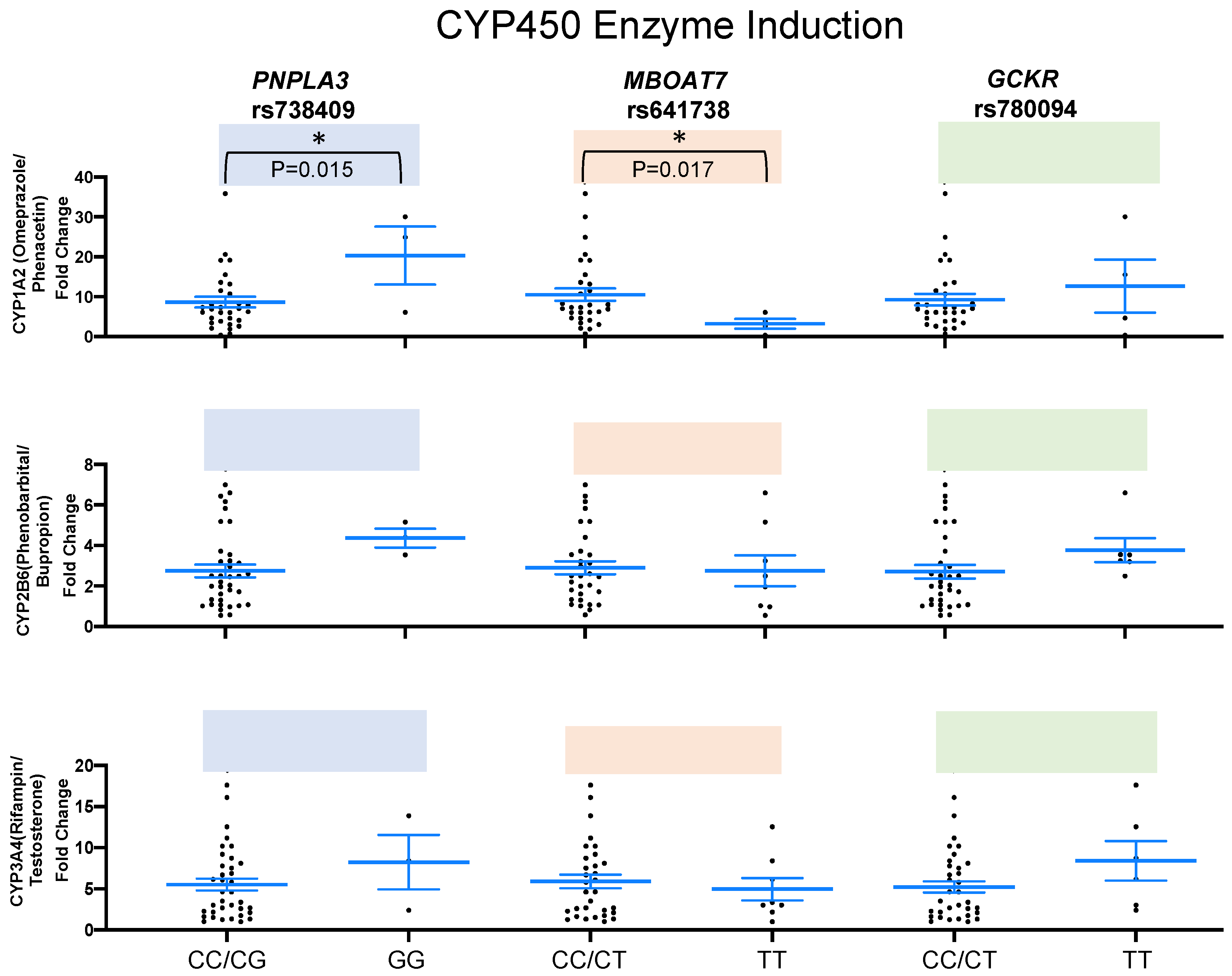
2.2. SNPs and Liver Enzymes
3. Discussion
4. Material and Methods
4.1. Human Primary Hepatocytes
4.2. DNA Isolation and Genotyping
4.3. Drug Metabolism Studies
4.4. Statistical Methods
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Sanyal, A.J. Epidemiology and Natural History of Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2015, 35, 221–235. [Google Scholar] [CrossRef]
- Pacana, T.; Sanyal, A.J. Recent advances in understanding/management of non-alcoholic steatohepatitis. F1000Prime Rep. 2015, 7, 28. [Google Scholar] [CrossRef]
- Jain, M.R.; Giri, S.R.; Trivedi, C.; Bhoi, B.; Rath, A.; Vanage, G.; Vyas, P.; Ranvir, R.; Patel, P.R. Saroglitazar, a novel PPARalpha/gamma agonist with predominant PPARalpha activity, shows lipid-lowering and insulin-sensitizing effects in preclinical models. Pharmacol. Res. Perspect. 2015, 3, e00136. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 3, e1001324. [Google Scholar] [CrossRef]
- Schulze, K.; Imbeaud, S.; Letouzé, E.; Alexandrov, L.B.; Calderaro, J.; Rebouissou, S.; Couchy, G.; Meiller, C.; Shinde, J.; Soysouvanh, F.; et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat. Genet. 2015, 47, 505–511. [Google Scholar] [CrossRef]
- Pingitore, P.; Pirazzi, C.; Mancina, R.M.; Motta, B.M.; Indiveri, C.; Pujia, A.; Montalcini, T.; Hedfalk, K.; Romeo, S. Recombinant PNPLA3 protein shows triglyceride hydrolase activity and its I148M mutation results in loss of function. Biochim. Biophys. Acta 2014, 1841, 574–580. [Google Scholar] [CrossRef]
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2021, 70, 940–950. [Google Scholar] [CrossRef]
- Fernandes Silva, L.; Vangipurapu, J.; Kuulasmaa, T.; Laakso, M. An intronic variant in the GCKR gene is associated with multiple lipids. Sci. Rep. 2019, 9, 10240. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Emdin, C.A.; Haas, M.E.; Khera, A.V.; Aragam, K.; Chaffin, M.; Klarin, D.; Hindy, G.; Jiang, L.; Wei, W.Q.; Feng, Q.; et al. A missense variant in Mitochondrial Amidoxime Reducing Component 1 gene and protection against liver disease. PLoS Genet. 2020, 16, e1008629. [Google Scholar] [CrossRef] [PubMed]
- Hudert, C.A.; Adams, L.A.; Alisi, A.; Anstee, Q.M.; Crudele, A.; Draijer, L.G.; EU-PNAFLD Investigators; Furse, S.; Hengstler, J.G.; Jenkins, B.; et al. Variants in mitochondrial amidoxime reducing component 1 and hydroxysteroid 17-beta dehydrogenase 13 reduce severity of nonalcoholic fatty liver disease in children and suppress fibrotic pathways through distinct mechanisms. Hepatol. Commun. 2022, 6, 1934–1948. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated with Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef] [PubMed]
- Motomura, T.; Amirneni, S.; Diaz-Aragon, R.; Faccioli, L.A.; Malizio, M.R.; Coard, M.C.; Kocas-Kilicarslan, Z.N.; Frau, C.; Haep, N.; Ostrowska, A.; et al. Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target. J. Pers. Med. 2021, 11, 619. [Google Scholar] [CrossRef]
- Trepo, E.; Valenti, L. Update on NAFLD genetics: From new variants to the clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef] [PubMed]
- Rotman, Y.; Koh, C.; Zmuda, J.M.; Kleiner, D.E.; Liang, T.J.; Nash, C.R.N. The association of genetic variability in patatin-like phospholipase domain-containing protein 3 (PNPLA3) with histological severity of nonalcoholic fatty liver disease. Hepatology 2010, 52, 894–903. [Google Scholar] [CrossRef]
- Buch, S.; Stickel, F.; Trepo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Barata, L.; Feitosa, M.F.; Bielak, L.F.; Halligan, B.; Baldridge, A.S.; Guo, X.; Yerges-Armstrong, L.M.; Smith, A.V.; Yao, J.; Palmer, N.D.; et al. Insulin Resistance Exacerbates Genetic Predisposition to Nonalcoholic Fatty Liver Disease in Individuals Without Diabetes. Hepatol. Commun. 2019, 3, 894–907. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Sveinbjornsson, G.; Ulfarsson, M.O.; Thorolfsdottir, R.B.; Jonsson, B.A.; Einarsson, E.; Gunnlaugsson, G.; Rognvaldsson, S.; Arnar, D.O.; Baldvinsson, M.; Bjarnason, R.G.; et al. Multiomics study of nonalcoholic fatty liver disease. Nat. Genet. 2022, 54, 1652–1663. [Google Scholar] [CrossRef]
- Wen, H.; Yang, H.J.; An, Y.J.; Kim, J.M.; Lee, D.H.; Jin, X.; Park, S.W.; Min, K.J.; Park, S. Enhanced phase II detoxification contributes to beneficial effects of dietary restriction as revealed by multi-platform metabolomics studies. Mol. Cell. Proteom. 2013, 12, 575–586. [Google Scholar] [CrossRef]
- Yang, J.; Trépo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouzé, E.; Imbeaud, S.; Bayard, Q.; Gustot, T.; Deviere, J.; et al. A 17-Beta-Hydroxysteroid Dehydrogenase 13 Variant Protects from Hepatocellular Carcinoma Development in Alcoholic Liver Disease. Hepatology 2019, 70, 231–240. [Google Scholar] [CrossRef]
- Tilson, S.G.; Morell, C.M.; Lenaerts, A.S.; Park, S.B.; Hu, Z.; Jenkins, B.; Koulman, A.; Liang, T.J.; Vallier, L. Modeling PNPLA3-Associated NAFLD Using Human-Induced Pluripotent Stem Cells. Hepatology 2021, 74, 2998–3017. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Fracanzani, A.L.; Dongiovanni, P. MBOAT7 down-regulation by genetic and environmental factors predisposes to MAFLD. EBioMedicine 2020, 57, 102866. [Google Scholar] [CrossRef]
- Ioannou, G.N. Epidemiology and risk-stratification of NAFLD-associated HCC. J. Hepatol. 2021, 75, 1476–1484. [Google Scholar] [CrossRef]
- Schneider, C.V.; Schneider, K.M.; Conlon, D.M.; Park, J.; Vujkovic, M.; Zandvakili, I.; Ko, Y.A.; Trautwein, C.; Carr, R.M.; Strnad, P.; et al. A genome-first approach to mortality and metabolic phenotypes in MTARC1 p.Ala165Thr (rs2642438) heterozygotes and homozygotes. Med 2021, 2, 851–863.e3. [Google Scholar] [CrossRef]
- Thorn, C.F.; Aklillu, E.; Klein, T.E.; Altman, R.B. PharmGKB summary: Very important pharmacogene information for CYP1A2. Pharmacogenet Genom. 2012, 22, 73–77. [Google Scholar] [CrossRef]
- Dai, D.; Zeldin, D.C.; Blaisdell, J.A.; Chanas, B.; Coulter, S.J.; Ghanayem, B.I.; Goldstein, J.A. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics 2001, 11, 597–607. [Google Scholar] [CrossRef]
- Zi, J.; Liu, D.; Ma, P.; Huang, H.; Zhu, J.; Wei, D.; Yang, J.; Chen, C. Effects of CYP2C9*3 and CYP2C9*13 on Diclofenac Metabolism and Inhibition-based Drug-Drug Interactions. Drug Metab. Pharmacokinet. 2010, 25, 343–350. [Google Scholar] [CrossRef]
- Pharmgkb. Available online: https://www.pharmgkb.org/clinicalAnnotation/1450931522 (accessed on 3 August 2023).
- Pharmgkb. Available online: https://www.pharmgkb.org/variantAnnotation/982044657 (accessed on 3 August 2023).
- Yoon, H.; Shaw, J.L.; Haigis, M.C.; Greka, A. Lipid metabolism in sickness and in health: Emerging regulators of lipotoxicity. Mol. Cell 2021, 81, 3708–3730. [Google Scholar] [CrossRef]
- Iyer, K.R.; Sinz, M.W. Characterization of Phase I and Phase II hepatic drug metabolism activities in a panel of human liver preparations. Chem. Biol. Interact. 1999, 118, 151–169. [Google Scholar] [CrossRef]
- Jancova, P.; Anzenbacher, P.; Anzenbacherova, E. Phase II drug metabolizing enzymes. Biomed. Pap. Med. Fac. Univ. Palacky. Olomouc Czech Repub. 2010, 154, 103–116. [Google Scholar] [CrossRef]
- Bessone, F.; Dirchwolf, M.; Rodil, M.A.; Razori, M.V.; Roma, M.G. Review article: Drug-induced liver injury in the context of nonalcoholic fatty liver disease—A physiopathological and clinical integrated view. Aliment. Pharmacol. Ther. 2018, 48, 892–913. [Google Scholar] [CrossRef]
- Danan, G.; Benichou, C. Causality assessment of adverse reactions to drugs--I. A novel method based on the conclusions of international consensus meetings: Application to drug-induced liver injuries. J. Clin. Epidemiol. 1993, 46, 1323–1330. [Google Scholar] [CrossRef]
- Danan, G.; Teschke, R. RUCAM in Drug and Herb Induced Liver Injury: The Update. Int. J. Mol. Sci. 2015, 17, 14. [Google Scholar] [CrossRef]
- Teschke, R. Top-ranking drugs out of 3312 drug-induced liver injury cases evaluated by the Roussel Uclaf Causality Assessment Method. Expert. Opin. Drug Metab. Toxicol. 2018, 14, 1169–1187. [Google Scholar] [CrossRef]
- Allard, J.; Le Guillou, D.; Begriche, K.; Fromenty, B. Drug-induced liver injury in obesity and nonalcoholic fatty liver disease. Adv. Pharmacol. 2019, 85, 75–107. [Google Scholar]
- David, S.; Hamilton, J.P. Drug-induced Liver Injury. US Gastroenterol. Hepatol. Rev. 2010, 6, 73–80. [Google Scholar]
- Fisher, C.D.; Lickteig, A.J.; Augustine, L.M.; Ranger-Moore, J.; Jackson, J.P.; Ferguson, S.S.; Cherrington, N.J. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab. Dispos. 2009, 37, 2087–2094. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Jager, L.R.; Leek, J.T. An estimate of the science-wise false discovery rate and application to the top medical literature. Biostatistics 2014, 15, 1–12. [Google Scholar] [CrossRef]
- Cleves, M.A. Exploratory Analysis of Single Nucleotide Polymorphism (SNP) for Quantitative Traits. Stata J. 2005, 5, 141–153. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metabolic Pathway | Substrate and Concentrations | Marker Metabolite Analyzed | Ion Mode Application | Mass Transitions Monitoring |
|---|---|---|---|---|
| CYP1A2 | Phenacetin (100 μM) | Acetaminophen | Positive | m/z 152.1 to 109.9 |
| CYP2A6 | Coumarin (50 μM) | 7-Hydroxycoumarin; 7-hydroxycoumarin glucuronide; 7-hydroxycoumarin sulfate | Negative | m/z 161.0 to 132.9 (7-hydroxycoumarin); m/z 336.9 to 160.9 (glucuronide); m/z 240.9 to 161.0 (sulfate) |
| CYP2B6 | Bupropion (500 μM) | Hydroxybupropion | Positive | m/z 250.1 to 130.1 |
| CYP2C8 | Paclitaxel (20 μM) | 6α-Hydroxypaclitaxel | Positive | m/z 870.4 to 525.2 |
| CYP2C9 | Diclofenac (25 μM) | 4-Hydroxydiclofenac | Negative | m/z 309.8 to 265.9 |
| CYP2C19 | S-Mephenytoin (250 μM) | 4-Hydroxy-S-mephenytoin | Positive | m/z 235.2 to 150.0 |
| CYP2D6 | Dextromethorphan (15 μM) | Dextrorphan | Positive | m/z 258.1 to 157.1 |
| CYP1A2 | Acetaminophen (100 μM) | Acetaminophen | Positive | m/z 152.1 to 109.9 |
| CYP2B6 | Bupropion (500 μM) | Hydroxybupropion | Positive | m/z 250.1 to 130.1 |
| CYP3A4 | Testosterone (200 μM) | 6β-Hydroxytestosterone | Positive | m/z 305.2 to 269.1 |
| CYP2E1 | Chlorzoxazone (250 μM) | 6-Hydroxychlorzoxazone | Negative | m/z 183.9 to 119.8 |
| CYP3A4 | Midazolam (20 μM) | 1′-Hydroxymidazolam | Positive | m/z 342.1 to 203.1 |
| CYP3A4 | Testosterone (200 μM) | 6β-Hydroxytestosterone | Positive | m/z 305.2 to 269.1 |
| ECOD | 7-Ethoxycoumarin (100 μM) | 7-Hydroxycoumarin | Negative | m/z 161.0 to 132.9 |
| UGT | 7-Hydroxycoumarin (100 μM) | 7-Hydroxycoumarin glucuronide | Negative | m/z 336.9 to 160.9 |
| SULT | 7-Hydroxycoumarin (100 μM) | 7-Hydroxycoumarin sulfate | Negative | m/z 240.9 to 161.0 |
| UGT | Acetaminophen (10, 100, 200 mM) | Acetaminophen glucuronide | Negative | m/z 326.0 to 150.0 |
| SULT | Acetaminophen (10, 100, 200 mM) | Acetaminophen sulfate | Negative | m/z 229.8 to 150.0 |
| GST | Acetaminophen (10, 100, 200 mM) | Acetaminophen glutathione | Negative | m/z 455.0 to 271.8 |
| FMO | Benzydamine hydrochloride (250 μM) | Benzydamine-N-oxide | Positive | m/z 326.4 to 102.1 |
| MAO | Kynuramine hydrobromide (160 μM) | 4-Hydroxyquinoline | Negative | m/z 144.1 to 102.1 |
| AO | Carbazeran (10 μM) | 4-Hydroxycarbazeran | Positive | m/z 377.0 to 234.2 |
| NAT1 | 4-Aminobenzoic acid (200 μM) | N-Acetyl-p-aminobenzoic acid | Negative | m/z 178.0 to 133.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faccioli, L.A.P.; Cetin, Z.; Kocas-Kilicarslan, Z.N.; Ortiz, K.; Sun, Y.; Hu, Z.; Kurihara, T.; Tafaleng, E.N.; Florentino, R.M.; Wang, Z.; et al. Evaluation of Human Hepatocyte Drug Metabolism Carrying High-Risk or Protection-Associated Liver Disease Genetic Variants. Int. J. Mol. Sci. 2023, 24, 13406. https://doi.org/10.3390/ijms241713406
Faccioli LAP, Cetin Z, Kocas-Kilicarslan ZN, Ortiz K, Sun Y, Hu Z, Kurihara T, Tafaleng EN, Florentino RM, Wang Z, et al. Evaluation of Human Hepatocyte Drug Metabolism Carrying High-Risk or Protection-Associated Liver Disease Genetic Variants. International Journal of Molecular Sciences. 2023; 24(17):13406. https://doi.org/10.3390/ijms241713406
Chicago/Turabian StyleFaccioli, Lanuza A. P., Zeliha Cetin, Zehra N. Kocas-Kilicarslan, Kimberly Ortiz, Yiyue Sun, Zhiping Hu, Takeshi Kurihara, Edgar N. Tafaleng, Rodrigo M. Florentino, Zi Wang, and et al. 2023. "Evaluation of Human Hepatocyte Drug Metabolism Carrying High-Risk or Protection-Associated Liver Disease Genetic Variants" International Journal of Molecular Sciences 24, no. 17: 13406. https://doi.org/10.3390/ijms241713406
APA StyleFaccioli, L. A. P., Cetin, Z., Kocas-Kilicarslan, Z. N., Ortiz, K., Sun, Y., Hu, Z., Kurihara, T., Tafaleng, E. N., Florentino, R. M., Wang, Z., Xia, M., Miedel, M. T., Taylor, D. L., Behari, J., Ostrowska, A., Constantine, R., Li, A., & Soto-Gutierrez, A. (2023). Evaluation of Human Hepatocyte Drug Metabolism Carrying High-Risk or Protection-Associated Liver Disease Genetic Variants. International Journal of Molecular Sciences, 24(17), 13406. https://doi.org/10.3390/ijms241713406