
JNK Cascade-Induced Apoptosis—A Unique Role in GqPCR Signaling

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Components and Activation of the JNK Cascade
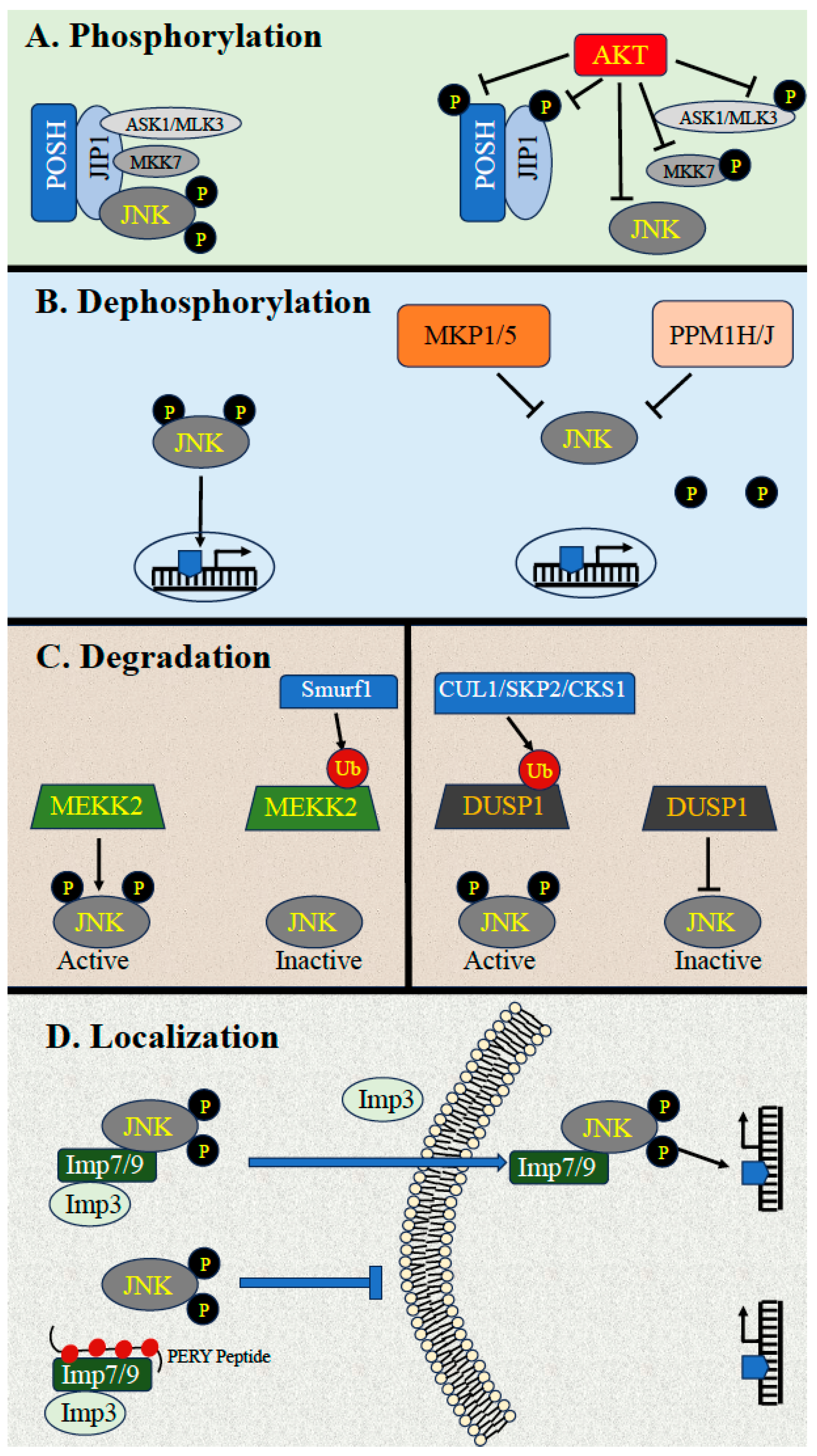
3. Downregulation of JNK Activity
3.1. Downregulation by Phosphorylation
3.2. Downregulation by Dephosphorylation
3.3. Dephosphorylation by MKPs
3.4. Downregulation by Scaffold Proteins and Ubiquitin-Dependent Degradation
3.5. Downregulation through Dynamic Subcellular Localizations
4. Cellular Processes Regulated by the JNK Cascade
4.1. Role of the JNK Cascade in Apoptosis
4.1.1. A Brief Review of Apoptosis
4.1.2. JNK in the Induction and Regulation of Apoptosis
4.1.3. Indirect Effects through the Regulation of Transcription
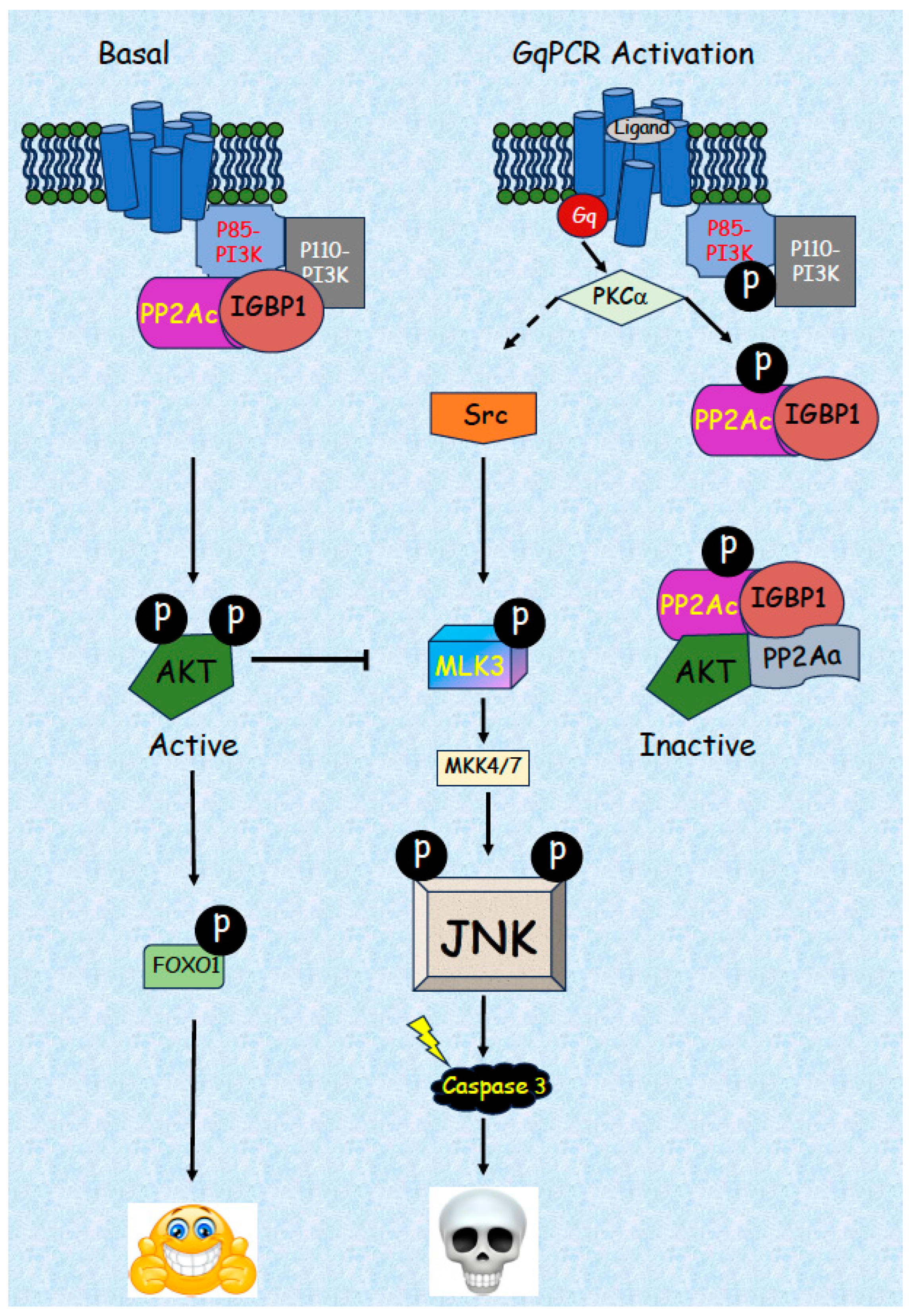
5. JNK-Related Induction of Apoptosis by GqPCRs
5.1. Activation of JNK by GqPCRs in Various Cell Types
5.2. Detailed Mechanism of JNK Activation by GqPCRs/PKCs
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Thorner, J.; Hunter, T.; Cantley, L.C.; Sever, R. Signal transduction: From the atomic age to the post-genomic era. Cold Spring Harb. Perspect. Biol. 2014, 6, a022913. [Google Scholar] [CrossRef]
- Keshet, Y.; Seger, R. The MAP kinase signaling cascades: A system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010, 661, 3–38. [Google Scholar] [CrossRef] [PubMed]
- Seger, R.; Krebs, E.G. The MAPK signaling cascade. FASEB J. 1995, 9, 726–735. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Wortzel, I.; Seger, R. Alternative Splicing of MAPKs in the Regulation of Signaling Specificity. Cells 2021, 10, 3466. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Davis, R.J. Cell Signaling and Stress Responses. Cold Spring Harb. Perspect. Biol. 2016, 8, a006072. [Google Scholar] [CrossRef]
- Martinez-Limon, A.; Joaquin, M.; Caballero, M.; Posas, F.; de Nadal, E. The p38 Pathway: From Biology to Cancer Therapy. Int. J. Mol. Sci. 2020, 21, 1913. [Google Scholar] [CrossRef]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Tubita, A.; Tusa, I.; Rovida, E. Playing the Whack-A-Mole Game: ERK5 Activation Emerges Among the Resistance Mechanisms to RAF-MEK1/2-ERK1/2- Targeted Therapy. Front. Cell Dev. Biol. 2021, 9, 647311. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Davis, R.J. TNF and MAP kinase signalling pathways. Semin. Immunol. 2014, 26, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed]
- Reinehr, R.; Haussinger, D. Hyperosmotic activation of the CD95 system. Methods Enzymol. 2007, 428, 145–160. [Google Scholar] [CrossRef]
- Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. [Google Scholar] [CrossRef]
- Huang, Y.; Wang, Y.; Li, X.; Chen, Z.; Li, X.; Wang, H.; Ni, M.; Li, J. Molecular mechanism of ER stress-induced gene expression of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) in macrophages. FEBS J. 2015, 282, 2361–2378. [Google Scholar] [CrossRef]
- Chadee, D.N.; Yuasa, T.; Kyriakis, J.M. Direct activation of mitogen-activated protein kinase kinase kinase MEKK1 by the Ste20p homologue GCK and the adapter protein TRAF2. Mol. Cell. Biol. 2002, 22, 737–749. [Google Scholar] [CrossRef]
- Dan, I.; Watanabe, N.M.; Kusumi, A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001, 11, 220–230. [Google Scholar] [CrossRef]
- Suddason, T.; Gallagher, E. A RING to rule them all? Insights into the Map3k1 PHD motif provide a new mechanistic understanding into the diverse roles of Map3k1. Cell Death Differ. 2015, 22, 540–548. [Google Scholar] [CrossRef]
- Prinz, E.; Aviram, S.; Aronheim, A. WDR62 mediates TNFalpha-dependent JNK activation via TRAF2-MLK3 axis. Mol. Biol. Cell 2018, 29, 2470–2480. [Google Scholar] [CrossRef] [PubMed]
- Coso, O.A.; Chiariello, M.; Yu, J.C.; Teramoto, H.; Crespo, P.; Xu, N.; Miki, T.; Gutkind, J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell 1995, 81, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, E.D.; Gutowski, S.; Sternweis, P.C.; Cobb, M.H. RhoA binds to the amino terminus of MEKK1 and regulates its kinase activity. J. Biol. Chem. 2004, 279, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Mita, H.; Tsutsui, J.; Takekawa, M.; Witten, E.A.; Saito, H. Regulation of MTK1/MEKK4 kinase activity by its N-terminal autoinhibitory domain and GADD45 binding. Mol. Cell. Biol. 2002, 22, 4544–4555. [Google Scholar] [CrossRef]
- Cheng, J.; Yu, L.; Zhang, D.; Huang, Q.; Spencer, D.; Su, B. Dimerization through the catalytic domain is essential for MEKK2 activation. J. Biol. Chem. 2005, 280, 13477–13482. [Google Scholar] [CrossRef]
- Fleming, Y.; Armstrong, C.G.; Morrice, N.; Paterson, A.; Goedert, M.; Cohen, P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem. J. 2000, 352 Pt 1, 145–154. [Google Scholar] [CrossRef]
- Ludwig, S.; Engel, K.; Hoffmeyer, A.; Sithanandam, G.; Neufeld, B.; Palm, D.; Gaestel, M.; Rapp, U.R. 3pK, a novel mitogen-activated protein (MAP) kinase-activated protein kinase, is targeted by three MAP kinase pathways. Mol. Cell. Biol. 1996, 16, 6687–6697. [Google Scholar] [CrossRef]
- Ha, J.; Kang, E.; Seo, J.; Cho, S. Phosphorylation Dynamics of JNK Signaling: Effects of Dual-Specificity Phosphatases (DUSPs) on the JNK Pathway. Int. J. Mol. Sci. 2019, 20, 6157. [Google Scholar] [CrossRef]
- Wei, W.; Jin, J.; Schlisio, S.; Harper, J.W.; Kaelin, W.G., Jr. The v-Jun point mutation allows c-Jun to escape GSK3-dependent recognition and destruction by the Fbw7 ubiquitin ligase. Cancer Cell 2005, 8, 25–33. [Google Scholar] [CrossRef]
- Barthwal, M.K.; Sathyanarayana, P.; Kundu, C.N.; Rana, B.; Pradeep, A.; Sharma, C.; Woodgett, J.R.; Rana, A. Negative regulation of mixed lineage kinase 3 by protein kinase B/AKT leads to cell survival. J. Biol. Chem. 2003, 278, 3897–3902. [Google Scholar] [CrossRef]
- Zehorai, E.; Seger, R. Beta-Like Importins Mediate the Nuclear Translocation of MAPKs. Cell. Physiol. Biochem. 2019, 52, 802–821. [Google Scholar] [CrossRef]
- Matsuura, H.; Nishitoh, H.; Takeda, K.; Matsuzawa, A.; Amagasa, T.; Ito, M.; Yoshioka, K.; Ichijo, H. Phosphorylation-dependent scaffolding role of JSAP1/JIP3 in the ASK1-JNK signaling pathway. A new mode of regulation of the MAP kinase cascade. J. Biol. Chem. 2002, 277, 40703–40709. [Google Scholar] [CrossRef]
- Nakano, R.; Nakayama, T.; Sugiya, H. Biological Properties of JNK3 and Its Function in Neurons, Astrocytes, Pancreatic beta-Cells and Cardiovascular Cells. Cells 2020, 9, 1802. [Google Scholar] [CrossRef] [PubMed]
- Whitmarsh, A.J. The JIP family of MAPK scaffold proteins. Biochem. Soc. Trans. 2006, 34, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.H.; Khursigara, G.; Sun, X.; Franke, T.F.; Chao, M.V. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol. Cell. Biol. 2001, 21, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Sondarva, G.; Viswakarma, N.; Nair, R.S.; Osipo, C.; Tzivion, G.; Rana, B.; Rana, A. Human Epidermal Growth Factor Receptor 2 (HER2) Impedes MLK3 Kinase Activity to Support Breast Cancer Cell Survival. J. Biol. Chem. 2015, 290, 21705–21712. [Google Scholar] [CrossRef]
- Kraus, S.; Levy, G.; Hanoch, T.; Naor, Z.; Seger, R. Gonadotropin-releasing hormone induces apoptosis of prostate cancer cells: Role of c-Jun NH2-terminal kinase, protein kinase B, and extracellular signal-regulated kinase pathways. Cancer Res. 2004, 64, 5736–5744. [Google Scholar] [CrossRef]
- Park, H.S.; Kim, M.S.; Huh, S.H.; Park, J.; Chung, J.; Kang, S.S.; Choi, E.J. Akt (protein kinase B) negatively regulates SEK1 by means of protein phosphorylation. J. Biol. Chem. 2002, 277, 2573–2578. [Google Scholar] [CrossRef]
- Kim, A.H.; Yano, H.; Cho, H.; Meyer, D.; Monks, B.; Margolis, B.; Birnbaum, M.J.; Chao, M.V. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron 2002, 35, 697–709. [Google Scholar] [CrossRef]
- Figueroa, C.; Tarras, S.; Taylor, J.; Vojtek, A.B. Akt2 negatively regulates assembly of the POSH-MLK-JNK signaling complex. J. Biol. Chem. 2003, 278, 47922–47927. [Google Scholar] [CrossRef]
- Zhang, Q.G.; Wang, X.T.; Han, D.; Yin, X.H.; Zhang, G.Y.; Xu, T.L. Akt inhibits MLK3/JNK3 signaling by inactivating Rac1: A protective mechanism against ischemic brain injury. J. Neurochem. 2006, 98, 1886–1898. [Google Scholar] [CrossRef] [PubMed]
- Abell, A.N.; Granger, D.A.; Johnson, G.L. MEKK4 stimulation of p38 and JNK activity is negatively regulated by GSK3beta. J. Biol. Chem. 2007, 282, 30476–30484. [Google Scholar] [CrossRef] [PubMed]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational prediction and experimental verification of new MAP kinase docking sites and substrates including Gli transcription factors. PLoS Comput. Biol. 2010, 6, e1000908. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Ito, K.; Kanda, A.; Tomoda, K.; Miller-Larsson, A.; Barnes, P.J.; Mercado, N. Protein tyrosine phosphatase PTP-RR regulates corticosteroid sensitivity. Respir. Res. 2016, 17, 30. [Google Scholar] [CrossRef]
- Yao, Z.; Seger, R. The molecular Mechanism of MAPK/ERK inactivation. Curr. Genom. 2004, 5, 385–393. [Google Scholar] [CrossRef]
- Caunt, C.J.; Keyse, S.M. Dual-specificity MAP kinase phosphatases (MKPs): Shaping the outcome of MAP kinase signalling. FEBS J. 2013, 280, 489–504. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.I.; Brummer, T.; O’Brien, P.M.; Daly, R.J. Dual-specificity phosphatases: Critical regulators with diverse cellular targets. Biochem. J. 2009, 418, 475–489. [Google Scholar] [CrossRef] [PubMed]
- Khoubai, F.Z.; Grosset, C.F. DUSP9, a Dual-Specificity Phosphatase with a Key Role in Cell Biology and Human Diseases. Int. J. Mol. Sci. 2021, 22, 1538. [Google Scholar] [CrossRef]
- Masuda, K.; Shima, H.; Katagiri, C.; Kikuchi, K. Activation of ERK induces phosphorylation of MAPK phosphatase-7, a JNK specific phosphatase, at Ser-446. J. Biol. Chem. 2003, 278, 32448–32456. [Google Scholar] [CrossRef]
- Musikacharoen, T.; Bandow, K.; Kakimoto, K.; Kusuyama, J.; Onishi, T.; Yoshikai, Y.; Matsuguchi, T. Functional involvement of dual specificity phosphatase 16 (DUSP16), a c-Jun N-terminal kinase-specific phosphatase, in the regulation of T helper cell differentiation. J. Biol. Chem. 2011, 286, 24896–24905. [Google Scholar] [CrossRef]
- Shillingford, S.; Zhang, L.; Surovtseva, Y.; Dorry, S.; Lolis, E.; Bennett, A.M. A novel site on dual-specificity phosphatase MKP7/DUSP16 is required for catalysis and MAPK binding. J. Biol. Chem. 2022, 298, 102617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Nallaparaju, K.C.; Liu, X.; Jiao, H.; Reynolds, J.M.; Wang, Z.X.; Dong, C. MAPK phosphatase 7 regulates T cell differentiation via inhibiting ERK-mediated IL-2 expression. J. Immunol. 2015, 194, 3088–3095. [Google Scholar] [CrossRef] [PubMed]
- Bordo, D.; Bork, P. The rhodanese/Cdc25 phosphatase superfamily. Sequence-structure-function relations. EMBO Rep. 2002, 3, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.Y.; Wu, J.W.; Wang, Z.X. Mitogen-activated protein kinase (MAPK) phosphatase 3-mediated cross-talk between MAPKs ERK2 and p38alpha. J. Biol. Chem. 2011, 286, 16150–16162. [Google Scholar] [CrossRef] [PubMed]
- Zeke, A.; Bastys, T.; Alexa, A.; Garai, A.; Meszaros, B.; Kirsch, K.; Dosztanyi, Z.; Kalinina, O.V.; Remenyi, A. Systematic discovery of linear binding motifs targeting an ancient protein interaction surface on MAP kinases. Mol. Syst. Biol. 2015, 11, 837. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, C.S.; Lu, C.; Lin, S.C.; Wu, J.W.; Wang, Z.X. A conserved motif in JNK/p38-specific MAPK phosphatases as a determinant for JNK1 recognition and inactivation. Nat. Commun. 2016, 7, 10879. [Google Scholar] [CrossRef]
- Martin-Blanco, E.; Gampel, A.; Ring, J.; Virdee, K.; Kirov, N.; Tolkovsky, A.M.; Martinez-Arias, A. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998, 12, 557–570. [Google Scholar] [CrossRef]
- Sanchez-Tillo, E.; Comalada, M.; Xaus, J.; Farrera, C.; Valledor, A.F.; Caelles, C.; Lloberas, J.; Celada, A. JNK1 Is required for the induction of Mkp1 expression in macrophages during proliferation and lipopolysaccharide-dependent activation. J. Biol. Chem. 2007, 282, 12566–12573. [Google Scholar] [CrossRef]
- Good, M.C.; Zalatan, J.G.; Lim, W.A. Scaffold proteins: Hubs for controlling the flow of cellular information. Science 2011, 332, 680–686. [Google Scholar] [CrossRef]
- Oehrl, W.; Cotsiki, M.; Panayotou, G. Differential regulation of M3/6 (DUSP8) signaling complexes in response to arsenite-induced oxidative stress. Cell. Signal. 2013, 25, 429–438. [Google Scholar] [CrossRef]
- Willoughby, E.A.; Collins, M.K. Dynamic interaction between the dual specificity phosphatase MKP7 and the JNK3 scaffold protein beta-arrestin 2. J. Biol. Chem. 2005, 280, 25651–25658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hao, J.; Zhao, Z.; Ben, P.; Fang, F.; Shi, L.; Gao, Y.; Liu, J.; Wen, C.; Luo, L.; et al. beta-Arrestins facilitate ubiquitin-dependent degradation of apoptosis signal-regulating kinase 1 (ASK1) and attenuate H2O2-induced apoptosis. Cell. Signal. 2009, 21, 1195–1206. [Google Scholar] [CrossRef] [PubMed]
- Vaishnav, M.; MacFarlane, M.; Dickens, M. Disassembly of the JIP1/JNK molecular scaffold by caspase-3-mediated cleavage of JIP1 during apoptosis. Exp. Cell Res. 2011, 317, 1028–1039. [Google Scholar] [CrossRef]
- Laine, A.; Ronai, Z. Ubiquitin chains in the ladder of MAPK signaling. Sci. STKE 2005, 2005, re5. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.H.; Kurie, J.M. MKK4/SEK1 is negatively regulated through a feedback loop involving the E3 ubiquitin ligase itch. J. Biol. Chem. 2009, 284, 29399–29404. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Abrams, B.; Grill, B.; Goncharov, A.; Huang, X.; Chisholm, A.D.; Jin, Y. Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 2005, 120, 407–420. [Google Scholar] [CrossRef]
- Baker, S.T.; Opperman, K.J.; Tulgren, E.D.; Turgeon, S.M.; Bienvenut, W.; Grill, B. RPM-1 uses both ubiquitin ligase and phosphatase-based mechanisms to regulate DLK-1 during neuronal development. PLoS Genet. 2014, 10, e1004297. [Google Scholar] [CrossRef]
- Yamashita, M.; Ying, S.X.; Zhang, G.M.; Li, C.; Cheng, S.Y.; Deng, C.X.; Zhang, Y.E. Ubiquitin ligase Smurf1 controls osteoblast activity and bone homeostasis by targeting MEKK2 for degradation. Cell 2005, 121, 101–113. [Google Scholar] [CrossRef]
- Chen, H.F.; Chuang, H.C.; Tan, T.H. Regulation of Dual-Specificity Phosphatase (DUSP) Ubiquitination and Protein Stability. Int. J. Mol. Sci. 2019, 20, 2668. [Google Scholar] [CrossRef]
- Gao, M.; Labuda, T.; Xia, Y.; Gallagher, E.; Fang, D.; Liu, Y.C.; Karin, M. Jun turnover is controlled through JNK-dependent phosphorylation of the E3 ligase Itch. Science 2004, 306, 271–275. [Google Scholar] [CrossRef]
- Caswell, P.T.; Dickens, M. JIP3 localises to exocytic vesicles and focal adhesions in the growth cones of differentiated PC12 cells. Mol. Cell. Biochem. 2018, 444, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Kadoya, T.; Khurana, A.; Tcherpakov, M.; Bromberg, K.D.; Didier, C.; Broday, L.; Asahara, T.; Bhoumik, A.; Ronai, Z. JAMP, a Jun N-terminal kinase 1 (JNK1)-associated membrane protein, regulates duration of JNK activity. Mol. Cell. Biol. 2005, 25, 8619–8630. [Google Scholar] [CrossRef]
- Yang, G.; Liu, Y.; Yang, K.; Liu, R.; Zhu, S.; Coquinco, A.; Wen, W.; Kojic, L.; Jia, W.; Cynader, M. Isoform-specific palmitoylation of JNK regulates axonal development. Cell Death Differ. 2012, 19, 553–561. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Zehorai, E.; Hanoch, T.; Blenis, J.; Seger, R. The nuclear translocation of the kinases p38 and JNK promotes inflammation-induced cancer. Sci. Signal. 2018, 11, eaao3428. [Google Scholar] [CrossRef] [PubMed]
- Satake, T.; Otsuki, K.; Banba, Y.; Suenaga, J.; Hirano, H.; Yamanaka, Y.; Ohno, S.; Hirai, S. The interaction of Kinesin-1 with its adaptor protein JIP1 can be regulated via proteins binding to the JIP1-PTB domain. BMC Cell Biol. 2013, 14, 12. [Google Scholar] [CrossRef] [PubMed]
- Wallbach, M.; Duque Escobar, J.; Babaeikelishomi, R.; Stahnke, M.J.; Blume, R.; Schroder, S.; Kruegel, J.; Maedler, K.; Kluth, O.; Kehlenbach, R.H.; et al. Distinct functions of the dual leucine zipper kinase depending on its subcellular localization. Cell. Signal. 2016, 28, 272–283. [Google Scholar] [CrossRef]
- Gibson, E.S.; Woolfrey, K.M.; Li, H.; Hogan, P.G.; Nemenoff, R.A.; Heasley, L.E.; Dell’Acqua, M.L. Subcellular Localization and Activity of the Mitogen-Activated Protein Kinase Kinase 7 (MKK7) gamma Isoform are Regulated through Binding to the Phosphatase Calcineurin. Mol. Pharmacol. 2019, 95, 20–32. [Google Scholar] [CrossRef]
- Wang, J.; Tang, R.; Lv, M.; Zhang, J.; Shen, B. Selective unresponsiveness to the inhibition of p38 MAPK activation by cAMP helps L929 fibroblastoma cells escape TNF-alpha-induced cell death. Mol. Cancer 2010, 9, 6. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK-signaling: A multiplexing hub in programmed cell death. Genes Cancer 2017, 8, 682–694. [Google Scholar] [CrossRef]
- Coffey, E.T. Nuclear and cytosolic JNK signalling in neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Kuan, C.Y.; Yang, D.D.; Samanta Roy, D.R.; Davis, R.J.; Rakic, P.; Flavell, R.A. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 1999, 22, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Karpac, J.; Jasper, H. Insulin and JNK: Optimizing metabolic homeostasis and lifespan. Trends Endocrinol. Metab. 2009, 20, 100–106. [Google Scholar] [CrossRef]
- Gan, T.; Fan, L.; Zhao, L.; Misra, M.; Liu, M.; Zhang, M.; Su, Y. JNK Signaling in Drosophila Aging and Longevity. Int. J. Mol. Sci. 2021, 22, 9649. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- de Los Reyes Corrales, T.; Losada-Perez, M.; Casas-Tinto, S. JNK Pathway in CNS Pathologies. Int. J. Mol. Sci. 2021, 22, 3883. [Google Scholar] [CrossRef]
- Garg, R.; Kumariya, S.; Katekar, R.; Verma, S.; Goand, U.K.; Gayen, J.R. JNK signaling pathway in metabolic disorders: An emerging therapeutic target. Eur. J. Pharmacol. 2021, 901, 174079. [Google Scholar] [CrossRef]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK expression by macrophages promotes obesity-induced insulin resistance and inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Schaan, A.P.; Cabral, G.F.; Santana-da-Silva, M.N.; Pinto, P.; Vidal, A.F.; Ribeiro-Dos-Santos, A. A Cell’s Fate: An Overview of the Molecular Biology and Genetics of Apoptosis. Int. J. Mol. Sci. 2019, 20, 4133. [Google Scholar] [CrossRef]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27 (Suppl. S1), S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Lossi, L. The concept of intrinsic versus extrinsic apoptosis. Biochem. J. 2022, 479, 357–384. [Google Scholar] [CrossRef] [PubMed]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Shakeri, R.; Kheirollahi, A.; Davoodi, J. Apaf-1: Regulation and function in cell death. Biochimie 2017, 135, 111–125. [Google Scholar] [CrossRef]
- Yamada, K.; Yoshida, K. Mechanical insights into the regulation of programmed cell death by p53 via mitochondria. Biochim Biophys. Acta Mol. Cell Res. 2019, 1866, 839–848. [Google Scholar] [CrossRef]
- Das, G.; Shravage, B.V.; Baehrecke, E.H. Regulation and function of autophagy during cell survival and cell death. Cold Spring Harb. Perspect. Biol. 2012, 4, a008813. [Google Scholar] [CrossRef]
- Doherty, J.; Baehrecke, E.H. Life, death and autophagy. Nat. Cell Biol. 2018, 20, 1110–1117. [Google Scholar] [CrossRef]
- Bjorkblom, B.; Vainio, J.C.; Hongisto, V.; Herdegen, T.; Courtney, M.J.; Coffey, E.T. All JNKs can kill, but nuclear localization is critical for neuronal death. J. Biol. Chem. 2008, 283, 19704–19713. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.; Franca, T.C.C.; Jacevic, V.; Wang, X.; Kuca, K. Immune Evasion, a Potential Mechanism of Trichothecenes: New Insights into Negative Immune Regulations. Int. J. Mol. Sci. 2018, 19, 3307. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Mi, Q.S.; Hardwick, J.M.; Longo, D.L. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc. Natl. Acad. Sci. USA 1999, 96, 3775–3780. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed]
- Inoshita, S.; Takeda, K.; Hatai, T.; Terada, Y.; Sano, M.; Hata, J.; Umezawa, A.; Ichijo, H. Phosphorylation and inactivation of myeloid cell leukemia 1 by JNK in response to oxidative stress. J. Biol. Chem. 2002, 277, 43730–43734. [Google Scholar] [CrossRef] [PubMed]
- Rath, S.; Das, L.; Kokate, S.B.; Pratheek, B.M.; Chattopadhyay, S.; Goswami, C.; Chattopadhyay, R.; Crowe, S.E.; Bhattacharyya, A. Regulation of Noxa-mediated apoptosis in Helicobacter pylori-infected gastric epithelial cells. FASEB J. 2015, 29, 796–806. [Google Scholar] [CrossRef]
- Donovan, N.; Becker, E.B.; Konishi, Y.; Bonni, A. JNK phosphorylation and activation of BAD couples the stress-activated signaling pathway to the cell death machinery. J. Biol. Chem. 2002, 277, 40944–40949. [Google Scholar] [CrossRef]
- Putcha, G.V.; Le, S.; Frank, S.; Besirli, C.G.; Clark, K.; Chu, B.; Alix, S.; Youle, R.J.; LaMarche, A.; Maroney, A.C.; et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 2003, 38, 899–914. [Google Scholar] [CrossRef]
- Kim, B.J.; Ryu, S.W.; Song, B.J. JNK- and p38 kinase-mediated phosphorylation of Bax leads to its activation and mitochondrial translocation and to apoptosis of human hepatoma HepG2 cells. J. Biol. Chem. 2006, 281, 21256–21265. [Google Scholar] [CrossRef]
- Lei, K.; Davis, R.J. JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef]
- Tsuruta, F.; Sunayama, J.; Mori, Y.; Hattori, S.; Shimizu, S.; Tsujimoto, Y.; Yoshioka, K.; Masuyama, N.; Gotoh, Y. JNK promotes Bax translocation to mitochondria through phosphorylation of 14-3-3 proteins. EMBO J. 2004, 23, 1889–1899. [Google Scholar] [CrossRef]
- Sunayama, J.; Tsuruta, F.; Masuyama, N.; Gotoh, Y. JNK antagonizes Akt-mediated survival signals by phosphorylating 14-3-3. J. Cell Biol. 2005, 170, 295–304. [Google Scholar] [CrossRef]
- Deng, Y.; Ren, X.; Yang, L.; Lin, Y.; Wu, X. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell 2003, 115, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Prakasam, A.; Ghose, S.; Oleinik, N.V.; Bethard, J.R.; Peterson, Y.K.; Krupenko, N.I.; Krupenko, S.A. JNK1/2 regulate Bid by direct phosphorylation at Thr59 in response to ALDH1L1. Cell Death Dis. 2014, 5, e1358. [Google Scholar] [CrossRef] [PubMed]
- Park, B. JNK1-mediated phosphorylation of Smac/DIABLO at the serine 6 residue is functionally linked to its mitochondrial release during TNF-alpha--induced apoptosis of HeLa cells. Mol. Med. Rep. 2014, 10, 3205–3210. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Kamata, H.; Solinas, G.; Luo, J.L.; Maeda, S.; Venuprasad, K.; Liu, Y.C.; Karin, M. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell 2006, 124, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Ibraheem, K.; Yhmed, A.M.A.; Nasef, M.M.; Georgopoulos, N.T. TRAF3/p38-JNK Signalling Crosstalk with Intracellular-TRAIL/Caspase-10-Induced Apoptosis Accelerates ROS-Driven Cancer Cell-Specific Death by CD40. Cells 2022, 11, 3274. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Han, D.; Petrovic, L.M.; Kaplowitz, N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J. Biol. Chem. 2011, 286, 35071–35078. [Google Scholar] [CrossRef]
- Kamata, H.; Honda, S.; Maeda, S.; Chang, L.; Hirata, H.; Karin, M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 2005, 120, 649–661. [Google Scholar] [CrossRef]
- Osawa, Y.; Nagaki, M.; Banno, Y.; Yamada, Y.; Imose, M.; Nozawa, Y.; Moriwaki, H.; Nakashima, S. Possible involvement of reactive oxygen species in D-galactosamine-induced sensitization against tumor necrosis factor-alpha-induced hepatocyte apoptosis. J. Cell. Physiol. 2001, 187, 374–385. [Google Scholar] [CrossRef]
- Win, S.; Than, T.A.; Min, R.W.; Aghajan, M.; Kaplowitz, N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology 2016, 63, 1987–2003. [Google Scholar] [CrossRef]
- Zhang, J.; Min, R.W.M.; Le, K.; Zhou, S.; Aghajan, M.; Than, T.A.; Win, S.; Kaplowitz, N. The role of MAP2 kinases and p38 kinase in acute murine liver injury models. Cell Death Dis. 2017, 8, e2903. [Google Scholar] [CrossRef]
- Zhou, Q.; Lam, P.Y.; Han, D.; Cadenas, E. c-Jun N-terminal kinase regulates mitochondrial bioenergetics by modulating pyruvate dehydrogenase activity in primary cortical neurons. J. Neurochem. 2008, 104, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Ameyar, M.; Wisniewska, M.; Weitzman, J.B. A role for AP-1 in apoptosis: The case for and against. Biochimie 2003, 85, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Bossy-Wetzel, E.; Bakiri, L.; Yaniv, M. Induction of apoptosis by the transcription factor c-Jun. EMBO J. 1997, 16, 1695–1709. [Google Scholar] [CrossRef]
- Fan, M.; Chambers, T.C. Role of mitogen-activated protein kinases in the response of tumor cells to chemotherapy. Drug Resist. Updat. 2001, 4, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.R.; McAtarsney, C.P.; Bredhold, K.E.; Kline, A.M.; Mayo, L.D. Mutant and wild-type p53 form complexes with p73 upon phosphorylation by the kinase JNK. Sci. Signal. 2018, 11, eaao4170. [Google Scholar] [CrossRef]
- Saha, M.N.; Jiang, H.; Yang, Y.; Zhu, X.; Wang, X.; Schimmer, A.D.; Qiu, L.; Chang, H. Targeting p53 via JNK pathway: A novel role of RITA for apoptotic signaling in multiple myeloma. PLoS ONE 2012, 7, e30215. [Google Scholar] [CrossRef]
- Yogosawa, S.; Yoshida, K. Tumor suppressive role for kinases phosphorylating p53 in DNA damage-induced apoptosis. Cancer Sci. 2018, 109, 3376–3382. [Google Scholar] [CrossRef]
- Weng, Q.; Liu, Z.; Li, B.; Liu, K.; Wu, W.; Liu, H. Oxidative Stress Induces Mouse Follicular Granulosa Cells Apoptosis via JNK/FoxO1 Pathway. PLoS ONE 2016, 11, e0167869. [Google Scholar] [CrossRef]
- Johnson, G.L.; Nakamura, K. The c-jun kinase/stress-activated pathway: Regulation, function and role in human disease. Biochim. Biophys. Acta 2007, 1773, 1341–1348. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- Jiang, H.; Galtes, D.; Wang, J.; Rockman, H.A. G protein-coupled receptor signaling: Transducers and effectors. Am. J. Physiol. Cell Physiol. 2022, 323, C731–C748. [Google Scholar] [CrossRef] [PubMed]
- Syrovatkina, V.; Alegre, K.O.; Dey, R.; Huang, X.Y. Regulation, Signaling, and Physiological Functions of G-Proteins. J. Mol. Biol. 2016, 428, 3850–3868. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, Z.G.; Dhanasekaran, D.N. G protein regulation of MAPK networks. Oncogene 2007, 26, 3122–3142. [Google Scholar] [CrossRef] [PubMed]
- Kraus, S.; Benard, O.; Naor, Z.; Seger, R. C-Src is Activated by the EGF Receptor in a Pathway that Mediates JNK and ERK Activation by Gonadotropin-Releasing Hormone in COS7 Cells. Int. J. Mol. Sci. 2020, 21, 8575. [Google Scholar] [CrossRef]
- Schattauer, S.S.; Bedini, A.; Summers, F.; Reilly-Treat, A.; Andrews, M.M.; Land, B.B.; Chavkin, C. Reactive oxygen species (ROS) generation is stimulated by kappa opioid receptor activation through phosphorylated c-Jun N-terminal kinase and inhibited by p38 mitogen-activated protein kinase (MAPK) activation. J. Biol. Chem. 2019, 294, 16884–16896. [Google Scholar] [CrossRef]
- Yamauchi, J.; Hirasawa, A.; Miyamoto, Y.; Itoh, H.; Tsujimoto, G. Beta2-adrenergic receptor/cyclic adenosine monophosphate (cAMP) leads to JNK activation through Rho family small GTPases. Biochem. Biophys. Res. Commun. 2001, 284, 1199–1203. [Google Scholar] [CrossRef]
- Nishida, M.; Tanabe, S.; Maruyama, Y.; Mangmool, S.; Urayama, K.; Nagamatsu, Y.; Takagahara, S.; Turner, J.H.; Kozasa, T.; Kobayashi, H.; et al. G alpha 12/13- and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J. Biol. Chem. 2005, 280, 18434–18441. [Google Scholar] [CrossRef]
- Smrcka, A.V.; Fisher, I. G-protein betagamma subunits as multi-functional scaffolds and transducers in G-protein-coupled receptor signaling. Cell. Mol. Life Sci. 2019, 76, 4447–4459. [Google Scholar] [CrossRef]
- Khan, S.M.; Sung, J.Y.; Hebert, T.E. Gbetagamma subunits-Different spaces, different faces. Pharmacol. Res. 2016, 111, 434–441. [Google Scholar] [CrossRef]
- Lai, X.; Ye, L.; Liao, Y.; Jin, L.; Ma, Q.; Lu, B.; Sun, Y.; Shi, Y.; Zhou, N. Agonist-induced activation of histamine H3 receptor signals to extracellular signal-regulated kinases 1 and 2 through PKC-, PLD-, and EGFR-dependent mechanisms. J. Neurochem. 2016, 137, 200–215. [Google Scholar] [CrossRef]
- Madukwe, J.C.; Garland-Kuntz, E.E.; Lyon, A.M.; Smrcka, A.V. G protein betagamma subunits directly interact with and activate phospholipase CE. J. Biol. Chem. 2018, 293, 6387–6397. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, J.; Kaziro, Y.; Itoh, H. Differential regulation of mitogen-activated protein kinase kinase 4 (MKK4) and 7 (MKK7) by signaling from G protein beta gamma subunit in human embryonal kidney 293 cells. J. Biol. Chem. 1999, 274, 1957–1965. [Google Scholar] [CrossRef]
- Gautam, J.; Banskota, S.; Shah, S.; Jee, J.G.; Kwon, E.; Wang, Y.; Kim, D.Y.; Chang, H.W.; Kim, J.A. 4-Hydroxynonenal-induced GPR109A (HCA(2) receptor) activation elicits bipolar responses, G(alphai) -mediated anti-inflammatory effects and G(betagamma) -mediated cell death. Br. J. Pharmacol. 2018, 175, 2581–2598. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yamauchi, J.; Kaziro, Y.; Itoh, H. Activation of c-fos promoter by Gbetagamma-mediated signaling: Involvement of Rho and c-Jun N-terminal kinase. J. Biochem. 1999, 125, 515–521. [Google Scholar] [CrossRef]
- Ubeysinghe, S.; Wijayaratna, D.; Kankanamge, D.; Karunarathne, A. Molecular regulation of PLCbeta signaling. Methods Enzymol. 2023, 682, 17–52. [Google Scholar] [CrossRef] [PubMed]
- Naor, Z.; Benard, O.; Seger, R. Activation of MAPK cascades by G-protein-coupled receptors: The case of gonadotropin-releasing hormone receptor. Trends Endocrinol. Metab. 2000, 11, 91–99. [Google Scholar] [CrossRef]
- Levi, N.L.; Hanoch, T.; Benard, O.; Rozenblat, M.; Harris, D.; Reiss, N.; Naor, Z.; Seger, R. Stimulation of Jun N-terminal kinase (JNK) by gonadotropin-releasing hormone in pituitary alpha T3-1 cell line is mediated by protein kinase C, c-Src, and CDC42. Mol. Endocrinol. 1998, 12, 815–824. [Google Scholar]
- Reiss, N.; Levi, L.N.; Shacham, S.; Harris, D.; Seger, R.; Naor, Z. Mechanism of mitogen-activated protein kinase activation by gonadotropin-releasing hormone in the pituitary of alphaT3-1 cell line: Differential roles of calcium and protein kinase C. Endocrinology 1997, 138, 1673–1682. [Google Scholar] [CrossRef]
- Roberson, M.S.; Zhang, T.; Li, H.L.; Mulvaney, J.M. Activation of the p38 mitogen-activated protein kinase pathway by gonadotropin-releasing hormone. Endocrinology 1999, 140, 1310–1318. [Google Scholar] [CrossRef]
- Harris, D.; Bonfil, D.; Chuderland, D.; Kraus, S.; Seger, R.; Naor, Z. Activation of MAPK cascades by GnRH: ERK and Jun N-terminal kinase are involved in basal and GnRH-stimulated activity of the glycoprotein hormone LHbeta-subunit promoter. Endocrinology 2002, 143, 1018–1025. [Google Scholar] [CrossRef]
- Kraus, S.; Naor, Z.; Seger, R. Intracellular signaling pathways mediated by the gonadotropin-releasing hormone (GnRH) receptor. Arch. Med. Res. 2001, 32, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Bonfil, D.; Chuderland, D.; Kraus, S.; Shahbazian, D.; Friedberg, I.; Seger, R.; Naor, Z. Extracellular signal-regulated kinase, Jun N-terminal kinase, p38, and c-Src are involved in gonadotropin-releasing hormone-stimulated activity of the glycoprotein hormone follicle-stimulating hormone beta-subunit promoter. Endocrinology 2004, 145, 2228–2244. [Google Scholar] [CrossRef] [PubMed]
- Sviridonov, L.; Dobkin-Bekman, M.; Shterntal, B.; Przedecki, F.; Formishell, L.; Kravchook, S.; Rahamim-Ben Navi, L.; Bar-Lev, T.H.; Kazanietz, M.G.; Yao, Z.; et al. Differential signaling of the GnRH receptor in pituitary gonadotrope cell lines and prostate cancer cell lines. Mol. Cell. Endocrinol. 2013, 369, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Dobkin-Bekman, M.; Rahamin-Ben Navi, L.; Shterntal, B.; Sviridonov, L.; Przedecki, F.; Naidich-Exler, M.; Brodie, C.; Seger, R.; Naor, Z. Differential role of PKC isoforms in GnRH and phorbol 12-myristate 13-acetate activation of extracellular signal-regulated kinase and Jun N-terminal kinase. Endocrinology 2010, 151, 4894–4907. [Google Scholar] [CrossRef]
- Nadel, G.; Yao, Z.; Ben-Ami, I.; Naor, Z.; Seger, R. Gq-Induced Apoptosis is Mediated by AKT Inhibition That Leads to PKC-Induced JNK Activation. Cell. Physiol. Biochem. 2018, 50, 121–135. [Google Scholar] [CrossRef]
- Markou, T.; Cieslak, D.; Gaitanaki, C.; Lazou, A. Differential roles of MAPKs and MSK1 signalling pathways in the regulation of c-Jun during phenylephrine-induced cardiac myocyte hypertrophy. Mol. Cell. Biochem. 2009, 322, 103–112. [Google Scholar] [CrossRef]
- Tanaka, Y.; Gavrielides, M.V.; Mitsuuchi, Y.; Fujii, T.; Kazanietz, M.G. Protein kinase C promotes apoptosis in LNCaP prostate cancer cells through activation of p38 MAPK and inhibition of the Akt survival pathway. J. Biol. Chem. 2003, 278, 33753–33762. [Google Scholar] [CrossRef]
- Kraus, S.; Naor, Z.; Seger, R. Gonadotropin-releasing hormone in apoptosis of prostate cancer cells. Cancer Lett. 2006, 234, 109–123. [Google Scholar] [CrossRef]
- Imai, A.; Tamaya, T. GnRH receptor and apoptotic signaling. Vitam. Horm. 2000, 59, 1–33. [Google Scholar] [CrossRef]
- Savulescu, D.; Feng, J.; Ping, Y.S.; Mai, O.; Boehm, U.; He, B.; O’Malley, B.W.; Melamed, P. Gonadotropin-releasing hormone-regulated prohibitin mediates apoptosis of the gonadotrope cells. Mol. Endocrinol. 2013, 27, 1856–1870. [Google Scholar] [CrossRef]
- Maudsley, S.; Davidson, L.; Pawson, A.J.; Chan, R.; Lopez de Maturana, R.; Millar, R.P. Gonadotropin-releasing hormone (GnRH) antagonists promote proapoptotic signaling in peripheral reproductive tumor cells by activating a Galphai-coupling state of the type I GnRH receptor. Cancer Res. 2004, 64, 7533–7544. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Zhang, Q.G.; Han, D.; Xu, J.; Lu, Q.; Zhang, G.Y. Inhibition of MLK3-MKK4/7-JNK1/2 pathway by Akt1 in exogenous estrogen-induced neuroprotection against transient global cerebral ischemia by a non-genomic mechanism in male rats. J. Neurochem. 2006, 99, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed]
- Siess, K.M.; Leonard, T.A. Lipid-dependent Akt-ivity: Where, when, and how. Biochem. Soc. Trans. 2019, 47, 897–908. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Vogel, R.O.; Puigserver, P. Clk2 and B56beta mediate insulin-regulated assembly of the PP2A phosphatase holoenzyme complex on Akt. Mol. Cell 2011, 41, 471–479. [Google Scholar] [CrossRef]
- Ruvolo, P.P.; Qui, Y.H.; Coombes, K.R.; Zhang, N.; Ruvolo, V.R.; Borthakur, G.; Konopleva, M.; Andreeff, M.; Kornblau, S.M. Low expression of PP2A regulatory subunit B55alpha is associated with T308 phosphorylation of AKT and shorter complete remission duration in acute myeloid leukemia patients. Leukemia 2011, 25, 1711–1717. [Google Scholar] [CrossRef]
- Shouse, G.; de Necochea-Campion, R.; Mirshahidi, S.; Liu, X.; Chen, C.S. Novel B55alpha-PP2A mutations in AML promote AKT T308 phosphorylation and sensitivity to AKT inhibitor-induced growth arrest. Oncotarget 2016, 7, 61081–61092. [Google Scholar] [CrossRef]
- Nadel, G.; Yao, Z.; Wainstein, E.; Cohen, I.; Ben-Ami, I.; Schajnovitz, A.; Maik-Rachline, G.; Naor, Z.; Horwitz, B.A.; Seger, R. GqPCR-stimulated dephosphorylation of AKT is induced by an IGBP1-mediated PP2A switch. Cell Commun. Signal. 2022, 20, 5. [Google Scholar] [CrossRef]
- Goldberg, Y. Protein phosphatase 2A: Who shall regulate the regulator? Biochem. Pharmacol. 1999, 57, 321–328. [Google Scholar] [CrossRef]
- Dhand, R.; Hiles, I.; Panayotou, G.; Roche, S.; Fry, M.J.; Gout, I.; Totty, N.F.; Truong, O.; Vicendo, P.; Yonezawa, K.; et al. PI 3-kinase is a dual specificity enzyme: Autoregulation by an intrinsic protein-serine kinase activity. EMBO J. 1994, 13, 522–533. [Google Scholar] [CrossRef]
- Sents, W.; Ivanova, E.; Lambrecht, C.; Haesen, D.; Janssens, V. The biogenesis of active protein phosphatase 2A holoenzymes: A tightly regulated process creating phosphatase specificity. FEBS J. 2013, 280, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Stanevich, V.; Satyshur, K.A.; Kong, M.; Watkins, G.R.; Wadzinski, B.E.; Sengupta, R.; Xing, Y. Structural basis of protein phosphatase 2A stable latency. Nat. Commun. 2013, 4, 1699. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.T.; Chiang, S.J.; Lai, T.Y.; Cheng, Y.L.; Chung, C.E.; Kuo, S.C.; Reece, K.M.; Chen, Y.C.; Chang, N.S.; Wadzinski, B.E.; et al. Visualization of subunit interactions and ternary complexes of protein phosphatase 2A in mammalian cells. PLoS ONE 2014, 9, e116074. [Google Scholar] [CrossRef] [PubMed]
- Prickett, T.D.; Brautigan, D.L. Overlapping binding sites in protein phosphatase 2A for association with regulatory A and alpha-4 (mTap42) subunits. J. Biol. Chem. 2004, 279, 38912–38920. [Google Scholar] [CrossRef]
- Carbone, A.M.; Borges, J.I.; Suster, M.S.; Sizova, A.; Cora, N.; Desimine, V.L.; Lymperopoulos, A. Regulator of G-Protein Signaling-4 Attenuates Cardiac Adverse Remodeling and Neuronal Norepinephrine Release-Promoting Free Fatty Acid Receptor FFAR3 Signaling. Int. J. Mol. Sci. 2022, 23, 5803. [Google Scholar] [CrossRef]
- Wang, Q.; Liu-Chen, L.Y.; Traynor, J.R. Differential modulation of mu- and delta-opioid receptor agonists by endogenous RGS4 protein in SH-SY5Y cells. J. Biol. Chem. 2009, 284, 18357–18367. [Google Scholar] [CrossRef]
- Bastin, G.; Dissanayake, K.; Langburt, D.; Tam, A.L.C.; Lee, S.H.; Lachhar, K.; Heximer, S.P. RGS4 controls Galphai3-mediated regulation of Bcl-2 phosphorylation on TGN38-containing intracellular membranes. J. Cell Sci. 2020, 133, jcs231034. [Google Scholar] [CrossRef]
- Kuhar, J.R.; Bedini, A.; Melief, E.J.; Chiu, Y.C.; Striegel, H.N.; Chavkin, C. Mu opioid receptor stimulation activates c-Jun N-terminal kinase 2 by distinct arrestin-dependent and independent mechanisms. Cell. Signal. 2015, 27, 1799–1806. [Google Scholar] [CrossRef]
- Kook, S.; Zhan, X.; Kaoud, T.S.; Dalby, K.N.; Gurevich, V.V.; Gurevich, E.V. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J. Biol. Chem. 2013, 288, 37332–37342. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nadel, G.; Maik-Rachline, G.; Seger, R. JNK Cascade-Induced Apoptosis—A Unique Role in GqPCR Signaling. Int. J. Mol. Sci. 2023, 24, 13527. https://doi.org/10.3390/ijms241713527
Nadel G, Maik-Rachline G, Seger R. JNK Cascade-Induced Apoptosis—A Unique Role in GqPCR Signaling. International Journal of Molecular Sciences. 2023; 24(17):13527. https://doi.org/10.3390/ijms241713527
Chicago/Turabian StyleNadel, Guy, Galia Maik-Rachline, and Rony Seger. 2023. "JNK Cascade-Induced Apoptosis—A Unique Role in GqPCR Signaling" International Journal of Molecular Sciences 24, no. 17: 13527. https://doi.org/10.3390/ijms241713527